

Abstract
Bisphosphonates stop bone loss by inhibiting the activity of bone resorbing osteoclasts. However, the effect of bisphosphonates on bone mass cannot completely explain the reduction in fracture incidence observed in patients treated with these agents. Recent research efforts provided an explanation to this dichotomy by demonstrating that part of the beneficial effect of bisphosphonates on the skeleton is due to prevention of osteoblast and osteocyte apoptosis. Work of our group, independently confirmed by other investigators, demonstrated that bisphosphonates are able to prevent osteoblast and osteocyte apoptosis in vitro and in vivo. This pro-survival effect is strictly dependent on the expression of connexin (Cx)43, as demonstrated in vitro using cells lacking Cx43 or expressing dominant negative mutants of the protein as well as in vivo using Cx43 osteoblast/osteocyte-specific conditional knock-out mice. Remarkably, this Cx43-dependent survival effect of bisphosphonates is independent of gap junctions and results from opening of Cx43 hemichannels. Hemichannel opening leads to activation of the kinases Src and extracellular signal-regulated kinases (ERKs), followed by phosphorylation of the ERK cytoplasmic target p90RSK kinase and its substrates BAD and C/EBPβ, resulting in inhibition of apoptosis. The anti-apoptotic effect of bisphosphonates is separate from the effect of the drugs on osteoclasts, as analogs that lack anti-resorptive activity are still able to inhibit osteoblast and osteocyte apoptosis in vitro. Furthermore, a bisphosphonate analog that does not inhibit osteoclast activity prevented osteoblast and osteocyte apoptosis and the loss of bone mass and strength induced by glucocorticoids in mice. Preservation of the bone forming function of mature osteoblasts and maintenance of the osteocytic network, in combination with lack anti-catabolic actions, open new therapeutic possibilities for bisphosphonates in the treatment of osteopenic conditions in which decreased bone resorption is not desired.
Keywords: bisphosphonate, osteoblast, osteocyte, apoptosis, ERKs, connexin43
Introduction
Bone fragility leading to fractures and disability is associated with the bone disease caused by glucocorticoid excess, sex steroid deficiency, and advanced age. Therefore, preservation of bone strength is key in the management of these conditions. Whereas the majority of bone strength is determined by the amount of mineral, about 40% is due to other factors including bone size, the architecture of cortical and cancellous compartments, organization of the mineral and organic components of the bone matrix, presence of microdamage, as well as the prevalence of osteocyte apoptosis [1, 2]. Anti-resorptive agents, best exemplified by bisphosphonates, maintain bone strength further than what is expected based on the moderate increase in bone mineral density (BMD) that they induce [3, 4]. The discovery that bisphosphonates have direct anti-apoptotic effects on osteoblasts and osteocytes, summarized in this review, provides the basis for the additional beneficial actions of these compounds beyond inhibiting osteoclast activity and halting the loss of bone mineral.
Effects of bisphosphonates on osteoblastic cell function
Bisphosphonates inhibit resorption by acting on osteoclasts to reduce their activity or to increase the rate of apoptosis [5]. As a consequence of this inhibitory effect on osteoclast function, bone formation coupled to resorption is also decreased resulting in an overall reduction in the rate of bone remodeling [6]. Several pieces of evidence support the notion that bisphosphonates also influence directly the function of cells of the osteoblastic lineage, and that this effect may in turn contribute to the reduction in osteoclast formation and activity (Table 1). Thus, early studies showed that osteoblastic cells derived from neonatal mice treated with the bisphosphonate incadronate inhibit the differentiation of bone marrow or spleen osteoclast precursors into mature osteoclasts [7]. Similarly, osteoblastic CRP10/30 cells pre-treated with various bisphosphonates inhibited resorption by rat mature osteoclasts [8]. These inhibitory effects could be reproduced by conditioned media from treated osteoblastic cells, suggesting that bisphosphonates promote the release of factor(s) that inhibit osteoclast formation and activity in vitro [7-9]. Consistent with these early findings, bisphosphonates have been recently shown to decrease the expression of the receptor activator of NF-kB ligand (RANKL) and increase the expression of the RANKL decoy receptor osteoprotegerin (OPG) in human osteoblastic cells [10, 11]. Taken together these findings suggest the possibility that the anti-resorptive effect of bisphosphonates mediated by osteoblastic cells is due to interference with the pro-differentiation and survival of osteoclasts induced by RANKL signaling.
Table 1. Effect of bisphosphonates on osteoblastic cells.
| effect | cell model | |
|---|---|---|
| osteoblast-mediated inhibition of osteoclast differentiation and resorption |
decreased osteoclasts and resorption |
murine calvaria cells [7], rat CRP 10/30 cells [8, 9] |
| reduced RANKL and increased OPG expression |
primary human osteoblastic cells [10, 11] | |
| osteoblast proliferation |
increased | primary human [19, 21, 22] and rat [23] osteoblastic cells, human MG-63 [22, 24] and rat ROS17/2.8 cells |
| decreased | murine MC3T3-E1 cells [13], human fetal osteoblastic cells [35], primary murine [27] and rat [28] calvaria cells, primary human adult [30, 31, 33] and fetal osteoblastic cells [32] |
|
| osteoblast differentiation and/or mineralization |
increased | chick periosteal calvaria explants [12], murine and human bone marrow cells [15], rat calvaria bone [14], primary human osteoblastic cells [10, 19, 21, 22, 25], rat organ cultures [17], mouse [20] and human [34] mesenchymal stem cells, human osteoblast-like periosteal cells [26], murine MC3T3-E1 [13, 16, 17] and ST2 [17] cells, human Saos-2 [18] and MG-63 [22, 24] cells |
| decreased | human mesenchymal stem cells [34], primary human osteoblastic cells [10, 30, 31, 33], murine [27] and rat [28] calvaria cells, human bone marrow stromal cells [29], chick periosteal calvaria explants [12] |
|
| osteoblast/osteocyte apoptosis |
inhibited | Murine MLO-Y4 osteocytic [58, 59, 62, 65], MC3T3E1 [64], Ob-6 [59] cells, embryonic fibroblasts [61], and primary calvaria cells [58, 61], human MG63 [63] and primary osteoblast-like [63] cells, rat ROS17/2.8 [61] and UMR106 [64] cells |
| increased | primary human adult [19, 25] and fetal osteoblastic [35] cells, human bone marrow stromal cells [29], primary murine calvaria cells [27] |
|
Several pieces of evidence indicate that bisphosphonates modulate the proliferation and differentiation rates of cells of the osteoblastic lineage (Table 1). Some studies reveal that culture of osteoblastic cells in the presence of bisphosphonates increases proliferation, stimulates differentiation towards the osteoblastic lineage, and enhances mineralization [12-25]. Other studies show that the drugs decrease proliferation and inhibit osteoblast differentiation and mineralization [26-34], or that they inhibit proliferation and increased differentiation [35]. This apparent contradiction is explained by the fact that growth and pro-differentiation effects are exerted at lower concentrations of bisphosphonates ranging from 10−9M to 10−6M, whereas the inhibitory effects are found at concentrations higher than 10−5M.
In vivo administration of bisphosphonates has been also shown to promote early osteoblastogenesis in mice [15] and to increase the expression of osteoblast differentiation markers and promote calvarial wound closure in rats [14]. In addition, suppression of bone turnover by bisphosphonates increases the mineral and matrix bone tissue maturity [36]. Moreover, long term administration of bisphosphonates to dogs, non-human primates, and humans increases wall thickness, an index of focally elevated osteoblast numbers or activity, not explained by a reduction in the remodeling space [37-40]. Consistent with an increase in osteoblast activity, humans treated for 3 years with zoledronic acid exhibit increased mineral apposition rate [41]. However, other reports failed to show a change in mineral apposition rate patients treated with alendronate or pamidronate [37, 42]. Overall, this evidence suggests that bisphosphonates have a positive effect on bone formation, even in the face of reduced overall bone remodeling; thus, where osteoblasts are present, more bone is made.
Apoptosis and bone homeostasis
Studies during the last decade have demonstrated that apoptosis plays a central role in the maintenance of skeletal mass and strength and have uncovered novel molecular mechanisms involved in its regulation in bone cells [43]. It is now known that all osteoclasts die by apoptosis after completing a bone resorption cycle; that the majority of osteoblasts also die whereas the remainders become lining cells or osteocytes; and that osteocytes also can die prematurely. Furthermore, it is now recognized that systemic hormones, local growth factors, cytokines and pharmacological agents, as well as physical stimuli such as mechanical forces regulate the rate of bone cell apoptosis.
Changes in the prevalence of osteoblast apoptosis could have a significant impact in the number of osteoblasts present at sites of bone formation and their function. Thus, it is now appreciated that increased osteoblast apoptosis is at least partially responsible for the decreased bone formation rate associated with the osteopenia induced by glucocorticoid excess [44]; and, conversely, inhibition of osteoblast apoptosis might contribute to the anabolic effect of intermittent administration of PTH [45, 46]. In addition, mice overexpressing the anti-apoptotic protein Bcl-2 under the control of the 2.3kb fragment of the collagen1a1 promoter (col2.3kb-Bcl-2 mice) exhibit increased BV/TV in the distal femur compared to WT mice and are protected from bone loss with age, adding evidence for a beneficial effect of preventing osteoblast apoptosis [47]. However, young mice lacking caspase-3, crucial molecule for apoptosis, exhibit decreased (instead of increased) BV/TV [48]. Therefore, further investigations are warranted to clarify the relationship between osteoblast apoptosis and bone mass in vivo. Studies using cultured rat calvaria cells demonstrate that apoptosis increases during bone-like tissue development in vitro [49], consisting with apoptosis being one of the fate of differentiating osteoblasts. Interestingly, calvaria cells isolated from col2.3kb-Bcl-2 mice exhibit increased adhesion and expression of osteoblast-specific genes, but reduced mineralization [50], suggesting a inverse relationship between osteoblast survival and their mineralization capability. Nevertheless, this appear not to be the case with bisphosphonates, as administration of the drugs induce an increase, rather than decrease bone mineralization [36].
In addition, osteocytes, mature osteoblasts embedded in the mineralized matrix and responsible for the adaptive response of the skeleton to both systemic and mechanical stimuli [51], also die by apoptosis [43]. Moreover, osteocyte apoptosis is increased in conditions of increased bone fragility, such as osteoporosis due to excess of glucocorticoids [44] and sex steroid deficiency [52, 53]. Furthermore, reduced mechanical loading in mice leads to osteocyte apoptosis, suggesting that physiologic levels of strain provide survival signals to osteocytes [54]. The progressive loss of bone mass and strength that ensues with age is also associated with increased osteocyte apoptosis [55]. Conversely, osteocyte apoptosis is prevented by mechanical stimulation and by sex steroids and PTH in vitro and in vivo [45, 52, 53, 56, 57].
Earlier work by our group revealed that the beneficial effects of bisphosphonates on bone are accompanied by enhanced viability of osteoblasts and osteocytes. Thus, the bisphosphonate alendronate prevented the increase in osteoblast and osteocyte apoptosis induced by glucocorticoids in mice [58, 59]. More recently, risedronate and alendronate were also shown to inhibit osteocyte apoptosis in this case induced by fatigue cyclic loading in rats [60]. Delayed osteoblast apoptosis might result in an increased work time of osteoblasts leading to the gradual increase in trabecular thickness observed in animals or patients upon long-term treatment with bisphosphonates [37-40]. Preservation of osteocyte viability might contribute to the maintenance of the osteocyte network. Both effects could explain part of the anti-fracture efficacy of bisphosphonates.
Bisphosphonates affect osteoblasts/osteocytes and osteoclasts by different mechanisms
Our earlier work [58] as well as more recent evidence by us and other investigators [59, 61-65] established that protection from apoptosis results from direct activation of survival signaling triggered by bisphosphonates on osteoblastic and osteocytic cells (Table 1). Mechanistic studies using osteocytic MLO-Y4 cells and primary murine calvaria cells demonstrated that bisphosphonates inhibit apoptosis induced by the glucocorticoid dexamethasone, the inhibitor of DNA repair etoposide that blocks topo-isomerase II activity, or TNFα, an activator of death receptors, [58] suggesting that bisphosphonates inhibits cell death, independently of the pro-apoptotic pathway activated.
As reviewed elsewhere in this issue, the mechanism by which bisphosphonates affect osteoclasts involves intracellular accumulation of high concentrations of the drugs. In the case of amino-bisphosphonates, this leads to inhibition of farnesyl pyrophosphate synthase, an enzyme crucial for osteoclast function [66]; whereas for non-amino-bisphosphonates, it results in the formation of toxic metabolites. In both cases, intracellular accumulation of the drugs inhibits osteoclast function and can lead to osteoclast apoptosis (Figure 1). In contrast to their actions on osteoclasts, the pro-survival effect of bisphosphonates on cells of the osteoblastic lineage is exerted at concentrations ranging from 10−11 to 10−6 M, which are several orders of magnitude lower than the ones required to inhibit osteoclast activity [66-69]. Even though bisphosphonates bind with high affinity to mineral, there is a continuous release of low levels of the drugs (as discussed elsewhere in this issue). This may result in a constant supply of bisphosphonates at low concentrations, which could maintain osteoblast and osteocyte viability. Remarkably, this survival effect is lost at high concentrations. Similar biphasic pattern of responsiveness has been shown for other responses of osteoblastic cells to the drugs, including the increases in proliferation, stimulation of formation of colony forming units for fibroblasts and osteoblasts, as well as stimulation of mineralization and alkaline phosphatase activity [12, 15, 23].
Figure 1. Bisphosphonates prevent osteoblast and osteocyte apoptosis and inhibit osteoclast activity via different mechanisms.
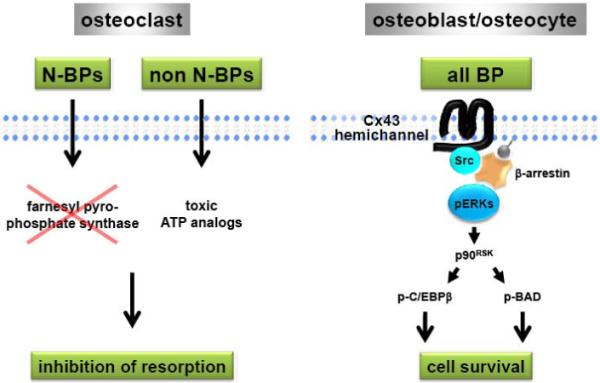
For simplicity, only one of the six molecules of Cx43 that form the hemichannel is shown. N-BPs, amino-bisphosphonates; non N-BPs, non amino-bisphosphonates. See text for details.
Another difference between the action of bisphosphonates on osteoblastic cells versus osteoclasts is that pro-survival of osteoblasts and osteocytes is exerted by all bisphosphonates, non-amino as well as amino-analogs, at similar effective doses (EC50 ~ 10−10M) [58, 69]. Remarkably, anti-apoptosis by bisphosphonates appear to be unrelated to the anti-resorptive properties of these agents, as IG9402, NE11809, and several other bisphosphonates that do not affect osteoclast activity, also exhibit anti-apoptotic properties on osteoblastic and osteocytic cells in vitro [58, 62, 67-69]. As discussed below, recent evidence demonstrates that administration to mice of IG9402, one of these compounds that do not affect osteoclasts, prevented the increase in osteoblast and osteocyte apoptosis induced by glucocorticoid excess as effectively as alendronate without decreasing resorption (refer to manuscript to be submitted for this issue).
All this evidence indicates that the pro-survival effect of bisphosphonates on cells of the osteoblastic lineage is mediated by a different mechanism of action than the one by which these drugs affect osteoclasts; and that the actions on osteoblasts and osteocytes can be dissociated from the ones on osteoclasts with novel analogs.
How do bisphosphonates trigger survival signaling in osteoblastic cells?
Studies using cultured cells showed that bisphosphonates induce the activation of the ERKs, recognized survival kinases (Figure 1). Thus, all bisphosphonates examined rapidly induce phosphorylation of ERKs in osteocytic cells as well as primary cultures of osteoblastic cells, independently of their structure, presence of amino residues in the molecule, or ability to inhibit osteoclast function [58]. Increased levels of phosphorylated ERKs were found also in bones of animals that had received a single injection of alendronate or IG9402 (shown elsewhere in this issue). Mechanistic studies have demonstrated that ERK activation is required for the prevention of osteoblast and osteoclast apoptosis by bisphosphonates, as pharmacological or genetic inhibition of MEK, the kinase that activates ERKs, abolished the anti-apoptotic effect of bisphosphonates [58, 61].
Similar to bisphosphonates, ligand-induced activation of sex steroid receptors also attenuates apoptosis of osteoblasts and osteocytes by a mechanism that requires ERK activation [53]. However, the molecular events that lead to activation of these survival kinases by estrogens or androgens are different from the ones involved in bisphosphonate action. Thus, the sex steroids activate ERKs via an extra-nuclear function of their receptors that can be mediated by the ligand binding domain. In addition, kinase-dependent activation of transcription factors and gene transcription is required for the anti-apoptotic effect of sex steroids on osteoblastic cells [70, 71]. On the other hand, anti-apoptosis induced by bisphosphonates requires neither transcription nor ERK-dependent transcription factors [70]. Instead, ERK-mediated anti-apoptosis induced by bisphosphonates requires the kinase activity of the cytoplasmic target of ERKs, p90RSK, which in turn phosphorylates the pro-apoptotic protein BAD and the CCAAT/enhancer binding protein C/EBPβ. Phosphorylation of BAD renders it inactive, whereas phosphorylation of C/EBPβ leads to binding of pro-caspases, thus inhibiting apoptosis independently of the transcriptional activity of this transcription factor (Figure 1). Consistent with the evidence that estrogens and bisphosphonates phosphorylate diverse targets of ERKs probably resulting from activation of spatially distinct pools of these kinases, the two agents had additive effects on osteocyte survival [70].
Cx43 hemichannel opening by bisphosphonates leads to activation of the Src/ERK signaling pathway in osteoblasts and osteocytes
Binding of respective ligands to receptors for growth factors, cytokines, and hormones as well as engagement of integrins by extracellular matrix proteins, trigger molecular events that culminate in ERK activation. In the case of bisphosphonates, no receptors have been identified. Therefore, we probed into the mechanism by which these drugs activate ERKs by exploring the possibility that bisphosphonates affected the permeability of channels formed by connexin (Cx) 43, the major member of the connexin family of proteins expressed in osteoblasts and osteocytes [72-75].
Connexin hexamers form plasma membrane channels, named hemichannels. Hemichannels of adjacent cells can align to form a gap junction channel. Intercellular communication among osteocytes and with osteoblasts is maintained through gap junctions: clusters of thousands of channels that open only transiently to allow the exchange of small (<1 kDalton) water-soluble molecules [76]. Hemichannels are also present in unopposed cell membranes and their opening allows exchange of cytoplasmic contents with the extracellular fluid [76, 77].
Using mouse embryonic fibroblasts, osteoblastic cell lines, and authentic osteoblasts expressing or deficient in Cx43, we demonstrated that Cx43 is required for induction of cell survival by bisphosphonates [61]. Furthermore, we found that Cx43 is present in non-junctional osteocyte membranes, and that exposure to bisphosphonates of osteoblastic and osteocytic cells results in opening of hemichannels without affecting gap junction communication. Whether bisphosphonates, due to their small molecular size, enter osteoblastic cells upon inducing opening of the hemichannels still remains unknown.
Transfection of Cx43, but not other connexins, into Cx43-naïve cells was also able to confer de novo responsiveness to bisphosphonates [61]. By transfecting mutants of Cx43, we found that the signal-transducing property of Cx43 requires the trans-membrane pore-forming region of the protein. In addition, survival by bisphosphonates depends on the presence of the C-terminal cytoplasmic domain of Cx43, by which the protein interacts with Src kinase, an upstream activator of ERKs. Using genetic and pharmacological approaches, we determined that Src expression, its kinase activity, and the SH2 and SH3 domains are required for ERK activation and prevention of osteoblast and osteocyte apoptosis by bisphosphonates. In more recent work, we have shown that Cx43 forms a complex with β-arrestin that retains activated ERKs in the cytoplasm, enabling exclusive activation of ERK cytoplasmic substrates, such as p90RSK [78]. Cx43 is also required for the protective effect of bisphosphonates on osteoblastic cells in vivo, as alendronate was not able to prevent apoptosis in mice lacking Cx43 in osteoblasts and osteocytes [59]. This evidence adds Cx43 to the list of trans-membrane molecules capable of transducing survival signals in response to extracellular cues and raises the possibility that Cx43 may serve in this capacity for endogenously produced molecules or even other drugs [61, 77].
Bisphosphonates have been shown to specifically bind to osteoblastic cells [79]. The requirement of Cx43 for anti-apoptosis by bisphosphonates have raised the possibility that interaction of bisphosphonates with Cx43 present in the cell membrane results in hemichannel opening, thereby initiating intracellular survival signaling. However, we found that, although Cx43 is necessary for bisphosphonate-induced survival of osteoblastic cells, this protein is dispensable for bisphosphonate cell binding [80]. Thus, bisphosphonates can bind to cells lacking Cx43 or cells in which connexin channels have been disrupted, suggesting that the drugs bind to another moiety that, in turn, interacts with Cx43. Because bisphosphonate binding to osteoblastic cells is displaced by protein phosphatase substrates, the unknown molecule which links bisphosphonates with Cx43 could be a phosphatase. Future studies need to be pursued towards the identification of bisphosphonate receptor proteins in osteoblastic cells.
A bisphosphonate that does not affect osteoclasts prevents osteoblast and osteocyte apoptosis and the loss of bone strength induced by glucocorticoids
We tested directly whether prevention of osteoblast and osteocyte apoptosis by bisphosphonates, in the absence of an anti-resorptive effect, had a beneficial effect on the skeleton. Towards this end, we examined the effect on glucocorticoid-induced bone disease of administration of the bisphosphonate analog IG9402, which we have previously shown in vitro that prevents osteoblast and osteocyte apoptosis but does not decrease osteoclast viability [69]. As shown in this issue in a separate manuscript (refer to manuscript submitted), unlike alendronate that decreases circulating levels of bone resorption and formation markers, administration of IG9402 did not affect bone remodeling. In full agreement with the in vitro data and in spite of its lack of anti-remodeling effect, IG9402 was as effective as alendronate in preventing glucocorticoid-induced osteoblast and osteocyte apoptosis. Moreover, the decrease in bone mineral density, bone volume, osteoblast number and bone formation rate induced by glucocorticoids in vertebral cancellous bone were all preserved by IG9402. In addition, the ability of IG9402 to protect osteoblasts and osteocytes from the pro-apoptotic effect of glucocorticoids was associated with preservation of bone strength as assessed by mechanical testing. These findings demonstrate that osteoblast and osteocyte apoptosis can be selectively prevented by bisphosphonate analogs, opening new avenues for the treatment of bone fragility without significantly affecting the rate of bone remodeling. The outcome of long-term administration of these analogs warrants further investigation.
Acknowledgements
The authors thank Stavros C. Manolagas, Robert S. Weinstein, A. Michael Parfitt, and other members of the UAMS Center for Osteoporosis and Metabolic Bone Diseases for advice and insightful discussions. This research was supported by grants from the National Institutes of Health (R01-AR053643, KO2-AR02127, R03 TW006919, R01-DK076007, and P01-AG13918).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Weinstein RS. True strength. J. Bone Min. Res. 2000;15:621–5. doi: 10.1359/jbmr.2000.15.4.621. [DOI] [PubMed] [Google Scholar]
- [2].Felsenberg D, Boonen S. The bone quality framework: determinants of bone strength and their interrelationships, and implications for osteoporosis management. Clin. Ther. 2005;27:1–11. doi: 10.1016/j.clinthera.2004.12.020. [DOI] [PubMed] [Google Scholar]
- [3].Cummings SR, Karpf DB, Harris F, Genant HK, Ensrud K, LaCroix AZ, Black DM. Improvement in spine bone density and reduction in risk of vertebral fractures during treatment with antiresorptive drugs. Am. J. Med. 2002;112:281–9. doi: 10.1016/s0002-9343(01)01124-x. [DOI] [PubMed] [Google Scholar]
- [4].Watts NB, Cooper C, Lindsay R, Eastell R, Manhart MD, Barton IP, Van Staa TP, Adachi JD. Relationship between changes in bone mineral density and vertebral fracture risk associated with risedronate: greater increases in bone mineral density do not relate to greater decreases in fracture risk. J Clin. Densitom. 2004;7:255–61. doi: 10.1385/jcd:7:3:255. [DOI] [PubMed] [Google Scholar]
- [5].Rogers MJ. From molds and macrophages to mevalonate: a decade of progress in understanding the molecular mode of action of bisphosphonates. Calcif. Tissue Int. 2004;75:451–61. doi: 10.1007/s00223-004-0024-1. [DOI] [PubMed] [Google Scholar]
- [6].Papapoulos SE. Bisphosphonates for Postmenopausal Osteoporosis. In: Rosen C, editor. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 2009. pp. 237–41. [Google Scholar]
- [7].Nishikawa M, Akatsu T, Katayama Y, Yasutomo Y, Kado S, Kugal N, Yamamoto M, Nagata N. Bisphosphonates act on osteoblastic cells and inhibit osteoclast formation in mouse marrow cultures. Bone. 1996;18:9–14. doi: 10.1016/8756-3282(95)00426-2. [DOI] [PubMed] [Google Scholar]
- [8].Sahni M, Guenther HL, Fleisch H, Collin P, Martin TJ. Bisphosphonates act on rat bone resorption through the mediation of osteoblasts. J. Clin. Invest. 1993;91:2004–11. doi: 10.1172/JCI116422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Vitte C, Fleisch H, Guenther HL. Bisphosphonates induce osteoblasts to secrete an inhibitor of osteoclast-mediated resorption. Endocrinology. 1996;137:2324–33. doi: 10.1210/endo.137.6.8641182. [DOI] [PubMed] [Google Scholar]
- [10].Viereck V, Emons G, Lauck V, Frosch KH, Blaschke S, Grundker C, Hofbauer LC. Bisphosphonates pamidronate and zoledronic acid stimulate osteoprotegerin production by primary human osteoblasts. Biochem. Biophys. Res. Commun. 2002;291:680–6. doi: 10.1006/bbrc.2002.6510. [DOI] [PubMed] [Google Scholar]
- [11].Pan B, Farrugia AN, To LB, Findlay DM, Green J, Lynch K, Zannettino AC. The nitrogen-containing bisphosphonate, zoledronic acid, influences RANKL expression in human osteoblast-like cells by activating TNF-alpha converting enzyme (TACE) J. Bone Miner. Res. 2004;19:147–54. doi: 10.1359/jbmr.2004.19.1.147. [DOI] [PubMed] [Google Scholar]
- [12].Tenenbaum HC, Torontali M, Sukhu B. Effects of bisphosphonates and inorganic pyrophosphate on osteogenesis in vitro. Bone. 1992;13:249–55. doi: 10.1016/8756-3282(92)90205-b. [DOI] [PubMed] [Google Scholar]
- [13].Igarashi K, Hirafuji M, Adachi H, Shinoda H, Mitani H. Effects of bisphosphonates on alkaline phosphatase activity, mineralization, and prostaglandin E2 synthesis in the clonal osteoblast-like cell line MC3T3-E1. Prostaglandins Leukot. Essent. Fatty Acids. 1997;56:121–5. doi: 10.1016/s0952-3278(97)90508-1. [DOI] [PubMed] [Google Scholar]
- [14].D’Aoust P, McCulloch CA, Tenenbaum HC, Lekic PC. Etidronate (HEBP) promotes osteoblast differentiation and wound closure in rat calvaria. Cell Tissue Res. 2000;302:353–63. doi: 10.1007/s004419900165. [DOI] [PubMed] [Google Scholar]
- [15].Giuliani N, Pedrazzoni M, Negri G, Passeri G, Impicciatore M, Girasole G. Bisphosphonates stimulate formation of osteoblast precursors and mineralizad nodules in murine and human bone marrow cultures in vitro and promote early osteoblastogenesis in young and aged mice in vivo. Bone. 1998;22:455–61. doi: 10.1016/s8756-3282(98)00033-7. [DOI] [PubMed] [Google Scholar]
- [16].Fujita T, Izumo N, Fukuyama R, Meguro T, Yasutomi C, Nakamuta H, Koida M. Incadronate and etidronate accelerate phosphate-primed mineralization of MC4 cells via ERK1/2-Cbfa1 signaling pathway in a Ras-independent manner: further involvement of mevalonate-pathway blockade for incadronate. Jpn. J. Pharmacol. 2001;86:86–96. doi: 10.1254/jjp.86.86. [DOI] [PubMed] [Google Scholar]
- [17].Itoh F, Aoyagi S, Furihata-Komatsu H, Aoki M, Kusama H, Kojima M, Kogo H. Clodronate stimulates osteoblast differentiation in ST2 and MC3T3-E1 cells and rat organ cultures. Eur. J Pharmacol. 2003;477:9–16. doi: 10.1016/j.ejphar.2003.08.011. [DOI] [PubMed] [Google Scholar]
- [18].Chaplet M, Detry C, Deroanne C, Fisher LW, Castronovo V, Bellahcene A. Zoledronic acid up-regulates bone sialoprotein expression in osteoblastic cells through Rho GTPase inhibition. Biochem. J. 2004;384:591–8. doi: 10.1042/BJ20040380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Frediani B, Spreafico A, Capperucci C, Chellini F, Gambera D, Ferrata P, Baldi F, Falsetti P, Santucci A, Bocchi L, Marcolongo R. Long-term effects of neridronate on human osteoblastic cell cultures. Bone. 2004;35:859–69. doi: 10.1016/j.bone.2004.06.001. [DOI] [PubMed] [Google Scholar]
- [20].Kim HK, Kim JH, Abbas AA, Yoon TR. Alendronate enhances osteogenic differentiation of bone marrow stromal cells: a preliminary study. Clin. Orthop. Relat Res. 2009;467:3121–8. doi: 10.1007/s11999-008-0409-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Fromigue O, Body JJ. Bisphosphonates influence the proliferation and the maturation of normal human osteoblasts. J. Endocrinol. Invest. 2002;25:539–46. doi: 10.1007/BF03345497. [DOI] [PubMed] [Google Scholar]
- [22].Im GI, Qureshi SA, Kenney J, Rubash HE, Shanbhag AS. Osteoblast proliferation and maturation by bisphosphonates. Biomaterials. 2004;25:4105–15. doi: 10.1016/j.biomaterials.2003.11.024. [DOI] [PubMed] [Google Scholar]
- [23].Mathov I, Plotkin LI, Sgarlata CL, Leoni J, Bellido T. Extracellular signal-regulated kinases and calcium channels are involved in the proliferative effect of bisphosphonates on osteoblastic cells in vitro. J. Bone Miner. Res. 2001;16:2050–6. doi: 10.1359/jbmr.2001.16.11.2050. [DOI] [PubMed] [Google Scholar]
- [24].Xiong Y, Yang HJ, Feng J, Shi ZL, Wu LD. Effects of alendronate on the proliferation and osteogenic differentiation of MG-63 cells. J. Int. Med. Res. 2009;37:407–16. doi: 10.1177/147323000903700216. [DOI] [PubMed] [Google Scholar]
- [25].Pan B, To LB, Farrugia AN, Findlay DM, Green J, Gronthos S, Evdokiou A, Lynch K, Atkins GJ, Zannettino AC. The nitrogen-containing bisphosphonate, zoledronic acid, increases mineralisation of human bone-derived cells in vitro. Bone. 2004;34:112–23. doi: 10.1016/j.bone.2003.08.013. [DOI] [PubMed] [Google Scholar]
- [26].Tsuchimoto M, Azuma Y, Higuchi O, Sugimoto I, Hirata N, Kiyoki M, Yamamoto I. Alendronate modulates osteogenesis of human osteoblastic cells in vitro. Jpn. J. Pharmacol. 1994;66:25–33. doi: 10.1254/jjp.66.25. [DOI] [PubMed] [Google Scholar]
- [27].Idris AI, Rojas J, Greig IR, van’t Hof RJ, Ralston SH. Aminobisphosphonates cause osteoblast apoptosis and inhibit bone nodule formation in vitro. Calcif. Tissue Int. 2008;82:191–201. doi: 10.1007/s00223-008-9104-y. [DOI] [PubMed] [Google Scholar]
- [28].Orriss IR, Key ML, Colston KW, Arnett TR. Inhibition of osteoblast function in vitro by aminobisphosphonates. J. Cell Biochem. 2009;106:109–18. doi: 10.1002/jcb.21983. [DOI] [PubMed] [Google Scholar]
- [29].Pozzi S, Vallet S, Mukherjee S, Cirstea D, Vaghela N, Santo L, Rosen E, Ikeda H, Okawa Y, Kiziltepe T, Schoonmaker J, Xie W, Hideshima T, Weller E, Bouxsein ML, Munshi NC, Anderson KC, Raje N. High-dose zoledronic acid impacts bone remodeling with effects on osteoblastic lineage and bone mechanical properties. Clin. Cancer Res. 2009;15:5829–39. doi: 10.1158/1078-0432.CCR-09-0426. [DOI] [PubMed] [Google Scholar]
- [30].Garcia-Moreno C, Serrano S, Nacher M, Farre M, Diez A, Marinoso ML, Carbonell J, Mellibovsky L, Nogues X, Ballester J, Aubia J. Effect of alendronate on cultured normal human osteoblasts. Bone. 1998;22:233–9. doi: 10.1016/s8756-3282(97)00270-6. [DOI] [PubMed] [Google Scholar]
- [31].Khokher MA, Dandona P. Diphosphonates inhibit human osteoblast secretion and proliferation. Metabolism. 1989;38:184–7. doi: 10.1016/0026-0495(89)90260-6. [DOI] [PubMed] [Google Scholar]
- [32].Reinholz GG, Getz B, Sanders ES, Karpeisky MY, Padyukova NS, Mikhailov SN, Ingle JN, Spelsberg TC. Distinct mechanisms of bisphosphonate action between osteoblasts and breast cancer cells: identity of a potent new bisphosphonate analogue. Breast Cancer Res Treat. 2002;71:257–68. doi: 10.1023/a:1014418017382. [DOI] [PubMed] [Google Scholar]
- [33].Greiner S, Kadow-Romacker A, Lubberstedt M, Schmidmaier G, Wildemann B. The effect of zoledronic acid incorporated in a poly(D,L-lactide) implant coating on osteoblasts in vitro. J. Biomed. Mater. Res. A. 2007;80:769–75. doi: 10.1002/jbm.a.30950. [DOI] [PubMed] [Google Scholar]
- [34].Kellinsalmi M, Monkkonen H, Monkkonen J, Leskela HV, Parikka V, Hamalainen M, Lehenkari P. In vitro comparison of clodronate, pamidronate and zoledronic acid effects on rat osteoclasts and human stem cell-derived osteoblasts. Basic Clin. Pharmacol. Toxicol. 2005;97:382–91. doi: 10.1111/j.1742-7843.2005.pto_176.x. [DOI] [PubMed] [Google Scholar]
- [35].Reinholz GG, Getz B, Pederson L, Sanders ES, Subramaniam M, Ingle JN, Spelsberg TC. Bisphosphonates directly regulate cell proliferation, differentiation, and gene expression in human osteoblasts. Cancer Res. 2000;60:6001–7. [PubMed] [Google Scholar]
- [36].Gourion-Arsiquaud S, Allen MR, Burr DB, Vashishth D, Tang SY, Boskey AL. Bisphosphonate treatment modifies canine bone mineral and matrix properties and their heterogeneity. Bone. 2010;46:666–72. doi: 10.1016/j.bone.2009.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Chavassieux PM, Arlot ME, Reda C, Wei L, Yates AJ, Meunier PJ. Histomorphometric assessment of the long-term effects of alendronate on bone quality and remodeling in patients with osteoporosis. J. Clin. Invest. 1997;100:1475–80. doi: 10.1172/JCI119668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Balena R, Toolan BC, Shea M, Markatos A, Myers ER, Lee SC, Opas EE, Seedor JG, Klein H, Frankenfield D. The effects of 2-year treatment with the aminobisphosphonate alendronate on bone metabolism, bone histomorphometry, and bone strength in ovariectomized nonhuman primates. J. Clin. Invest. 1993;92:2577–86. doi: 10.1172/JCI116872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Storm T, Steiniche T, Thamsborg G, Melsen F. Changes in bone histomorphometry after long-term treatment with intermittent, cyclic etidronate for postmenopausal osteoporosis. J. Bone Min. Res. 1993;8:199–208. doi: 10.1002/jbmr.5650080211. [DOI] [PubMed] [Google Scholar]
- [40].Boyce RW, Paddock CL, Gleason JR, Sletsema WK, Eriksen EF. The effects of risedronate on canine cancellous bone remodeling: three-dimensional kinetic reconstruction of the remodeling site. J. Bone Min. Res. 1995;10:211–21. doi: 10.1002/jbmr.5650100207. [DOI] [PubMed] [Google Scholar]
- [41].Recker RR, Delmas PD, Halse J, Reid IR, Boonen S, Garcia-Hernandez PA, Supronik J, Lewiecki EM, Ochoa L, Miller P, Hu H, Mesenbrink P, Hartl F, Gasser J, Eriksen EF. Effects of intravenous zoledronic acid once yearly on bone remodeling and bone structure. J Bone Miner. Res. 2008;23:6–16. doi: 10.1359/jbmr.070906. [DOI] [PubMed] [Google Scholar]
- [42].Bravenboer N, Papapoulos SE, Holzmann P, Hamdy NA, Netelenbos JC, Lips P. Bone histomorphometric evaluation of pamidronate treatment in clinically manifest osteoporosis. Osteoporos. Int. 1999;9:489–93. doi: 10.1007/s001980050175. [DOI] [PubMed] [Google Scholar]
- [43].Jilka RL, Bellido T, Almeida M, Plotkin LI, O’Brien CA, Weinstein RS, Manolagas SC. Apoptosis in bone cells. In: Bilezikian JP, Raisz LG, Martin TJ, editors. Principles of Bone Biology. Academic Press; San Diego, San Francisco, New York, London, Sydney, Tokyo: 2008. pp. 237–61. [Google Scholar]
- [44].Weinstein RS, Jilka RL, Parfitt AM, Manolagas SC. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids: potential mechanisms of their deleterious effects on bone. J. Clin. Invest. 1998;102:274–82. doi: 10.1172/JCI2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Jilka RL, Weinstein RS, Bellido T, Roberson P, Parfitt AM, Manolagas SC. Increased bone formation by prevention of osteoblast apoptosis with parathyroid hormone. J. Clin. Invest. 1999;104:439–46. doi: 10.1172/JCI6610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Bellido T, Ali AA, Plotkin LI, Fu Q, Gubrij I, Roberson PK, Weinstein RS, O’Brien CA, Manolagas SC, Jilka RL. Proteasomal degradation of Runx2 shortens parathyroid hormone-induced anti-apoptotic signaling in osteoblasts. A putative explanation for why intermittent administration is needed for bone anabolism. J. Biol. Chem. 2003;278:50259–72. doi: 10.1074/jbc.M307444200. [DOI] [PubMed] [Google Scholar]
- [47].Pantschenko AG, Zhang W, Nahounou M, McCarthy MB, Stover ML, Lichtler AC, Clark SH, Gronowicz GA. Effect of osteoblast-targeted expression of bcl-2 in bone: differential response in male and female mice. J. Bone Miner. Res. 2005;20:1414–29. doi: 10.1359/JBMR.050315. [DOI] [PubMed] [Google Scholar]
- [48].Miura M, Chen XD, Allen MR, Bi Y, Gronthos S, Seo BM, Lakhani S, Flavell RA, Feng XH, Robey PG, Young M, Shi S. A crucial role of caspase-3 in osteogenic differentiation of bone marrow stromal stem cells. J. Clin. Invest. 2004;114:1704–13. doi: 10.1172/JCI20427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Lynch MP, Capparelli C, Stein JL, Stein GS, Lian JB. Apoptosis during bone-like tissue development in vitro. J Cell Biochem. 1998;68:31–49. [PubMed] [Google Scholar]
- [50].Zhang W, Pantschenko AG, McCarthy MB, Gronowicz G. Bone-targeted overexpression of Bcl-2 increases osteoblast adhesion and differentiation and inhibits mineralization in vitro. Calcif. Tissue Int. 2007;80:111–22. doi: 10.1007/s00223-006-0168-2. [DOI] [PubMed] [Google Scholar]
- [51].Marotti G, Cane V, Palazzini S, Palumbo C. Structure-function relationships in the osteocyte. Ital. J. Min. Electrol. Metab. 1990;4:93–106. [Google Scholar]
- [52].Tomkinson A, Reeve J, Shaw RW, Noble BS. The death of osteocytes via apoptosis accompanies estrogen withdrawal in human bone. J. Clin. Endocrinol. Metab. 1997;82:3128–35. doi: 10.1210/jcem.82.9.4200. [DOI] [PubMed] [Google Scholar]
- [53].Kousteni S, Bellido T, Plotkin LI, O’Brien CA, Bodenner DL, Han L, Han K, Di Gregorio GB, Katzenellenbogen JA, Katzenellenbogen BS, Roberson PK, Weinstein RS, Jilka RL, Manolagas SC. Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: dissociation from transcriptional activity. Cell. 2001;104:719–30. [PubMed] [Google Scholar]
- [54].Aguirre JI, Plotkin LI, Stewart SA, Weinstein RS, Parfitt AM, Manolagas SC, Bellido T. Osteocyte apoptosis is induced by weightlessness in mice and precedes osteoclast recruitment and bone loss. J. Bone Min. Res. 2006;21:605–15. doi: 10.1359/jbmr.060107. [DOI] [PubMed] [Google Scholar]
- [55].Almeida M, Han L, Martin-Millan M, Plotkin LI, Stewart SA, Roberson PK, Kousteni S, O’Brien CA, Bellido T, Parfitt AM, Weinstein RS, Jilka RL, Manolagas SC. Skeletal involution by age-associated oxidative stress and its acceleration by loss of sex steroids. J. Biol. Chem. 2007;282:27285–97. doi: 10.1074/jbc.M702810200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Plotkin LI, Mathov I, Aguirre JI, Parfitt AM, Manolagas SC, Bellido T. Mechanical stimulation prevents osteocyte apoptosis: requirement of integrins, Src kinases and ERKs. Am. J. Physiol. Cell Physiol. 2005;289:C633–C643. doi: 10.1152/ajpcell.00278.2004. [DOI] [PubMed] [Google Scholar]
- [57].Noble BS, Peet N, Stevens HY, Brabbs A, Mosley JR, Reilly GC, Reeve J, Skerry TM, Lanyon LE. Mechanical loading: biphasic osteocyte survival and the targeting of osteoclasts for bone destruction in rat cortical bone. Am. J. Physiol. Cell Physiol. 2003;284:C934–C943. doi: 10.1152/ajpcell.00234.2002. [DOI] [PubMed] [Google Scholar]
- [58].Plotkin LI, Weinstein RS, Parfitt AM, Roberson PK, Manolagas SC, Bellido T. Prevention of osteocyte and osteoblast apoptosis by bisphosphonates and calcitonin. J. Clin. Invest. 1999;104:1363–74. doi: 10.1172/JCI6800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Plotkin LI, Lezcano V, Thostenson J, Weinstein RS, Manolagas SC, Bellido T. Connexin 43 is required for the anti-apoptotic effect of bisphosphonates on osteocytes and osteoblasts in vivo. J. Bone Miner. Res. 2008;23:1712–21. doi: 10.1359/JBMR.080617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Follet H, Li J, Phipps RJ, Hui S, Condon K, Burr DB. Risedronate and alendronate suppress osteocyte apoptosis following cyclic fatigue loading. Bone. 2007;40:1172–7. doi: 10.1016/j.bone.2006.12.052. [DOI] [PubMed] [Google Scholar]
- [61].Plotkin LI, Manolagas SC, Bellido T. Transduction of cell survival signals by connexin-43 hemichannels. J. Biol. Chem. 2002;277:8648–57. doi: 10.1074/jbc.M108625200. [DOI] [PubMed] [Google Scholar]
- [62].Kogianni G, Mann V, Ebetino F, Nuttall M, Nijweide P, Simpson H, Noble B. Fas/CD95 is associated with glucocorticoid-induced osteocyte apoptosis. Life Sci. 2004;75:2879–95. doi: 10.1016/j.lfs.2004.04.048. [DOI] [PubMed] [Google Scholar]
- [63].Abe Y, Kawakami A, Nakashima T, Ejima E, Fujiyama K, Kiriyama T, Ide A, Sera N, Usa T, Tominaga T, Ashizawa K, Yokoyama N, Eguchi K. Etidronate inhibits human osteoblast apoptosis by inhibition of pro-apoptotic factor(s) produced by activated T cells. J. Lab. Clin. Med. 2000;136:344–54. doi: 10.1067/mlc.2000.109757. [DOI] [PubMed] [Google Scholar]
- [64].Gangoiti MV, Cortizo AM, Arnol V, Felice JI, McCarthy AD. Opposing effects of bisphosphonates and advanced glycation end-products on osteoblastic cells. Eur. J. Pharmacol. 2008;600:140–7. doi: 10.1016/j.ejphar.2008.10.031. [DOI] [PubMed] [Google Scholar]
- [65].Bivi N, Bereszczak JZ, Romanello M, Zeef LA, Delneri D, Quadrifoglio F, Moro L, Brancia FL, Tell G. Transcriptome and proteome analysis of osteocytes treated with nitrogen-containing bisphosphonates. J. Proteome. Res. 2009;8:1131–42. doi: 10.1021/pr8005606. [DOI] [PubMed] [Google Scholar]
- [66].Dunford JE, Thompson K, Coxon FP, Luckman SP, Hahn FM, Poulter CD, Ebetino FH, Rogers MJ. Structure-activity relationships for inhibition of farnesyl diphosphate synthase in vitro and inhibition of bone resorption in vivo by nitrogen-containing bisphosphonates. J. Pharmacol. Exp. Ther. 2001;296:235–42. [PubMed] [Google Scholar]
- [67].Van Beek E, Lowik C, Que I, Papapoulos S. Dissociation of binding and antiresorptive properties of hydroxybisphosphonates by substitution of the hydroxyl with an amino group. J. Bone Min. Res. 1996;11:1492–7. doi: 10.1002/jbmr.5650111016. [DOI] [PubMed] [Google Scholar]
- [68].Brown RJ, Van Beek E, Watts DJ, Lowik CW, Papapoulos SE. Differential effects of aminosubstituted analogs of hydroxy bisphosphonates on the growth of Dictyostelium discoideum. J. Bone Min. Res. 1998;13:253–8. doi: 10.1359/jbmr.1998.13.2.253. [DOI] [PubMed] [Google Scholar]
- [69].Plotkin LI, Manolagas SC, Bellido T. Dissociation of the pro-apoptotic effects of bisphosphonates on osteoclasts from their anti-apoptotic effects on osteoblasts/osteocytes with novel analogs. Bone. 2006;39:443–52. doi: 10.1016/j.bone.2006.02.060. [DOI] [PubMed] [Google Scholar]
- [70].Plotkin LI, Aguirre JI, Kousteni S, Manolagas SC, Bellido T. Bisphosphonates and estrogens inhibit osteocyte apoptosis via distinct molecular mechanisms downstream of ERK activation. J. Biol. Chem. 2005;280:7317–25. doi: 10.1074/jbc.M412817200. [DOI] [PubMed] [Google Scholar]
- [71].Kousteni S, Han L, Chen JR, Almeida M, Plotkin LI, Bellido T, Manolagas SC. Kinase-mediated regulation of common transcription factors accounts for the bone-protective effects of sex steroids. J. Clin. Invest. 2003;111:1651–64. doi: 10.1172/JCI17261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Civitelli R, Beyer EC, Warlow PM, Robertson AJ, Geist ST, Steinberg TH. Connexin43 mediates direct intercellular communication in human osteoblastic cell networks. J. Clin. Invest. 1993;91:1888–96. doi: 10.1172/JCI116406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Kato Y, Windle JJ, Koop BA, Mundy GR, Bonewald LF. Establishment of an osteocyte-like cell line, MLO-Y4. J. Bone Miner. Res. 1997;12:2014–23. doi: 10.1359/jbmr.1997.12.12.2014. [DOI] [PubMed] [Google Scholar]
- [74].Su M, Borke JL, Donahue HJ, Li Z, Warshawsky NM, Russell CM, Lewis JE. Expression of connexin 43 in rat mandibular bone and periodontal ligament (PDL) cells during experimental tooth movement. J. Dent. Res. 1997;76:1357–66. doi: 10.1177/00220345970760070501. [DOI] [PubMed] [Google Scholar]
- [75].Yellowley CE, Li Z, Zhou Z, Jacobs CR, Donahue HJ. Functional gap junctions between osteocytic and osteoblastic cells. J. Bone Miner. Res. 2000;15:209–17. doi: 10.1359/jbmr.2000.15.2.209. [DOI] [PubMed] [Google Scholar]
- [76].Evans WH, Martin PE. Gap junctions: structure and function. Mol. Membr. Biol. 2002;19:121–36. doi: 10.1080/09687680210139839. [DOI] [PubMed] [Google Scholar]
- [77].Goodenough DA, Paul DL. Beyond the gap: functions of unpaired connexon channels. Nat. Rev. Mol Cell Biol. 2003;4:285–94. doi: 10.1038/nrm1072. [DOI] [PubMed] [Google Scholar]
- [78].Plotkin LI, Vyas K, Gortazar AR, Manolagas SC, Bellido T. β Arrestin Complexes with Connexin (Cx) 43 and Anchors ERKs Outside the Nucleus: A Requirement for the Cx43/ERK-Mediated Anti-Apoptotic Effect of Bisphosphonates in Osteocytes. J. Bone Miner. Res. 2006;21:S65. [Google Scholar]
- [79].Boland R, Santillan G, Katz S, Morelli S, Stockman G, Roldan E. Modulation of [Ca2+] in osteoblastic cells by olpadronate and amino-olpadronate. Protein phosphatases as possible target of bisphosphonates. BIOCELL. 2003;27:132. [Google Scholar]
- [80].Boland RL, Morelli S, Santillan G, Scodelaro P, Colicheo A, Boland AR, Vyas K, Plotkin LI, Bellido T. Connexin 43 Is Required for Bisphosphonate-Induced Survival of Osteoblastic Cells but Not for Bisphosphonate Binding. J. Bone Miner. Res. 2006;21 [Google Scholar]