


Abstract
It has long been known that trypanosomes regulate mitochondrial biogenesis during the life cycle of the parasite; however, the mitochondrial protein inventory (MitoCarta) and its regulation remain unknown. We present a novel computational method for genome-wide prediction of mitochondrial proteins using a support vector machine-based classifier with ∼90% prediction accuracy. Using this method, we predicted the mitochondrial localization of 468 proteins with high confidence and have experimentally verified the localization of a subset of these proteins. We then applied a recently developed parallel sequencing technology to determine the expression profiles and the splicing patterns of a total of 1065 predicted MitoCarta transcripts during the development of the parasite, and showed that 435 of the transcripts significantly changed their expressions while 630 remain unchanged in any of the three life stages analyzed. Furthermore, we identified 298 alternatively splicing events, a small subset of which could lead to dual localization of the corresponding proteins.
INTRODUCTION
Trypanosoma brucei, a unicellular, flagellated protozoan parasite, is the causative agent of human African trypanosomiasis and the cattle-wasting disease Nagana in sub-Saharan Africa. T. brucei has a digenetic life cycle alternating between the tsetse fly and a mammal host. Besides being a human and veterinary pathogen, it has played a key role in the discovery and understanding of a number of general biological principles such as RNA editing, antigenic variation, GPI anchoring and trans-splicing (1–6). The later is a mechanism by which T. brucei processes the 5′-ends of its polycistronic mRNAs through the addition of a 39 nucleotide leader sequence. A unique aspect of the trypanosome biology is the development and regulation of its single, large mitochondrion. In the insect vector, the parasite has a fully developed organelle containing numerous cristae, electron transport complexes, where energy production is performed through electron transportation coupled with oxidative phosphorylation (7). In contrast, the parasite in bloodstream exclusively uses glycolysis in a specialized organelle, the glycosome, to meet its energy needs [reviewed in (8)]. This is also reflected in the size and the structure of the mitochondrion with a reduced number of cristae and reduced volume (9). As of today, the precise protein composition of the organelle largely remains unknown. However, recently, a comprehensive study identified mitochondrial proteins in insect form T. brucei using a shotgun proteomics approach, and assigned 597 proteins to the mitochondrion with high quality (10). In T. brucei, only 18 proteins have been predicted to be encoded in its mitochondrial genome (11,12), and thus the vast majority of the identified mitochondrial proteins are nuclear encoded, translated in the cytoplasm, and imported into the mitochondrion. Many proteins that are imported into the mitochondrion have targeting signals, typically at the N-termini (13–17), and in some cases at the C-termini (18) or protein internal (19,20). However, numerous mitochondrial proteins were found to be lacking such signal peptides including proteins that have been shown to be imported into mitochondria in trypanosomes (13,21–23). This poses a major challenge in the identification of mitochondrial proteins.
To date, a number of computational methods have been developed to predict mitochondrial proteins. These methods use different protein features, such as amino acid composition [AAC; e.g. MitPred (24), Predotar (25), PSORTII (26), PLOC (27), NNPSL (28)], physicochemical properties [e.g. Predotar (25), Proteome Analyst (29), LOCSVMPSI (30), ESLpred (31), LOCtree (32)], N-terminal signal peptides [e.g. TargetP (33), iPSORT (34), PredSL (35)] and Pfam domain occurrences [e.g. MITOPRED (36,37), MitPred (24)]. They employ different computational techniques, such as support vector machine [SVM; e.g. ELSpred (31), LOCSVMPSI (30), MitPred (24), PLOC (27), SubLoc (38)], artificial neural network [ANN; e.g. Predotar (25), PredSL (35), TargetP (33)], hidden Markov model [HMM; e.g. MitPred (24), PredSL (35)], k-nearest neighbor [k-NN; e.g. PSORTII (26), WoLF PSORT (39,40)] and expert systems [e.g. iPSORT (34), MitoProtII (41)]. While these methods have greatly advanced our knowledge about mitochondrial localization, they have a number of key limitations. For example, the N-terminal signal peptide-based approaches, though very popular, have intrinsic limitations in recognizing mitochondrial outer and inner membrane proteins as well as transmembrane proteins that do not seem to have any apparent targeting peptides. The prediction performance by Pfam domain-based methods, not based on targeting signals, is limited by the reality that there are no detectable Pfam domains that can clearly distinguish mitochondrial from non-mitochondrial proteins (24). One common issue with all these prediction methods is that while some of them can make accurate predictions of several subcellular compartments such as nucleus or cytoplasm, none of them can predict the mitochondrial localization very accurately.
We present a novel computational approach for prediction of mitochondrial proteins in T. brucei. By applying this approach, we added 468 new proteins to the pool of mitochondrial proteins. We verified a subset of these proteins using biological and biochemical assays, and then analyzed the regulation of MitoCarta gene expression and splicing patterns using a novel sequencing approach.
MATERIALS AND METHODS
Datasets
Positive data. 597 proteins identified as targeted to mitochondria with high (401) or moderate (196) confidence in a recent proteomic study on T. brucei (10) were chosen as our positive dataset. This dataset is available as Supplementary Table S1.
Negative data. 1318 proteins found in subcellular locations of T. brucei other than mitochondria in the same study (10) were collected. An additional 1353 potential non-mitochondrial proteins were compiled from the T. brucei proteome (9192 proteins from ftp://ftp.sanger.ac.uk/pub/databases/T.brucei_sequences/T.brucei_genome_v4, www.genedb.org) by removing entries either annotated as mitochondrial or as ‘hypothetical’, ‘potential’ or ‘putative’ proteins. Proteins with BLAST sequence similarities to any positive samples or any potential mitochondrial proteins in the MitoP2 database (E-value cutoff = 0.5) (www.mitop.de:8080/mitop2/) were removed. 1290 non-mitochondrial proteins remained and were selected as our negative set. This dataset is available as Supplementary Table S2.
Training and test sets. an SVM-based classifier requires both training and test sets. The collected positive and negative sets were split by randomly selecting 3/4th of the data from each set as the training sets and the remaining as the test data. To evaluate the accuracy and stability of our trained classifiers, this random sampling process was repeated fifty times, resulting in 50 groups of training and test sets.
Computational method
Protein features. We examined various features of proteins, including those widely used for protein localization prediction, in order to identify a list of features that can distinguish well mitochondrial from non-mitochondrial proteins. The initial features can be grouped into three categories: basic sequence features, physicochemical properties and signal peptide and transmembrane topology. Details are given in Supplementary Table S3.
Support vector machine. SVM has been successfully used to solve a wide range of biological data mining problems (30,42,43), due to its effectiveness to deal with multidimensional datasets with complex relationships among the data elements. In this study, LIBSVM (http://www.csie.ntu.edu.tw/∼cjlin/libsvm) with the popular non-linear Gaussian Radial Basis Function kernel (RBF) (44) was used for our classification.
Feature selection. LIBSVM was used to select discriminative features between the positive and the negative training sets. Given a set of candidate features, an F-score is calculated for each feature element to measure its discriminative power between the positive and the negative sets, and for the i-th feature, the F-score is calculated by
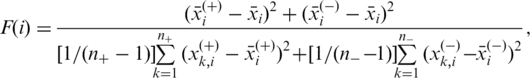 |
(1) |
where 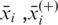 and
and 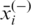 are the average value of the ith feature over the whole, the positive and negative dataset, respectively;
are the average value of the ith feature over the whole, the positive and negative dataset, respectively; 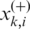 is the i-th feature of the k-th positive data and
is the i-th feature of the k-th positive data and 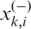 is the i-th feature of the k-th negative data;
is the i-th feature of the k-th negative data; 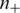 and
and 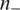 are the numbers of positive and negative data, respectively. The larger an F-score is, the more discriminative power the feature has. As our goal is to use the fewest number of features to achieve the best classification performance, a subset of feature elements with high F-score and having the smallest balanced error rate (BER) in 5-fold cross validation are selected:
are the numbers of positive and negative data, respectively. The larger an F-score is, the more discriminative power the feature has. As our goal is to use the fewest number of features to achieve the best classification performance, a subset of feature elements with high F-score and having the smallest balanced error rate (BER) in 5-fold cross validation are selected:
| (2) |
Where tp and tn are the numbers of true positives and true negatives, respectively, and fp and fn are the numbers of false positives and false negatives, respectively.
In a final assessment of the discriminative power of each selected feature element, we used the mean and the standard deviation in an analysis of variance (ANOVA) to compare the distributions of the feature between the positive and the negative samples.
Performance evaluation
For each group of training and testing sets, a prediction model is obtained that optimizes its performance on both the training and test data. Four common measures were used to evaluate the performance, namely prediction sensitivity: 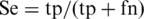 , prediction specificity:
, prediction specificity: 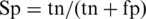 , prediction accuracy:
, prediction accuracy: 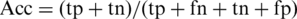 and the Matthews correlation coefficient of the predictions:
and the Matthews correlation coefficient of the predictions: 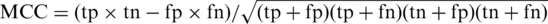 (45), with MCC = 1 representing a perfect prediction and 0 a random prediction. The trained model with the highest MCC is selected as the final prediction model in our study, henceforth referred to as Trypanosoma mitochondrial protein predictor (TMPP).
(45), with MCC = 1 representing a perfect prediction and 0 a random prediction. The trained model with the highest MCC is selected as the final prediction model in our study, henceforth referred to as Trypanosoma mitochondrial protein predictor (TMPP).
For a given query protein, the following function score (s) was used in TMPP to predict if the protein is mitochondrial:
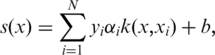 |
(3) |
where k is the RBF kernel function that measures the distance between the input vector x and the stored training vector xi; yi∈{1,–1} indicates a positive or negative sample for the i-th training vector; αi is the Lagrange multiplier for the i-th training vector, which is computed by quadratic programming, and b is the threshold for defining the hyperplane. A positive score predicts a mitochondrial protein. To evaluate the reliability of an assigned class, we introduce prediction precision (PP) as follows:
| (4) |
Based on the prediction on 597 positive and 1290 negative proteins, we define three levels of prediction confidence, namely high when PP > 0.9, medium when 0.8 ≤ PP < 0.9 and low when PP ≤ 0.8.
Cell lines and localization
T. brucei brucei AnTat 1.1 and MITat 1.2 (221) were used for the expression and splicing profile study. Late procyclic forms of AnTat 1.1 were cultivated in SDM79 supplemented with 10% FBS. For short stumpy bloodstream forms of AnTat 1.1, mice were immunosuppressed with 260mg kg-1 cyclophosphoamide (Sigma) 24 hours prior to intraperitoneal injection of 106 parasites. Short stumpy parasites were harvested 5 days post-infection at a density of 2–4 × 108 cells ml−1 blood. 75–80% of the cells showed a short stumpy phenotype as determined by light microscopy of blood smears after methanol fixation. Long slender forms were harvested from untreated mice at day three post-infection at a density of <5 × 107 cells ml−1 blood.
Genes were amplified by PCR from genomic DNA of T. brucei Mitat 1.2. Procyclic cells of T. brucei Mitat 1.2 were grown in semi-defined medium (SDM-79) supplemented with 10% fetal bovine serum to a density of 5 × 106 cells ml−1. For the localization study, cells were transiently transformed with 20 µg of plasmid pG-EGFPβ-ΔLII containing the gene of interest with a C-terminal green fluorescent protein tag as described in (46). For fluorescence microscopy, cells were stained with Mitotracker for 15 min at 37°C subsequently washed in PBS pH 7, and then fixed first for 1 min with 1% Paraformaldehyde and then for 5 min in ice cold methanol on a microscopic slide. For Western blotting, 107 cells were fractionated using a detergent lysis procedure followed by differential centrifugation steps as described in (47). Fractions were probed with polyclonal HSP70 antibody (kind gift from J. Bangs of Madison, Wisconsin), monoclonal iron sulfur protein antibody (kind gift from S.L. Hajduk, University of Georgia) and anti-GFP (Invitrogen).
PCR and cloning
PCR was done using the gene specific oligonucleotides and genomic DNA from T. brucei Mitat 1.2 in 25 µl (1 unit PFU Polymerase (NEB, USA), 2,5µl 10× buffer (NEB, USA), 2 mM MgCl2, 400 µM dNTPs, 30ng gDNA). After restriction digest 200ng of each the PCR product and the vector pG-EGFPβ-ΔLII were ligated in 20 µl using T4 DNA ligase (NEB, USA) at 14°C overnight.
RNA extraction, library construction and sequencing
First strand cDNA was synthesized from polyA RNA using random hexamers and Superscript II reverse transcriptase. Adapters were ligated to the purified cDNA using a customized bar-coded paired-end Illumina adapter (Fasteris, Geneva, Switzerland). After purification, each cDNA library was amplified using the standard Illumina mRNA-SEQ library amplification protocol. Fragments in the range 120–160 bp were gel purified and sequencing on the Illumina Genome Analyzer was carried out using the following sequencing primer (spliced leader primer 5′-ACCGAGATCTACAGTTTCTGTACTATATTG-3′).
Base calling was performed using the Genome Analyzer Pipeline (Illumina). Sequence reads of at least 24 nt were separated according to identified barcodes. The reads were mapped to the genome sequence of T. brucei TREU927 using maq (48) (http://maq.sourceforge.net) with n = 3 and an effective first read length of 24. Tag counts were normalized to the library size and scaled linearly to reflect counts of tags per million (TPM). Mapped tags were assigned to the downstream annotated protein-encoding gene. Tags mapping internally to a coding sequence (CDS) were assigned to it as internal splice sites. Alternatively, spliced leader addition sites were cataloged for each gene. Genes with ≤60% tags in the assigned major splice site were designated alternatively spliced. Differences in expression levels of a gene in two different libraries were tested for significance according to Audic and Claverie with a threshold of P < 10−5 (49). A test for statistically significant difference (Fisher two-tailed test, P < 10−5) between the normalized counts of sites was made for genes with different major splice sites in two life cycle stages. Significant differences were termed differential trans-splicing events. Expression levels were visualized as heatmaps after log2 transformation and hierarchical average linkage clustering of Euclidean distances using MeV (50) (http://www.tm4.org/mev.html).
RESULTS AND DISCUSSION
Features selected for distinguishing mitochondrial proteins
In order to identify a list of features that can well distinguish mitochondrial from non-mitochondrial proteins, we have examined 18 protein features, represented as 605 feature elements for each protein sequence (see Supplementary Table S3). From the original list of 605 feature elements, 66 discriminative feature elements were extracted by feature selection algorithm in LIBSVM. Details are given in Supplementary Table S4.
Basic sequence attributes. Mitochondrial proteins tend to have targeting signals in their N-termini, which may have a distinctive AAC. Hence, we decided to consider the AAC of the N-terminal residues and the remaining residues separately. We examined the conventional AAC and different types of split-AAC, which is calculated for the first n N-terminal residues for n = 15, 20, 25, 30, 35, respectively, and the remaining residues separately, by estimating the distinguishing power of these feature vectors through SVM based on 5-fold cross validation. Di-amino acid composition is also considered in our study. As shown in Supplementary Table S5, the conventional AAC gave the worst accuracy, while the split-AAC with n = 30 gave the best result and showed strong biases between mitochondrial proteins and non-mitochondrial proteins. As shown in Supplementary Figure S1, AAC in the first 30 N-terminal residues differs substantially from that in the remaining residues of mitochondrial proteins, while the difference is not nearly as remarkable as in non-mitochondrial proteins. Specifically, the N-terminal residues of mitochondrial proteins are rich in positively charged residue, arginine, while poor in negatively charged residues, aspartic acid and glutamic acid, reconfirming the discovery made in previous studies in trypanosomes and other systems (34,35,51). We also observed that the N-terminal mitochondrial sequences have more phenylalanine and tryptophan, but less lysine and in the remaining residues mitochondrial proteins have relatively high percentages of aromatic residues, namely histidine, tryptophan and tyrosine, but lower of hydrophilic residues, i.e. cysteine and asparagine, compared to non-mitochondrial proteins. All the aforementioned discriminative features were successfully selected by the feature selection program (Supplementary Table S4).
Physicochemical properties. Dubchak and coworkers have developed three general descriptors: composition, transition and distribution, to measure various structural and physicochemical properties of proteins, such as hydrophobicity, Van der Waals volume, polarity, polarizability, charge, secondary structure and solvent accessibility (52). We have adopted the three descriptors to represent these properties in our feature selection. In addition, we have also considered some structural properties such as secondary structural content (53,54), estimated radius of gyration (55), unfoldability (56) and disordered regions (56) of candidate proteins, considering the potential relevance of these properties to identification of mitochondrial proteins. Some physicochemical properties, namely normalized Van der Waals volume, polarity, polarizability, charge and solvent accessibility, showed strong biases to mitochondrial proteins compared to the non-mitochondrial proteins. Details are given in Supplementary Table S4.
Signal peptide and transmembrane topology. Signal peptides are important for targeting proteins from the cytosol to the endoplasmic reticulum (ER) membrane and for subsequent transport through the secretory pathways (57,58). Both alpha-helical and beta-barrel trans-membrane regions were considered. Our analysis revealed that signal peptides and trans-membrane regions, particularly beta-barrel trans-membrane regions are present less frequently in mitochondrial proteins than in non-mitochondrial proteins. A similar observation is made in other organisms, including yeast, mouse and human. Detailed results are given in Supplementary Table S6.
Evaluation of prediction performance
Training and test. The prediction performance of our trained SVM-based classifier was stable across all 50 test sets, on which the MCC ranges from 0.656 to 0.752, and both sensitivity and specificity are above 80%, as detailed in Table 1, strongly suggesting the effectiveness and stability of our trained classifiers.
Table 1.
Prediction performance on 50 testing sets
| MCC | Se | Sp | Acc | |
|---|---|---|---|---|
| Best performance | 0.752 | 0.823 | 0.926 | 0.894 |
| Worst performance | 0.656 | 0.801 | 0.874 | 0.852 |
| Mean | 0.709 | 0.816 | 0.901 | 0.875 |
| Std | 0.029 | 0.014 | 0.018 | 0.014 |
Prediction reliability. To evaluate the reliability of the predicted mitochondrial proteins, we have analyzed the relationship between the output score (s) and the prediction precision (PP). As shown in Figure 1, a prediction score higher than 0.8 indicates a mitochondrial protein with high confidence 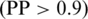 , a score lower than 0.8 but not lower than 0.4 indicates a mitochondrial protein with medium confidence
, a score lower than 0.8 but not lower than 0.4 indicates a mitochondrial protein with medium confidence 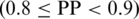 and a positive score lower than 0.4 assigns a protein to mitochondrial with low confidence
and a positive score lower than 0.4 assigns a protein to mitochondrial with low confidence 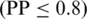 .
.
Figure 1.

Statistical relationship between the prediction accuracy and the SVM score. The relationship is based on the analysis of 597 (positive set) and 1290 (negative set). The x-axis is the absolute SVM score and the y-axis represents prediction precision.
Our prediction program correctly predicted 535 mitochondrial and 1238 non-mitochondrial proteins among the 597 positive and 1290 negative samples, respectively. Of the 535 predicted mitochondrial proteins, 82% are predicted with high confidence, while 11% and 7% are predicted with medium and low confidence, respectively.
Comparison with other methods. The performance of program TMPP has been compared with publicly available programs (see Supplementary Table S7), including MitPred (24), Predotar v1.03 (25), PSORTII (26), PLOC (27), Proteome Analyst (29), ESLpred (31), TargetP v1.1 (33), iSPORT (34), PredSL (35), MITOPRED (36,37), SubLoc v1.0 (38), WoLF PSORT (39,40) and MitoProtII (41). All these programs have been applied to the same test set containing 147 mitochondrial and 325 non-mitochondrial proteins. The prediction results of these programs are summarized in Table 2. TMPP outperforms all the other programs in terms of both the overall prediction accuracy and MCC on the independent test set. Currently MCC is considered to be the most reliable and robust criterion for assessing trained classifiers (24,36).
Table 2.
Comparison of prediction performance of 14 methods applied on 147 mitochondrial and 325 non-mitochondrial proteins
| Method | MCC | Se | Sp | Acc |
|---|---|---|---|---|
| TMPP | 0.752 | 0.823 | 0.926 | 0.894 |
| WoLF PSORT | 0.577 | 0.605 | 0.926 | 0.826 |
| Predotar | 0.543 | 0.612 | 0.901 | 0.811 |
| MitoProt | 0.539 | 0.735 | 0.821 | 0.794 |
| PredSL | 0.532 | 0.558 | 0.923 | 0.809 |
| PSORT | 0.474 | 0.497 | 0.920 | 0.788 |
| TargetP | 0.459 | 0.442 | 0.938 | 0.784 |
| Mitpred | 0.439 | 0.469 | 0.914 | 0.775 |
| MITOPRED | 0.422 | 0.667 | 0.772 | 0.739 |
| iPSORT | 0.419 | 0.497 | 0.886 | 0.765 |
| Proteome Analyst | 0.383 | 0.231 | 0.990 | 0.754 |
| ELSpred | 0.356 | 0.476 | 0.855 | 0.737 |
| PLOC | 0.320 | 0.429 | 0.862 | 0.727 |
| SubLoc | 0.243 | 0.517 | 0.732 | 0.665 |
A common limitation among the N-terminal information-based methods is that a significant number of mitochondrial proteins do not have targeting signals at N terminus (23,59). TMPP outperforms the existing prediction methods through utilizing more features, as well as through better treatment of previously used features such the AAC in the N terminal regions (see Supplementary Table S5 and Figure S1). TMPP was specifically trained for trypanosome sequences and may have learned domain knowledge that could lead to an advantage over other methods, potentially at the expense of generality.
Prediction of mitochondrial proteins in T. brucei
We have applied TMPP to T. brucei proteome and assigned 1065 proteins to mitochondrion with  including 597 proteins previously shown to be associated with the mitochondrion and 468 proteins without any previous mitochondrial association, covering 12% of the whole proteome of T. brucei. Similar numbers have been found for the yeast (876), and mouse (1098) MitoCarta, respectively (60,61). Using a reciprocal best BLAST hit approach we identified 137 homologs of the T. brucei MitoCarta in the human MitoCarta, 47 of which are disease associated, indicating the potential usefulness of T. brucei in studying human disease genes (Supplementary Table S8). When we compared the T. brucei to the yeast MitoCarta, we identified 120 homologs mostly in metabolism (33), protein synthesis (14) and iron sulfur cluster assembly (10).
including 597 proteins previously shown to be associated with the mitochondrion and 468 proteins without any previous mitochondrial association, covering 12% of the whole proteome of T. brucei. Similar numbers have been found for the yeast (876), and mouse (1098) MitoCarta, respectively (60,61). Using a reciprocal best BLAST hit approach we identified 137 homologs of the T. brucei MitoCarta in the human MitoCarta, 47 of which are disease associated, indicating the potential usefulness of T. brucei in studying human disease genes (Supplementary Table S8). When we compared the T. brucei to the yeast MitoCarta, we identified 120 homologs mostly in metabolism (33), protein synthesis (14) and iron sulfur cluster assembly (10).
Experimental validation of predicted mitochondrial proteins in T. brucei
In order to verify the prediction accuracy we experimentally localized a subset of proteins in the procyclic T. brucei cells by immunofluorescence microscopy and Western blotting (Figure 2). Using a C-terminal green fluorescent protein tag, we showed that 9 out of 12 randomly selected proteins from our prediction co-localized with the mitochondrion specific dye Mitotracker (Invitrogen, USA), while two proteins mainly localized to a structure surrounding the nucleus resembling the ER (Supplementary Figure S2). For one protein, we were unable to detect any GFP signal, likely due to very low expression or rapid degradation of the protein. Between the nine mitochondrially localized proteins, we could distinguish two localization patterns. The majority showed an even distribution of the GFP signal throughout the mitochondrial structure (Figure 2), while two proteins had a more punctuated localization (Figure 2, Tb09.160.3050/Tb09.211.3170) similar to the recently described gRNA associated proteins GAP1 and GAP2 (62). In our cases, the GFP signal could reflect the true localization or be due to the overexpression and C-terminal tagging of the protein. We also used an established biochemical assay of detergent lysis followed by differential centrifugation steps SDS–PAGE and western blotting in order to verify the localization (47). Using an antibody to GFP, we detected the majority of the tagged protein in the mitochondrial fraction, which was verified using antibodies to known mitochondrial (Rieske iron-sulfur protein, anti-ISP) and cytosolic (Heat shock protein 70, anti-HSP70) proteins (Figure 2). In all cases, the biochemical assay supported the localization detected by immunofluorescence microscopy verifying that the localization is not limited to the individual cells analyzed by microscopy.
Figure 2.

Localization of predicted MitoCarta proteins. The fluorescence microscopy images from nine GFP tagged proteins that were randomly picked from our prediction are shown. First column GFP, second column Mitotracker, third column, overlay including the DAPI and differential interference contrast image, fourth column western blotts of total cell protein fractionated in a mitochondrial fraction (M) and a cytosolic fraction (C) and probed with antibodies against the cytosolic HSP70, the mitochondrial iron sulfur protein and GFP, fifth column the SDS–PAGE from the fractionated protein extract.
The localization of two proteins predicted to be mitochondrial to an ER like structure suggested that there could be a common feature in the localization between the two compartments. Also, since we have investigated only the procyclic form of the parasite, there remains the possibility of mitochondrial localization of the proteins in other life cycle stages of the parasite. Overall, the mitochondrial localization of 75% of the randomly selected proteins from our prediction supports the strong performance of the presented prediction approach, especially since we have only analyzed one life cycle stage of the parasite.
Regulation of the MitoCarta genes during development
We have recently developed a new method to simultaneously map the 5′-splice sites and the expression profile of T. brucei that we named spliced leader trapping (SLT) (63). SLT uses the fact that trypansomes employ trans-splicing of a leader sequence onto the 5′-UTR of every mRNA. It allowed us to sequence a short stretch of the 5′-UTR immediate downstream of the spliced leader acceptor site using high throughput parallel sequencing. We obtained more than 4 million sequence tags from the proliferative long slender bloodstream form, the insect form of the parasite as well as from the quiescent short stumpy form. Using this information we were able to monitor the expression profiles of processed mRNAs and the corresponding splicing patterns. We detected transcripts for ≥95% of the 1065 MitoCarta genes that had a mean number of 75–87 TPM depending on the stage of the life cycle (Table 3). The number of expressed MitoCarta genes was ∼1000 independent of the life cycle stage of the parasite (Table 3), which is rather surprising given the large changes in morphology and function of the mitochondrion during development. Of the expressed genes, 630 transcripts did not significantly change in abundance while 435 showed a significant change in expression levels in at least one of the three life cycle stages (Supplementary Table S9). From this data, it appears that ∼60% of the MitoCarta is a core set for the mitochondrial biogenesis, while ∼40% are life cycle stage specific. However, a significant number of genes thought to be associated with insect form specific physiology are also expressed in the bloodstream form, suggesting that they are not regulated at the level of transcription but rather at the level of translation and protein stability, as shown for cytochrome c reductase (64). Alternatively, expressions of these transcripts could also indicate novel or previously undetected functions in the bloodstream form mitochondrion. One of the most prominent examples was the succinyl Co-A ligase beta subunit, which is one of the most highly expressed transcripts in cells of the bloodstream form (4344 TPM). Using RNAi, we showed that the loss of transcripts is not only lethal in the insect form of the parasite as had been shown previously but also in the bloodstream form, indicating that the corresponding protein performs an unknown function in the bloodstream form mitochondrion [(65), Supplementary Figure S3].
Table 3.
Expression and splicing profile of the MitoCarta in long slender, short stumpy and procyclic form T. brucei
| Long slender (LS) | Short stumpy (SS) | Procyclic (PC) | Total | |
|---|---|---|---|---|
| MitoCarta genes | – | – | – | 1065 |
| Differentially regulated | 109 (LS to SS) | 357 (SS to PC) | 350 (LS to PC) | 435a |
| Unchanged expression | – | – | – | 630 |
| Alternatively spliced | 174 | 186 | 154 | 298b |
| Differentially spliced | 11 (LS to SS) | 50 (SS/PC) | 45 (LS to PC) | – |
| Major internal sites | 88 | 90 | 74 | – |
| Only internal sites | 55 | 51 | 52 | – |
| N-terminal extensionsc | – | – | – | 23 |
| Genes expressed | 934 | 932 | 945 | – |
| TPM (mean) | 75 | 81 | 87 | – |
| TPM (median) | 24 | 22 | 29 | – |
aTotal number of genes that significantly change expression levels.
bThe major splice site contains less than 60% of the tags.
cSplice site would allow for N-terminal extension of the reading frame.
Splicing
The analysis of our SLT data revealed 298 alternative splicing events in the MitoCarta transcriptome (Supplementary Table S10). We called a transcript alternatively spliced if the major splice site contained less than 60% of the sequence tags. For 51–55 transcripts, depending on the life-cycle stage, the splicing occurred downstream of the annotated AUG start codon potentially leading to a loss or gain of mitochondrial targeting sequences as predicted by TargetP [(33), Figure 3, Supplementary Table S11]. We also detected 23 splicing events that would allow for an N-terminal extension of the encoded protein depending on the splice variant. Five of these transcripts were alternatively spliced such that different splicing events could lead to a change in localization of the corresponding protein products (Supplementary Table S11), which has previously been observed for alternative splice variants of A-kinase anchoring protein 1 in mouse fibroblasts (66). Interestingly, the N-terminal extensions could also lead to a change in localization from mitochondrial to non-mitochondrial by masking the targeting signal through the addition of amino acids to the N-terminus.
Figure. 3.

Two examples of alternative splice variants potentially leading to changes in localization. Screenshot of a region from chromosome of 2.5 kbp of two genes with SLT data in TPM from procyclic (green), bloodstream long slender (red), short stumpy forms (orange) and potential mitochondrial targeting signals (MTS, white box) and signal peptide sequence (SP, grey box) on the corresponding protein sequences (black line). (A) Tb927.4.4910, the upstream splice site potentially lead to a N-terminal extension of the open reading frame now containing a MTS of 58 amino acids as predicted by MITOPROT (confidence 0.75). The downstream splice site excludes the MTS, instead leading to a SP of 34 amino acids (confidence 0.98, SingnalP). (B) Tb11.01.1040, the uptstream splice site would allow translation from the currently annotated AUG leading to a protein containing a 30 amino acid MTS (confidence 0.99, MITOPROT), while the downstream, internal splice site would exclude the MTS leading to a 345 amino acid protein.
CONCLUSION
We have identified 66 feature elements to distinguish mitochondrial from non-mitochondrial proteins, and developed an SVM-based classifier to predict mitochondrial proteins in T. brucei. Using this approach we predicted the T. brucei MitoCarta to contain 1065 proteins, 468 of which did not have assigned cellular components. Using cell biology and biochemical methods, we have verified the localization of 75% of a randomly chosen subset of the predicted proteins supporting the performance of the prediction approach. The regulation of the total mitochondrial proteome indicates a minimal subset of proteins that have to be present to maintain the mitochondrial organelle and the alternative splicing patterns of a subset of MitoCarta transcripts indicate the dual use of the corresponding proteins in other parts of the cell.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Science Foundation (DEB-0830024 and DBI-0542119 to J.C. and Y.X., partial); China Scholarship Council Postgraduate Scholarship Program (to X.B.Z., partial); Swiss National Science foundation (31003A_125194 to T.O., K.G., A.C. and D.N.). Funding for open access charge: National Science Foundation (DEB-0830024).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Drs V. Olman, F. Zhou and Y. Ou at University of Georgia for inspiring discussions, and would like to acknowledge Isabel Roditi at University of Bern for providing RNA and critically reading the manuscript. This study was supported in part by computer clusters provided by the University of Georgia Research Computing Center, a partnership between the Office of the Vice President for Research and the Office of the Chief Information Officer.
REFERENCES
- 1.Benne R, Van den Burg J, Brakenhoff JP, Sloof P, Van Boom JH, Tromp MC. Major transcript of the frameshifted coxII gene from trypanosome mitochondria contains four nucleotides that are not encoded in the DNA. Cell. 1986;46:819–826. doi: 10.1016/0092-8674(86)90063-2. [DOI] [PubMed] [Google Scholar]
- 2.Horvath A, Berry EA, Maslov DA. Translation of the edited mRNA for cytochrome b in trypanosome mitochondria. Science. 2000;287:1639–1640. doi: 10.1126/science.287.5458.1639. [DOI] [PubMed] [Google Scholar]
- 3.Borst P, Cross GA. Molecular basis for trypanosome antigenic variation. Cell. 1982;29:291–303. doi: 10.1016/0092-8674(82)90146-5. [DOI] [PubMed] [Google Scholar]
- 4.Ferguson MA, Cross GA. Myristylation of the membrane form of a Trypanosoma brucei variant surface glycoprotein. J. Biol. Chem. 1984;259:3011–3015. [PubMed] [Google Scholar]
- 5.de Almeida ML, Turner MJ, Stambuk BB, Schenkman S. Identification of an acid-lipase in human serum which is capable of solubilizing glycophosphatidylinositol-anchored proteins. Biochem. Biophys. Res. Commun. 1988;150:476–482. doi: 10.1016/0006-291x(88)90545-1. [DOI] [PubMed] [Google Scholar]
- 6.Sutton RE, Boothroyd JC. Evidence for trans splicing in trypanosomes. Cell. 1986;47:527–535. doi: 10.1016/0092-8674(86)90617-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Besteiro S, Barrett MP, Riviere L, Bringaud F. Energy generation in insect stages of Trypanosoma brucei: metabolism in flux. Trends Parasitol. 2005;21:185–191. doi: 10.1016/j.pt.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 8.Michels P, Bringaud F, Herman M, Hannaert V. Metabolic functions of glycosomes in trypanosomatids. BBA-Molecular Cell Res. 2006;1763:1463–1477. doi: 10.1016/j.bbamcr.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 9.Vickerman K. Polymorphism and mitochondrial activity in sleeping sickness trypanosomes. Nature. 1965;208:762–766. doi: 10.1038/208762a0. [DOI] [PubMed] [Google Scholar]
- 10.Panigrahi AK, Ogata Y, Zikova A, Anupama A, Dalley RA, Acestor N, Myler PJ, Stuart KD. A comprehensive analysis of Trypanosoma brucei mitochondrial proteome. Proteomics. 2009;9:434–450. doi: 10.1002/pmic.200800477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sloof P, de Haan A, Eier W, van Iersel M, Boel E, van Steeg H, Benne R. The nucleotide sequence of the variable region in Trypanosoma brucei completes the sequence analysis of the maxicircle component of mitochondrial kinetoplast DNA. Mol. Biochem. Parasitol. 1992;56:289–299. doi: 10.1016/0166-6851(92)90178-m. [DOI] [PubMed] [Google Scholar]
- 12.Simpson L. The mitochondrial genome of kinetoplastid protozoa: genomic organization, transcription, replication, and evolution. Annu. Rev. Microbiol. 1987;41:363–382. doi: 10.1146/annurev.mi.41.100187.002051. [DOI] [PubMed] [Google Scholar]
- 13.Herrmann J, Neupert W. Protein transport into mitochondria. Curr. Opin. Microbiol. 2000;3:210–214. doi: 10.1016/s1369-5274(00)00077-1. [DOI] [PubMed] [Google Scholar]
- 14.Bertrand KI, Hajduk SL. Import of a constitutively expressed protein into mitochondria from procyclic and bloodstream forms of Trypanosoma brucei. Mol. Biochem. Parasitol. 2000;106:249–260. doi: 10.1016/s0166-6851(99)00218-2. [DOI] [PubMed] [Google Scholar]
- 15.Long S, Jirku M, Ayala FJ, Lukes J. Mitochondrial localization of human frataxin is necessary but processing is not for rescuing frataxin deficiency in Trypanosoma brucei. Proc. Natl Acad. Sci. USA. 2008;105:13468–13473. doi: 10.1073/pnas.0806762105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tetaud E, Giroud C, Prescott AR, Parkin DW, Baltz D, Biteau N, Baltz T, Fairlamb AH. Molecular characterisation of mitochondrial and cytosolic trypanothione-dependent tryparedoxin peroxidases in Trypanosoma brucei. Mol. Biochem. Parasitol. 2001;116:171–183. doi: 10.1016/s0166-6851(01)00320-6. [DOI] [PubMed] [Google Scholar]
- 17.Brown BS, Stanislawski A, Perry QL, Williams N. Cloning and characterization of the subunits comprising the catalytic core of the Trypanosoma brucei mitochondrial ATP synthase. Mol. Biochem. Parasitol. 2001;113:289–301. doi: 10.1016/s0166-6851(01)00233-x. [DOI] [PubMed] [Google Scholar]
- 18.Lee C, Sedman J, Neupert W, Stuart R. The DNA helicase, Hmi1p, is transported into mitochondria by a C-terminal cleavable targeting signal. J. Biol. Chem. 1999;274:20937–20942. doi: 10.1074/jbc.274.30.20937. [DOI] [PubMed] [Google Scholar]
- 19.Folsch H, Guiard B, Neupert W, Stuart RA. Internal targeting signal of the BCS1 protein: a novel mechanism of import into mitochondria. EMBO J. 1996;15:479–487. [PMC free article] [PubMed] [Google Scholar]
- 20.Chaudhuri M, Nargang FE. Import and assembly of Neurospora crassa Tom40 into mitochondria of Trypanosoma brucei in vivo. Curr. Genet. 2003;44:85–94. doi: 10.1007/s00294-003-0427-y. [DOI] [PubMed] [Google Scholar]
- 21.Priest JW, Hajduk SL. Trypanosoma brucei cytochrome c1 is imported into mitochondria along an unusual pathway. J. Biol. Chem. 2003;278:15084–15094. doi: 10.1074/jbc.M212956200. [DOI] [PubMed] [Google Scholar]
- 22.Priest JW, Wood ZA, Hajduk SL. Cytochromes c1 of kinetoplastid protozoa lack mitochondrial targeting presequences. Biochim. Biophys. Acta. 1993;1144:229–231. doi: 10.1016/0005-2728(93)90178-i. [DOI] [PubMed] [Google Scholar]
- 23.Tasker M, Timms M, Hendriks E, Matthews K. Cytochrome oxidase subunit VI of Trypanosoma brucei is imported without a cleaved presequence and is developmentally regulated at both RNA and protein levels. Mol. Microbiol. 2001;39:272–285. doi: 10.1046/j.1365-2958.2001.02252.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kumar M, Verma R, Raghava G. Prediction of mitochondrial proteins using support vector machine and hidden Markov model. J. Biol. Chem. 2006;281:5357–5363. doi: 10.1074/jbc.M511061200. [DOI] [PubMed] [Google Scholar]
- 25.Small I, Peeters N, Legeai F, Lurin C. Predotar: A tool for rapidly screening proteomes for N-terminal targeting sequences. Proteomics. 2004;4:1581–1590. doi: 10.1002/pmic.200300776. [DOI] [PubMed] [Google Scholar]
- 26.Nakai K, Horton P. PSORT: a program for detecting sorting signals in proteins and predicting their subcellular localization. Trends Biochem. Sci. 1999;24:34–35. doi: 10.1016/s0968-0004(98)01336-x. [DOI] [PubMed] [Google Scholar]
- 27.Park K, Kanehisa M. Prediction of protein subcellular locations by support vector machines using compositions of amino acids and amino acid pairs. Bioinformatics. 2003;19:1656–1663. doi: 10.1093/bioinformatics/btg222. [DOI] [PubMed] [Google Scholar]
- 28.Reinhardt A, Hubbard T. Using neural networks for prediction of the subcellular location of proteins. Nucleic Acids Res. 1998;26:2230–2236. doi: 10.1093/nar/26.9.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu Z, Szafron D, Greiner R, Lu P, Wishart D, Poulin B, Anvik J, Macdonell C, Eisner R. Predicting subcellular localization of proteins using machine-learned classifiers. Bioinformatics. 2004;20:547–556. doi: 10.1093/bioinformatics/btg447. [DOI] [PubMed] [Google Scholar]
- 30.Xie D, Li A, Wang M, Fan Z, Feng H. LOCSVMPSI: a web server for subcellular localization of eukaryotic proteins using SVM and profile of PSI-BLAST. Nucleic Acids Res. 2005;33:W105. doi: 10.1093/nar/gki359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhasin M, Raghava G. ESLpred: SVM-based method for subcellular localization of eukaryotic proteins using dipeptide composition and PSI-BLAST. Nucleic Acids Res. 2004;32:W414. doi: 10.1093/nar/gkh350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nair R, Rost B. Mimicking cellular sorting improves prediction of subcellular localization. J. Mol. Biol. 2005;348:85–100. doi: 10.1016/j.jmb.2005.02.025. [DOI] [PubMed] [Google Scholar]
- 33.Emanuelsson O, Nielsen H, Brunak S, von Heijne G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J. Mol. Biol. 2000;300:1005–1016. doi: 10.1006/jmbi.2000.3903. [DOI] [PubMed] [Google Scholar]
- 34.Bannai H, Tamada Y, Maruyama O, Nakai K, Miyano S. Extensive feature detection of N-terminal protein sorting signals. Bioinformatics. 2002;18:298–305. doi: 10.1093/bioinformatics/18.2.298. [DOI] [PubMed] [Google Scholar]
- 35.Petsalaki E, Bagos P, Litou Z, Hamodrakas S. PredSL: a tool for the N-terminal sequence-based prediction of protein subcellular localization. Genom., Proteom. Bioinform. 2006;4:48–55. doi: 10.1016/S1672-0229(06)60016-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guda C, Fahy E, Subramaniam S. MITOPRED: a genome-scale method for prediction of nucleus-encoded mitochondrial proteins. Bioinformatics. 2004;20:1785–1794. doi: 10.1093/bioinformatics/bth171. [DOI] [PubMed] [Google Scholar]
- 37.Guda C, Guda P, Fahy E, Subramaniam S. MITOPRED: a web server for the prediction of mitochondrial proteins. Nucleic Acids Res. 2004;32:W372. doi: 10.1093/nar/gkh374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hua S, Sun Z. Support vector machine approach for protein subcellular localization prediction. Bioinformatics. 2001;17:721–728. doi: 10.1093/bioinformatics/17.8.721. [DOI] [PubMed] [Google Scholar]
- 39.Horton P, Park K, Obayashi T, Fujita N, Harada H, Adams-Collier C, Nakai K. WoLF PSORT: protein localization predictor. Nucleic Acids Res. 2007;35:W585. doi: 10.1093/nar/gkm259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Horton P, Park K, Obayashi T, Nakai K. Proceedings of the 4th Annual Asia Pacific Bioinformatics Conference APBC06. Taipei, Taiwan: 2006. Protein subcellular localization prediction with WoLF PSORT; pp. 39–48. [Google Scholar]
- 41.Claros M, Vincens P. Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur. J. Biochem. 1996;241:779–786. doi: 10.1111/j.1432-1033.1996.00779.x. [DOI] [PubMed] [Google Scholar]
- 42.Cui J, Liu Q, Puett D, Xu Y. Computational prediction of human proteins that can be secreted into the bloodstream. Bioinformatics. 2008;24:2370–2375. doi: 10.1093/bioinformatics/btn418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guo Y, Yu L, Wen Z, Li M. Using support vector machine combined with auto covariance to predict protein-protein interactions from protein sequences. Nucleic Acids Res. 2008;36:3025–3030. doi: 10.1093/nar/gkn159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burges C. A tutorial on support vector machines for pattern recognition. Data mining knowledge discovery. 1998;2:121–167. [Google Scholar]
- 45.Matthews B. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochimica et Biophys. Acta. 1975;405:442–451. doi: 10.1016/0005-2795(75)90109-9. [DOI] [PubMed] [Google Scholar]
- 46.Vassella E, Acosta-Serrano A, Studer E, Lee S, Englund P, Roditi I. Multiple procyclin isoforms are expressed differentially during the development of insect forms of Trypanosoma brucei. J. Mol. Biol. 2001;312:597–607. doi: 10.1006/jmbi.2001.5004. [DOI] [PubMed] [Google Scholar]
- 47.Pusnik M, Small I, Read LK, Fabbro T, Schneider A. Pentatricopeptide repeat proteins in Trypanosoma brucei function in mitochondrial ribosomes. Mol. Cell. Biol. 2007;27:6876–6888. doi: 10.1128/MCB.00708-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li H, Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Audic S, Claverie J. The significance of digital gene expression profiles. Genome Res. 1997;7:986–995. doi: 10.1101/gr.7.10.986. [DOI] [PubMed] [Google Scholar]
- 50.Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Uboldi AD, Lueder FB, Walsh P, Spurck T, McFadden GI, Curtis J, Likic VA, Perugini MA, Barson M, Lithgow T, et al. A mitochondrial protein affects cell morphology, mitochondrial segregation and virulence in Leishmania. Int. J. Parasitol. 2006;36:1499–1514. doi: 10.1016/j.ijpara.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 52.Dubchak I, Muchnik I, Holbrook S, Kim S. Prediction of protein folding class using global description of amino acid sequence. Proc. Natl Acad. Sci. USA. 1995;92:8700–8704. doi: 10.1073/pnas.92.19.8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eisenhaber F, Imperiale F, Argos P, Fr mmel C. Prediction of secondary structural content of proteins from their amino acid composition alone. I. New analytic vector decomposition methods. Proteins. 1996;25:157–168. doi: 10.1002/(SICI)1097-0134(199606)25:2<157::AID-PROT2>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 54.Eisenhaber F, Fr mmel C, Argos P. Prediction of secondary structural content of proteins from their amino acid composition alone. II. The paradox with secondary structural class. Proteins. 1996;25:169–179. doi: 10.1002/(SICI)1097-0134(199606)25:2<169::AID-PROT3>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 55.Sabatini R, Hajduk S. RNA ligase and its involvement in guide RNA/mRNA chimera formation. Evidence for a cleavage-ligation mechanism of Trypanosoma brucei mRNA editing. J. Biol. Chem. 1995;270:7233–7240. doi: 10.1074/jbc.270.13.7233. [DOI] [PubMed] [Google Scholar]
- 56.Prilusky J, Felder CE, Zeev-Ben-Mordehai T, Rydberg EH, Man O, Beckmann JS, Silman I, Sussman JL. FoldIndex: a simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics. 2005;21:3435–3438. doi: 10.1093/bioinformatics/bti537. [DOI] [PubMed] [Google Scholar]
- 57.von Heijne G. The signal peptide. J. Mem. Biol. 1990;115:195–201. doi: 10.1007/BF01868635. [DOI] [PubMed] [Google Scholar]
- 58.Halic M, Beckmann R. The signal recognition particle and its interactions during protein targeting. Curr. Opin. Struct. Biol. 2005;15:116–125. doi: 10.1016/j.sbi.2005.01.013. [DOI] [PubMed] [Google Scholar]
- 59.Mokranjac D, Neupert W. Protein import into mitochondria. Biochem. Soc. Transac. 2005;33:1019–1023. doi: 10.1042/BST20051019. [DOI] [PubMed] [Google Scholar]
- 60.Perocchi F, Jensen L, Gagneur J, Ahting U, von Mering C, Bork P, Prokisch H, Steinmetz L. Assessing systems properties of yeast mitochondria through an interaction map of the organelle. PLoS Genet. 2006;2:e170. doi: 10.1371/journal.pgen.0020170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, Walford GA, Sugiana C, Boneh A, Chen WK, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hashimi H, Cicova Z, Novotna L, Wen YZ, Lukes J. Kinetoplastid guide RNA biogenesis is dependent on subunits of the mitochondrial RNA binding complex 1 and mitochondrial RNA polymerase. RNA. 2009;15:588–599. doi: 10.1261/rna.1411809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nilsson D, Gunasekera K, Mani J, Osteras M, Farinelli L, Baerlocher L, Roditi I, Ochsenreiter T. Spliced leader trapping reveals widespread alternative splicing patterns in the highly dynamic transcriptome of Trypanosoma brucei. PLoS Path. 2010 doi: 10.1371/journal.ppat.1001037. (In press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Priest J, Hajduk S. Developmental regulation of Trypanosoma brucei cytochrome c reductase during bloodstream to procyclic differentiation. Mol. Biochem. Parasitol. 1994;65:291–304. doi: 10.1016/0166-6851(94)90080-9. [DOI] [PubMed] [Google Scholar]
- 65.Bochud-Allemann N, Schneider A. Mitochondrial substrate level phosphorylation is essential for growth of procyclic Trypanosoma brucei. J. Biol. Chem. 2002;277:32849–32854. doi: 10.1074/jbc.M205776200. [DOI] [PubMed] [Google Scholar]
- 66.Huang LJ, Wang L, Ma Y, Durick K, Perkins G, Deerinck TJ, Ellisman MH, Taylor SS. NH2-Terminal targeting motifs direct dual specificity A-kinase-anchoring protein 1 (D-AKAP1) to either mitochondria or endoplasmic reticulum. J. Cell. Biol. 1999;145:951–959. doi: 10.1083/jcb.145.5.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.