

Abstract
The effects of a thiazolidinone derivative, 3-[(3-trifluoromethyl)phenyl]-5-[(4-carboxyphenyl)methylene]-2-thioxo-4-thiazolidinone (or CFTRinh-172), on cystic fibrosis transmembrane conductance regulator (CFTR) gating were studied in excised inside-out membrane patches from Chinese hamster ovary cells transiently expressing wild-type and mutant CFTR. We found that the application of CFTRinh-172 results in an increase of the mean closed time and a decrease of the mean open time of the channel. A hyperbolic relationship between the closing rate and [CFTRinh-172] suggests that CFTRinh-172 does not act as a simple pore blocker. Interestingly, the potency of inhibition increases as the open time of the channel is increased with an IC50 in the low nanomolar range for CFTR channels locked in an open state for tens of seconds. Our studies also provide evidence that CFTRinh-172 can bind to both the open state and the closed state. However, at least one additional step, presumably reflecting inhibitor-induced conformational changes, is required to shut down the conductance after the binding of the inhibitor to the channel. Using the hydrolysis-deficient mutant E1371S as a tool as the closing rate of this mutant is dramatically decreased, we found that CFTRinh-172–dependent inhibition of CFTR channel gating, in two aspects, mimics the inactivation of voltage-dependent cation channels. First, similar to the recovery from inactivation in voltage-gated channels, once CFTR is inhibited by CFTRinh-172, reopening of the channel can be seen upon removal of the inhibitor in the absence of adenosine triphosphate (ATP). Second, ATP induced a biphasic current response on inhibitor-bound closed channels as if the ATP-opened channels “inactivate” despite a continuous presence of ATP. A simplified six-state kinetic scheme can well describe our data, at least qualitatively. Several possible structural mechanisms for the effects of CFTRinh-172 will be discussed.
INTRODUCTION
CFTR, a member of the ATP-binding cassette (ABC) transporter superfamily, is an epithelial chloride channel that plays a critical role in fluid absorption and secretion. Loss-of-function mutations of CFTR result in the lethal hereditary disease cystic fibrosis (Riordan et al., 1989; Welsh and Smith, 1993; Welsh et al., 2001), whereas hyperfunction of CFTR chloride channels, usually resulted from bacterial enterotoxins, constitutes the basic cause for secretory diarrhea (Bhattacharya, 1995; Barrett and Keely, 2000). Therefore, the development of pharmacological reagents that can modulate CFTR function bears practical implications in clinical medicine.
There has been tremendous progress in developing reagents that potentiate CFTR activity in recent years (Thiagarajah and Verkman, 2003; Van Goor et al., 2008). In fact, one of the compounds is now in stage III clinical trial (Van Goor et al., 2009). However, progress in discovering CFTR inhibitors has been slow. Several well-studied CFTR inhibitors lack specificity and assume low potency (Hwang and Sheppard, 1999; Schultz et al., 1999). These include glibenclamide, diphenylamine-2-carboxylate, 5-nitro-2(3-phenylpropyl-amino) benzoate, and niflumic acid. They seem to share a common mechanism of action, plugging the pore from the cytoplasmic side of the channel. High-throughput screening of diverse small molecules has made possible the discovery of two more selective inhibitors, a thiazolidinone derivative, or CFTRinh-172 (Ma et al., 2002), and glycine hydrazide, or GlyH-101 (Muanprasat et al., 2004), for CFTR. Unlike aforementioned pore blockers, GlyH-101 appears to act on the anion permeation pathway from the extracellular side of the channel. On the other hand, CFTRinh-172 works from the cytoplasmic side by inhibiting CFTR gating (Taddei et al., 2004).
Like other members of the ABC protein family, CFTR possesses two nucleotide-binding domains (NBDs), NBD1 and NBD2, in addition to two transmembrane domains (TMDs) that form the permeation pathway for chloride ions. Although the transporter members of this family use ATP binding and hydrolysis to drive the movement of substrate across the membrane, CFTR proteins use the same energy-harvesting machinery to drive the conformational changes involved in the opening and closing of the gate (Chen and Hwang, 2008; Hwang and Sheppard, 2009). The molecular mechanism underlying CFTR gating has been extensively studied with numerous nucleotide and phosphate analogues (e.g., Aleksandrov et al., 2002; Vergani et al., 2003; Zhou et al., 2005; Cai et al., 2006; Tsai et al., 2009). The major advantage of these reagents is that the site of action is in the ATP-binding pockets. However, this same feature also limits the mechanistic insights within the role of NBDs in CFTR gating. In contrast, CFTRinh-172 does not compete with ATP (Taddei et al., 2004). Thus, unraveling how CFTRinh-172 works and identifying its site of action could shed light on the gating mechanism beyond NBDs.
Despite the high specificity of CFTRinh-172 on CFTR gating, very limited studies have been conducted to understand how it works (Taddei et al., 2004; Caci et al., 2008). The inhibition was found to be potent, rapid, reversible, and voltage independent, despite the fact that CFTRinh-172 carries a negative charge. Patch clamp experiments (Taddei et al., 2004) have shown that CFTRinh-172 reduces the open probability (Po) but does not change the unitary conductance. It was also reported that the decrease in Po was caused by an increase of the mean closed time with the mean open time being unchanged. Based on the lack of voltage dependence and an absence of changes in the open time, it was proposed that CFTRinh-172 affects channel gating rather than occludes the pore, and that the inhibitor probably acts during the closed phase of the gating cycle. However, it is strange that the degree of inhibition for G551D and G1349D was of the same order of magnitude as that of wild-type (WT)-CFTR, despite a very different closed time among these three channels (Bompadre et al., 2007).
Another set of experiments was performed to test the possibility that CFTRinh-172 interacts with the pore region of CFTR proteins (Caci et al., 2008). The effect of mutations in amino acid residues of the sixth transmembrane helix of CFTR on the apparent affinity of CFTRinh-172 has been examined. Fluorescence assays and whole cell patch clamp experiments have shown that mutations of several residues in this segment alter the sensitivity to CFTRinh-172. However, the residue in this region of CFTR, when mutated, that exhibits the most changes in the apparent affinity for CFTRinh-172 is R347, which does not line the permeation pathway (Cotten and Welsh, 1999; Bai et al., 2010).
The purpose of the current work was to elucidate the kinetic mechanism of CFTR inhibition by CFTRinh-172. Contrary to previous reports, we found that the mean open time of the CFTR channel is decreased by CFTRinh-172, and that prolongation of the channel open time leads to more potent inhibition. Although these findings support the idea that the inhibitor acts when the channel is opened, the nonlinear relationship between the closing rate and [CFTRinh-172] implies a more complicated mechanism of inhibition than a simple pore occlusion. Our data suggest that the binding of CFTRinh-172 to either the open or the closed state induces conformational changes that “inactivate” the channel. This inactivation emulates the well-established inactivation mechanism for voltage-gated cation channels. The structural and functional implications of our results are discussed.
MATERIALS AND METHODS
Cell culture and transient expression of CFTR
NIH3T3 cells stably expressing WT-CFTR channels and Chinese hamster ovary (CHO) cells were grown at 37°C and 5% CO2 in Dulbecco’s modified Eagle’s medium supplemented with 10% FBS. WT or mutant CFTR channels were expressed transiently in CHO cells. The cDNA constructs were cotransfected with pEGFP-C3 (Takara Bio Inc.) encoding green fluorescent protein using transfection reagent (PolyFect; QIAGEN) according to the manufacturer’s protocols. Before patch clamp recordings, cells were plated on sterile glass chips in 35-mm tissue culture dishes and incubated at 27°C for 1–2 d.
Inside-out patch clamp recordings
Patch clamp pipettes were made from borosilicate capillary glass using a two-stage vertical puller (Narishige). The pipette tips were fire polished with a homemade microforge to yield a pipette resistance of 3–5 MΩ measured in the working solution. Before the recording, a glass chip with cells grown on it was placed into a continuously perfused chamber on the stage of an inverted microscope (Olympus). CFTR currents were recorded at room temperature (22–24°C) with an amplifier (EPC9; HEKA). Data were filtered at 100 Hz with an eight-pole Bessel filter (Warner Instruments) and captured onto a hard disk at a sampling rate of 500 Hz. The membrane potential was held at −60 mV. Downward deflections in the current trace indicate channel opening. The current traces were reversed in polarity for data presentation.
The standard pipette solution contained (in mM): 140 NMDG-Cl, 2 MgCl2, 5 CaCl2, and 10 HEPES, pH 7.4 with NMDG. The bath solution contained (in mM): 145 NaCl, 5 KCl, 2 MgCl2, 1 CaCl2, 5 glucose, 5 HEPES, and 20 sucrose, pH 7.4 with NaOH. The perfusion solution after patch excision contained (in mM): 150 NMDG-Cl, 10 EGTA, 10 HEPES, 8 TRIS, and 2 MgCl2, pH 7.4 with NMDG.
Data analysis
The steady-state mean currents used for calculation of the degree of inhibition were calculated with IGOR software (WaveMetrics). The mean baseline currents were subtracted before the data were used for calculations. The degree of inhibition by CFTRinh-172 was calculated according to the equation:
where I0 and Iinh-172 represent uninhibited and inhibited current, respectively.
Kinetic parameters were extracted from patches with five or fewer simultaneous channel opening steps using a program developed by L. Csanády (for details, see Csanády, 2000). For a given [CFTRinh-172], the mean open (closed) time in the presence and absence of CFTRinh-172 was calculated as an average over 5–10 patches.
All averaged data are presented as means ± SEM; n represents the number of patches. Student’s t tests were performed using SigmaPlot (version 4.0; Systat Software Inc.), and effects were considered significant if P < 0.05.
Reagents
MgATP, PPi (tetrasodium salt), and PKA were purchased from Sigma-Aldrich. N6-(2-phenylethyl)-ATP (P-ATP) was purchased from BioLog Life Science Institute. MgATP and PPi were stored as a 250-mM stock solution at −20°C. 10 mM P-ATP stock solution was stored at −80°C. The [PKA] used in this study was 1.6 µg/ml.
Online supplemental material
We performed Monte Carlo simulations based on the proposed Scheme 3 to reproduce some of our major findings. The online supplemental material includes simulated results mimicking experimental data shown in Figs. 1, 3, 5, 6, 8, and 10. It is available at http://www.jgp.org/cgi/content/full/jgp.201010518/DC1.
Figure 1.

Voltage-independent inhibition of WT-CFTR by CFTRinh-172. (A) Continuous WT-CFTR current traces showing reversible inhibition by 1 or 10 µM CFTRinh-172. In an inside-out patch clamp experiment, after the channels in the patch were activated with 1 mM MgATP plus PKA, we applied 1 mM MgATP, followed by 1 mM MgATP plus 1 or 10 µM CFTRinh-172. The current recovery phase upon removal of 1 µM CFTRinh-172 was fitted with a single-exponential function, yielding a rate constant of 0.031 s−1 (0.024 ± 0.003 s−1; n = 17). (B) The concentration dependence of CFTR inhibition fitted with the Hill equation with IC50 = 1.16 ± 0.09 µM and Hill coefficient n = 1.31 ± 0.12. Each data point represents values determined from 6–14 patches. The error bars are smaller than the symbols. (Inset) The structure of CFTRinh-172. (C) Representative I-V curves in the presence (red) or absence (blue) of 1 µM CFTRinh-172. A voltage ramp over ±100 mV was applied to a patch yielding macroscopic currents. (D) Percent inhibition at different voltages was obtained from C. Data between −10 and 10 mV were discarded because of the small size of the signal. Similar observations were made in five patches. Inh-172 is the abbreviation for CFTRinh-172 in this and subsequent figures.
Figure 3.

The relationship between [CFTRinh-172] and the closing rate. Mean open times at different concentrations of CFTRinh-172 were obtained from single-channel kinetic analysis, as described in Materials and methods. Each data point represents values determined from 7–10 patches. This relationship follows a saturation function, suggesting the existence of an additional step after the binding of the inhibitor to the open channel.
Figure 5.

Inhibition of locked-open CFTR by CFTRinh-172. CFTR channels were locked into an open state by using ATP plus PPi for WT-CFTR (A) or ATP for E1371S-CFTR (B). Once a steady-state current was attained, CFTRinh-172 was applied. In either case, the current was inhibited nearly completely. During the activation period, the short flat and vertical lines are a result of saturation of the amplifier and subsequent changes in the amplifier gain. (C) Dose–response relationships of CFTRinh-172 for WT-CFTR locked open by PPi (green symbols) and E1371S-CFTR locked open with ATP (blue symbols). Each data point represents values determined from four to nine patches. The fitted dose–response curve for WT channels gated with ATP was shown for comparison (red line). Although an accurate curve fit for the dose response cannot be obtained for PPi locked-open channels, a rough estimate of the IC50 of ∼10 nM seems reasonable.
Figure 6.

Slow recovery from inhibition for the locked-open CFTR. (A–C) Representative traces showing recovery after inhibition of locked-open CFTR channels by CFTRinh-172. (D) The diagram shows the average rate of recovery from inhibition by CFTRinh-172 for WT and E1371S CFTR locked in an open state. The rate of recovery for ATP-gated WT-CFTR was plotted for comparison. The mean rate constants of recovery are: 0.024 ± 0.003 s−1 (n = 17) for ATP-gated WT-CFTR; 0.0042 ± 0.0011 s−1 (n = 10) for WT-CFTR locked in an open state with ATP plus PPi; 0.0026 ± 0.0004 s−1 (n = 18) for E1371S; and 0.0039 ± 0.0005 s−1 (n = 18) for E1371S/ΔR.
Figure 8.

The binding and inhibition of CFTRinh-172 in the closed state of E1371S/ΔR-CFTR channels. The inhibitor was applied for ∼20 s in the absence of ATP and was removed from the perfusion solution for 2 s before the application of ATP. Immediately after the application of ATP, one can observe a transient current increase. However, subsequent spontaneous inactivation of open channels without the inhibitor in the perfusion solution suggests that the binding of the inhibitor does happen in the closed state. Subsequent recovery of the current in the presence of ATP took place very slowly. Fitting the recovery phase with an exponential function generates a rate constant of 0.0035 ± 0.0005 s−1, n = 12, which is not different from the rate constant for inhibition that took place during the open state (Fig. 6).
Figure 10.

Inactivation of WT-CFTR by CFTRinh-172. A continuous recording of WT-CFTR channels exposed to CFTRinh-172 during the closed state. Phosphorylated channels were first exposed to 5 µM CFTRinh-172 in the absence of ATP. In the continuous presence of the inhibitor, the application of 1 mM ATP elicited a biphasic current response with an apparent inactivation phase. In the same patch, when ATP was applied after the inhibitor was removed, a different biphasic current response appeared (see supplemental text for simulated results based on Scheme 3).
RESULTS
Voltage-independent inhibition of CFTR current by CFTRinh-172
We studied the effect of CFTRinh-172 on CFTR Cl− channels in excised inside-out membrane patches from CHO cells transfected with cDNA encoding WT or mutant CFTR. The channels were first activated by phosphorylation to a steady state with 1.6 µg/ml PKA and 1 mM MgATP. After activation, patches were exposed to 1 mM MgATP, and then to 1 mM MgATP plus different concentrations of CFTRinh-172. Fig. 1 A shows representative recordings where 1 and 10 µM CFTRinh-172 cause ∼50 and ∼90% reduction of the original current, respectively, and this inhibition is reversible, as removal of the inhibitor recovers all the current. The current recovery phase upon washout of the inhibitor can be fitted with a single-exponential function, yielding a time constant of ∼40 s (42.0 ± 5.3 s; n = 17). The concentration dependence of the effect of CFTRinh-172 is shown in Fig. 1 B. Fitting the dose–response relationship with the Hill equation yields an IC50 of ∼1.1 µM and a Hill coefficient of ∼1, results very similar to those reported by Taddei et al. (2004). It should be noted that even at 10 µM (>6 × IC50), the inhibitor did not abolish all the current. Also as reported by Taddei et al. (2004), we found that the effect of CFTRinh-172 on CFTR is not sensitive to the membrane potential (Fig. 1, C and D).
Effects of CFTRinh-172 on single-channel kinetics
Although the results shown in Fig. 1 basically agree with what has been reported by Taddei et al. (2004), some differences emerged when we examined single-channel kinetics of CFTR in the presence of CFTRinh-172. The major kinetic effect of CFTRinh-172 on CFTR was reported to be an increase of the closed time without changes of the open time. For an inhibitor that only prolongs the closed time, a simple three-state scheme, Cinh↔C↔O, can be used to explain the results. However, this scheme will not be sufficient if the open time is altered by the inhibitor.
Fig. 2 shows recordings of WT-CFTR in inside-out membrane patches containing few channels so that individual openings and closings can be discerned and microscopic kinetic parameters can be obtained. Even by eye inspection, one can observe a decrease of the open time accompanying a prolonged closed time in the presence of CFTRinh-172. This effect of CFTRinh-172 on the open time is even more prominent on P-ATP–opened channels, as this high-affinity ATP analogue increases the open time of WT-CFTR (Fig. 2, B and C). At a macroscopic level, P-ATP–gated channels also seem more sensitive to CFTRinh-172 (66% inhibition for P-ATP– vs. 51% inhibition for ATP-gated channels at 1 µM CFTRinh-172). Shortening of the open time implies that the inhibitor works when the channel is in the open state and is typically one of the main characteristics of an open-channel blocker. Therefore, it is important to first examine the possibility that CFTRinh-172 acts by occluding the pore as a simple open-channel blocker.
Figure 2.

Effects of CFTRinh-172 on the open time of WT-CFTR. (A and B) Single-channel traces showing a reversible decrease of the open time in the presence of CFTRinh-172 for (A) ATP- and (B) P-ATP–gated channels. Dashed lines represent the current level of the closed state. (C) The mean open time (τo) decrease as a result of 5 µM CFTRinh-172 for channels opened with ATP (n = 10) or P-ATP (n = 7). The mean closed time (τc) was prolonged by CFTRinh-172 (τc = 637.3 ± 59.2 ms in the absence of CFTRinh-172 and 2,580.5 ± 623.4 ms in the presence of 5 µM CFTRinh-172). For channels opened with P-ATP, τc = 670.8 ± 55.8 ms and 2,058.3 ± 254.2 ms in the absence and presence of CFTRinh-172, respectively.
The mechanism of action for simple pore blockers can be described by the minimum scheme C↔O↔B, where C↔O and O↔B represent the ATP-dependent gating and inhibitor-dependent blocking steps, respectively. This scheme predicts a linear relationship between the closing rate of the channel and the concentration of the inhibitor as demonstrated for glibenclamide blockade of CFTR (Zhou et al., 2002). We measured the closing rate (i.e., 1/τo) at different [CFTRinh-172]. Interestingly, instead of a simple linear relationship between the closing rate and [CFTRinh-172], our results show a hyperbolic relationship (Fig. 3). Because shortening of the open time itself suggests that the inhibitor can bind to the open state (also see below), this saturation function can be described by a more expanded scheme: C↔O↔Oinh↔Iinh (Scheme 1). Here, the binding of the inhibitor to the open state (Oinh) by itself does not abolish the current, but it may induce some conformational changes that result in a nonconducting or “inactivated” state, Iinh. Alternatively, the inhibitor may bind to the closed state, but the inhibitor-bound channels open with a shorter open time; i.e., O’inh↔Cinh↔C↔O (Scheme 2).
Effects of the open time on the apparent affinity of CFTRinh-172
The observation that, at a submaximal [CFTRinh-172], the degree of inhibition is higher for WT-CFTR opened with P-ATP than that with ATP suggests a relationship between the stability of the open state and IC50. As shown in Fig. 2 C, the decrease of the mean open time by CFTRinh-172 is greater if CFTR channels are opened with P-ATP instead of ATP.
To further explore this relationship between the mean open time and the potency of CFTRinh-172, we tested CFTRinh-172 on a CFTR mutant, D1370N-CFTR, which exhibits an open time of ∼1 s (Bompadre et al., 2005). Fig. 4 A shows a representative current trace with two opening steps. Microscopic kinetic analysis revealed a mean open time of 1,017.1 ± 45.4 ms (n = 5) for this mutant CFTR. Fig. 4 B demonstrates the effects of 1 µM CFTRinh-172 on the macroscopic current of D1370N-CFTR. Compared with WT-CFTR (∼50% inhibition), >80% inhibition was observed at this concentration of CFTRinh-172. This more efficacious inhibition is accompanied by an even slower rate of recovery upon washout of CFTRinh-172 in the continuous presence of ATP. The time course of current recovery can be fitted well with a single-exponential function (Fig. 4 B, red line), yielding a rate constant of 0.006 ± 0.001 s−1 (n = 15), which is approximately fivefold slower than the rate of recovery for WT-CFTR (Fig. 1). This decrease in IC50 for CFTRinh-172 is best seen with the dose–response relationships (Fig. 4 C) that show a graded leftward shift as the mean open time is prolonged.
Figure 4.

Prolonging the mean open time of CFTR increases the potency of CFTRinh-172. (A) Representative single-channel trace for D1370N-CFTR, a hydrolysis-deficient mutant with a mean open time of ∼1 s. (B) A continuous current recording showing a reversible inhibition of macroscopic D1370N-CFTR currents by 1 µM CFTRinh-172. (C) Dose–response relationships of CFTRinh-172 for WT-CFTR gated by ATP or P-ATP and D1370N-CFTR gated by ATP. Note a graded leftward shift of the dose–response relationship as the mean open time is prolonged. The experimental data were fitted with the Hill equation. Fitting parameters: for P-ATP–gated WT channels: IC50 = 0.37 ± 0.22 µM and Hill coefficient, n = 0.69 ± 0.38; for D1370N-CFTR: IC50 = 0.064 ± 0.007 µM and n = 1.26 ± 0.17. Data points represent the average values determined from 4–14 patches.
The results shown in Fig. 4 suggest that the potency of CFTRinh-172 is determined by the open time: the longer the open time, the lower the IC50. For D1370N-CFTR with an open time of ∼1 s, the IC50 is already in the nanomolar range. We wondered if the potency can be increased additionally if we further prolong the open time, which can be accomplished by locking open WT-CFTR with ATP and PPi (Tsai et al., 2009). If the hypothesis that the increase in the open time leads to more potent inhibition is correct, one can expect a significant increase in the potency of CFTRinh-172 for channels locked in a stable open state for tens or hundreds of seconds. Fig. 5 A shows a continuous current trace of WT-CFTR activated with PKA, ATP, and PPi. Once the current reached a steady state, 2 µM CFTRinh-172 was applied. The macroscopic current immediately declined to an indiscernible level. A similar observation was made with E1371S-CFTR, a hydrolysis-deficient mutant with an open time of ∼100 s (Fig. 5 B). Fig. 5 C shows the dose–response relationships of CFTRinh-172 for WT-CFTR locked open with ATP and PPi (green circles), or E1371S-CFTR locked open with ATP (blue squares). Indeed, the dose–response curve shifts further to the left. It is technically difficult to assess the effect of CFTRinh-172 at a concentration <5 nM because the inhibition at very low inhibitor concentrations is extremely slow, so that the time course of CFTR inhibition by CFTRinh-172 is easily distorted by current decrease as a result of channel rundown. Therefore, we were not able to obtain a more complete dose response for locked-open WT channels. Nevertheless, the data shown in Fig. 5 C suggest an IC50 of ≤∼10 nM. Besides a difference in IC50 for the inhibitor, there is also a difference in maximal percent inhibition as seen in Fig. 5 C (>99% inhibition for locked-open channels vs. ∼90% for ATP-gated WT-CFTR).
The extremely high stability of the inhibited state for locked-open channels is also represented in the washout phase of the experiment. Fig. 6 A shows that once the locked-open WT channels are inhibited, removal of CFTRinh-172 in the continuous presence of ATP results in an extremely slow recovery of the current. When we fitted the time course of the current recovery phase with a single exponential function, the time constant was on the order of hundreds of seconds. A similar observation was made with E1371S-CFTR channels locked open with ATP (Fig. 6 B). One technical difficulty with this type of long-lasting experiments is the possibility of rundown induced by membrane-associated phosphatases (e.g., Weinreich et al.,1999), although during the slow current recovery phase, we often added PKA into the system to ensure that the observed slow current recovery is not a result of dephosphorylation of CFTR. To further verify this slow recovery, we quantified this recovery phase using a phosphorylation-independent construct characterized previously (i.e., E1371S mutation in a construct without the R domain, or E1371S/ΔR; see Bompadre et al., 2005). Again, the time constant of the current recovery for E1371S/ΔR is ∼6 min (Fig. 6, C and D). Because the activity of E1371S/ΔR-CFTR is independent of phosphorylation, in the following sections we will use this construct to study kinetics of current recovery from inhibition when dephosphorylation is a concern.
The results in Figs. 5 and 6 are important in several aspects. First, the immediate decrease of the macroscopic currents upon the addition of CFTRinh-172 indicates that the channel can be readily inhibited from the open state as proposed in Scheme 1. Although the observation that the open time of ATP-gated WT-CFTR is shortened by CFTRinh-172 (Fig. 2) suggests a direct effect of CFTRinh-172 on the open state, one can still argue that CFTRinh-172 binds only to the closed state, but the inhibitor-bound closed channel opens very slowly, and once it opens, the open time is short (Scheme 2). Although it remains possible that inhibitor-bound channels may open for a shorter time, Scheme 2 by itself cannot explain the immediate decrease of the current generated from locked-open channels, as the mean open time for the locked-open channels is tens to hundreds of seconds. Second, the dramatic leftward shift of the dose–response relationship for locked-open channels, however, also disapproves Scheme 1 because this scheme predicts that IC50 should be inversely proportional to Po (IC50 ∝ 1/Po). As the Po of ATP-gated WT-CFTR is ∼40–50% of that for channels locked open with ATP and PPi, a twofold change of IC50 is expected if Scheme 1 is correct. Instead, we observed a >100-fold change of IC50. Third, that IC50 for CFTRinh-172 is decreased to 10 nM as the open time is prolonged to tens of seconds suggests that stabilizing the open state somehow stabilizes the binding of CFTRinh-172. Fourth, the extremely stable inhibited state for channels locked in the open state provides a unique time window to address several kinetic questions concerning how CFTRinh-172 works, as described in the following sections.
Can the inhibited channel reopen without ATP?
We have shown above that CFTRinh-172 is unlikely to be a simple open pore blocker. One possible mechanism for CFTR inhibition by CFTRinh-172 could be promotion of ATP dissociation and/or separation of the NBD dimer, as ATP-triggered NBD dimerization is responsible for channel opening (Vergani et al., 2005). In other words, CFTRinh-172 works by promoting the closure of the gate opened by ATP. This idea can be tested by removing CFTRinh-172 in the absence of ATP once the locked-open channels are inhibited. If the inhibitor works by disengaging NBD dimers and thereby promoting ATP dissociation, one would predict that, once inhibited, channels will need ATP to open again. Fig. 7 shows such an experiment. Once E1371S-CFTR channels were activated with PKA and ATP, the channels were subsequently inhibited by applying 5 µM CFTRinh-172. The current was diminished within a few seconds. The inhibitor was then removed in the absence of ATP. One can clearly discern the reopening of at least four channels in this patch. It can also be seen that there are at least two types of openings in the absence of ATP. Some of those brief openings (Fig. 7, blue inset) probably represent ATP-independent events observed normally in the absence of ATP for E1371S-CFTR (Bompadre et al., 2005). On the other hand, the locked-open state is discernable by its characteristic long-lasting events with flickery closures (Fig. 7, red inset). Based on the idea that the open state represents an NBD dimer (Vergani et al., 2005; Zhou et al., 2006; Tsai et al., 2009), we conclude that CFTRinh-172 does not promote the closing of the gate by disrupting the NBD dimer. More likely, the two NBDs remain engaged (i.e., the ligand ATPs remain bound), whereas the current is inhibited. It is interesting to note that this reopening in the absence of ATP is reminiscent of one well-known phenomenon for the voltage-gated cation channels that inactivate after activation by depolarization. These inactivated channels can reopen transiently when the membrane potential is stepped to hyperpolarizing voltages, as the inactivated channel will sojourn to the open state before the activation gate closes (Ruppersberg et al., 1991).
Figure 7.

Reopening of inhibited E1371S-CFTR channels upon removing CFTRinh-172. A continuous recording of E1371S-CFTR, a hydrolysis-deficient mutant with a prolonged open time, shows reopening of the inhibited channels in the absence of ATP upon removal of CFTRinh-172. Note that the channels can reopen into the locked-open state. Because some of the reopened channels are expected to close during such a long recording in the absence of ATP, the number of reopened channels is underestimated if one simply counts the opening events. Similar observations were made in more than 30 patches.
The other important kinetic information from data shown in Fig. 7 is the rate of reopening upon removal of CFTRinh-172. It takes tens of seconds to observe the first reopening of the channels in Fig. 7. Thus, the inactivated state with both CFTRinh-172 and ATP bound is exceedingly stable and can be considered an absorbing state.
Does CFTRinh-172 bind to the closed state?
As described above, a lack of an inverse relationship between IC50 and Po suggests that Scheme 1 where CFTRinh-172 only binds to the open state is incorrect, despite the fact that this scheme can explain the reopening of the inhibited channels in the absence of ATP (Fig. 7). More likely, CFTRinh-172 can also bind to the closed state. Again, E1371S-CFTR allows us to address this question. Fig. 8 shows a recording where a super-maximal concentration of CFTRinh-172 was applied for 20 s in the absence of ATP when the channels are mostly closed. The inhibitor was removed for ∼2 s before ATP was added. If the inhibitor can only bind to the open state, ATP will lock open all of the channels because the inhibitor is no longer in the superfusion solution. On the other hand, if the inhibitor can bind to the closed state and the inhibitor-bound closed channels do not respond to ATP, one expects that ATP will not be able to elicit any current. Neither of these two scenarios holds. As seen in Fig. 8, the application of ATP induced a biphasic current change: an initial increase of the current, followed by step-wise shutdown of individual channels. This surprising result indicates that CFTRinh-172 can bind to the closed state, as ATP fails to lock open the channel. The extremely slow recovery of the current after the biphasic response indicates that at the beginning of the recovery phase, nearly all channels are absorbed in the stable inactivated state (see below for details). For the biphasic response itself, we propose that the CFTRinh-172–bound closed channel can be opened by ATP. However, unlike inhibitor-free E1371S channels that are locked open by ATP, the current from inhibitor-bound channels, first elicited by ATP, dropped by itself within a few seconds, as the inhibitor-bound open channels are now inactivated. Again, this observation is somewhat analogous to the closing of the inactivation gate of voltage-gated ion channels—the inactivation gate is not closed until the activation gate is opened (Armstrong, 1971, 1974; Armstrong and Bezanilla, 1977).
Although the biphasic current response to ATP after the channels are exposed to CFTRinh-172 during the closed state was seen consistently, the relative magnitude of the initial current induced by ATP varies. In 12 patches, the ratio of the peak amplitude of the transient current to the original current is 0.38 ± 0.03. Nevertheless, in the continuous presence of ATP, the whole current recovery takes place very slowly, as those shown in Fig. 6, where the inhibitor was applied to the open channels. Fitting the time course of current recovery with a single-exponential function produces a rate constant of 0.0035 ± 0.0005 s−1, which is not different from the rate of current recovery for channels inhibited at an open state (i.e., Fig. 6). This result suggests that even when the inhibitor hits the closed state, ATP binding (which presumably engages the two NBDs) will render nearly all the channels absorbed in the energetically stable inactivated state.
Three implications from the results shown in Figs. 7 and 8 are worth mentioning here. First, the similarity of these data to the inactivation (Fig. 8) and recovery from inactivation (Fig. 7) of voltage-gated channels implies the operation of two different gates, one controlled by ATP-induced NBD dimerization and a second “inactivation” gate that closes after CFTRinh-172 binds to the channel. Second, the direct demonstration that CFTRinh-172 can bind to the closed state necessitates a modification of Scheme 1. Third, the relatively small amplitude of the peak current in the biphasic response shown in Fig. 8 suggests that some of the inhibitor-bound closed channels do not respond to ATP. In other words, the inhibitor-bound closed state can travel to a different inactivated state. We therefore added to Scheme 1 two additional states, Cinh and I’inh, representing CFTRinh-172–bound closed state and inactivated CFTRinh-172–bound closed state, respectively (Scheme 3 below). It should also be noted that Iinh and I’inh are fundamentally different in that, as described above, Iinh has both ATP and inhibitor that remain bound, whereas the I’inh state has not been exposed to ATP.
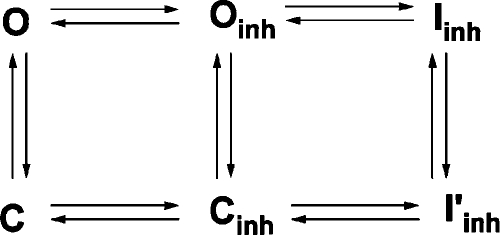 |
(SCHEME 3) |
In this scheme (Scheme 3), three vertical transitions represent ATP-dependent gating. In theory, there should be at least ATP-free and ATP-bound closed states, but they are lumped together because millimolar concentrations of ATP were used in our experiments so that the binding step is mostly saturated. The two horizontal transitions on the left represent binding and unbinding of CFTRinh-172 for the open state and the closed state, respectively, whereas the two horizontal steps on the right represent inactivation and recovery from inactivation of the open and closed channels.
More detailed examination of this scheme can be found in the supplemental text. Here, we will briefly summarize how the results shown above lead to this scheme, and then demonstrate a few experiments to further support the validity of the scheme. First, our experiments demonstrate that CFTRinh-172 can bind to both the open state (Fig. 5) and the closed state (Fig. 8); i.e., O↔Oinh and C↔Cinh. The binding of CFTRinh-172 to either the open state (Fig. 5) or the closed state (Fig. 8) does not lead to inhibition directly; an additional step is required; i.e., Oinh↔Iinh and Cinh↔I’inh. Fig. 8 also shows that the inhibitor-bound closed channels can be opened by ATP; i.e., Cinh↔Oinh. Because the Iinh→Oinh transition takes at least tens of seconds (Fig. 7), it is likely that Iinh, a state in which both ATP and inhibitor remain bound, is energetically very stable. In contrast, I’inh, a state with only inhibitor bound, is probably less stable because even after exposure of the closed channels to CFTRinh-172 for 20 s, many channels can be still opened by ATP (Fig. 8). Thus, in Cinh↔I’inh, I’inh does not serve as an absorbing state. Perhaps when ATP is applied, I’inh will travel to the absorbing state, Iinh.
Scheme 3 depicts two exit pathways for the captive inactivated state, Iinh: one “visible” pathway (Iinh→Oinh) where inactivated channels reopen first before they dissipate into other states, and the other “invisible” pathway (Iinh→I’inh), so called because no current will be seen during the recovery of the channels from Iinh. Although the experimental results in Fig. 7 demonstrate the reopening upon washout of CFTRinh-172 in the absence of ATP, the next set of experiments will address specifically the invisible pathway.
Unlike the visible pathway, the invisible one involves only transitions through nonconducting states. Because we cannot “see” these events directly, the existence of this pathway can only be inferred if the visible one fails to explain the experimental data. If Scheme 3 is correct, once the channels are fully inhibited to Iinh, not all channels recover visibly. This can be examined with the locked-open channels, as their reopenings are readily discernible. Fig. 9 A shows such an experiment in patches containing only a few channels so that the number of reopening events can be counted manually. Because the experimental protocol demands long stationary recordings, the phosphorylation-independent ΔR construct was used. E1371S/ΔR CFTR channels were opened with 1 mM ATP and then inhibited with 2 µM CFTRinh-172. After removal of ATP and the inhibitor from the solution, only one channel reopened in ∼200 s of recording. However, subsequent application of ATP opened three channels. Thus, the total number of channels recovered from inactivation exceeds the number of channels recovered from the reopening (or visible) pathway. Thus, some channels recover without going through the visible pathway.
Figure 9.

Recovery of inhibited channels bypasses the reopening step. (A) A continuous current trace of E1371S/ΔR-CFTR channels showing that some of the inhibited channels recover from inhibition without reopening. Similar observations were made in seven patches containing two to four channels. (B) Faster recovery of inhibited E1371S/ΔR-CFTR channels in the absence of ATP. 2 min after the removal of CFTRinh-172, more current can be elicited by ATP than that with ATP in the washout solution (Fig. 6 C).
In patches that generate macroscopic currents, one can test the existence of the pathway other than reopening by comparing current recovery upon removal of CFTRinh-172 in the presence or absence of ATP. Our experiments show that recovery from inhibition is paradoxically slowed down by the presence of ATP. In Fig. 9 B, one can see that ∼2 min after the inhibitor was removed, 45.6 ± 3.6% (n = 16) of the initial current was obtained with ATP when the washout solution contained no ATP. On the other hand, if the washout solution contained ATP, 25.4 ± 2.8% (n = 10) of current recovery occurred (Fig. 6 C). The phenomenon can be explained based on Scheme 3. In the absence of ATP, there are several ways of dissipation from the absorptive state (Iinh): Iinh→Oinh→O→C, Iinh→Oinh→Cinh→C, and Iinh→I’inh→Cinh→C. Because both ATP and CFTRinh-172 are removed from the system, most dissipative pathways are irreversible. In contrast, when the washout phase occurs in the presence of ATP, the reactions Iinh→I’inh and Oinh→Cinh are reversed and therefore two of the dissipative pathways are greatly slowed down. Thus, only one pathway is left for the recovery after inhibition.
DISCUSSION
In this study, we investigated the mechanism of CFTR inhibition by the thiazolidinone derivative, CFTRinh-172, a specific inhibitor identified by high-throughput screening assays (Ma et al., 2002). Here, we will first draw several model-independent conclusions based on our data and then discuss the validity and potential limitations of the proposed kinetic scheme. Finally, we will speculate on the possible structural mechanism for the action of CFTRinh-172.
CFTRinh-172 as a gating modulator
Contrary to a previous report (Taddei et al., 2004), we found that CFTRinh-172 causes both a decrease in the open time and an increase in the closed time for WT-CFTR gated by ATP. This finding, together with the observation that CFTRinh-172 inhibits locked-open CFTR channels without a significant delay, indicates that the inhibitor can bind to the open state. As the channels preexposed to CFTRinh-172 during the closed state respond to ATP differently than the channels without preexposure, one has to conclude that CFTRinh-172 can also bind to the closed state. Thus, the binding site for CFTRinh-172 is accessible in both the open state and the closed state.
Interestingly, unlike a simple pore-plugging mechanism, the binding of CFTRinh-172 to either the open state or the closed state does not cause immediate inhibition of the current. The hyperbolic relationship between [CFTRinh-172] and the closing rate suggests that an additional step is required for the inhibitor-bound open state to travel to the inactivated state. This additional step may reflect a conformational change initiated by the binding of the inhibitor. Our studies do not provide any information on the exact conformational changes involved. It should also be noted that we cannot completely rule out the possibility that CFTRinh-172 binds to the pore, as a more complicated mechanism of blockade that involves a docking and a blocking step may explain some of our data (Zhou et al., 2001; Zhang et al., 2009). However, a lack of voltage dependence (Fig. 1) (Ma et al., 2002; Taddei et al., 2004) is more in line with an allosteric mechanism rather than simply plugging the pore.
One of the most striking observations is the correlation between the potency of CFTRinh-172 and the open time: the longer the open time, the lower the IC50. This result suggests that the action of CFTRinh-172 on the open state is different from its effect on the closed state, although, as described above, both states can bind the inhibitor. A major difference between inhibitor-bound open state and inhibitor-bound closed state is noted in our experiments where CFTRinh-172 was applied at different times of the gating cycle. Although the binding of CFTRinh-172 to the locked-open channels leads to an energetically captive inactivated “open” state, the inactivated “closed” state is not as stable because even after a long exposure of the closed channels to CFTRinh-172, a significant amount of the inhibitor-bound closed channels is not inactivated, as they can still be opened by ATP (Fig. 8).
The conclusion that the inactivated open state, Iinh, is extremely stable energetically is also supported by experimental results on the recovery of inactivation. It takes tens of minutes for the current to recover upon washout of the inhibitor in the presence of ATP for the locked-open channels. Even for WT-CFTR under normal ATP-dependent gating, a time constant of ∼40 s for current recovery in the presence of ATP is surprisingly long considering that the open states (inhibitor-bound or inhibitor-free) are allowed to close rapidly. Perhaps the most important observation underscoring this line of reasoning is the long duration before reopening is seen upon washout of CFTRinh-172 in the absence of ATP for locked-open channels. Although these events are difficult to quantify because of the small size of the signal, raw data shown in Figs. 7 and 9 A indicate that it takes tens to hundreds of seconds to see the reopening events. Observing these reopening events in the absence of ATP allows us to conclude that the NBDs of the inactivated open state remain engaged even though the channel is no longer conducting. In other words, the opening of the channels in the absence of ATP suggests that ATP has yet to dissociate from NBDs. The long-lasting opening events also suggest that the NBD dimer is not separated (Vergani et al., 2005). Thus, the inhibitor does not work by disrupting NBD dimers.
A minimum kinetic model for the action of CFTRinh-172
As described in the Results, our experimental data lead to a six-state kinetic model (Scheme 3). It should be noted that although the existence of each state in Scheme 3 is supported by experimental observations, this model should be considered a minimum scheme. For example, it does not include steps portraying the dissociation of CFTRinh-172 from the inhibited states (Iinh and I’inh). However, including those steps will have to imply that both the open state and the closed state can be inactivated in the absence of CFTRinh-172. Although this could be the case, so far we do not have direct evidence supporting this notion. We have also lumped together many closed states because we are using a millimolar concentration of ATP so that the ATP-binding step is nearly saturated.
In the supplemental text, we demonstrate at least semi-quantitatively how Scheme 3 can duplicate most of our data. Here, we will qualitatively describe some interesting features of this kinetic model. Although there are overall seven transitions between states in Scheme 3, they can be divided into three groups: three gating transitions between the open state and the closed state, two steps for the binding and unbinding of CFTRinh-172, and two steps for inactivation of inhibitor-bound open and closed channels. Because our functional assay does not directly measure binding and unbinding of the inhibitor, we do not know if the binding affinity differs between the open state and the closed state. Our data, however, do indicate that the inactivated open state (Iinh) is more stable than the inactivated closed state. Thus, considering the four states on the right of Scheme 3, we can conclude that the opening of the activation gate by ATP facilitates inactivation, a phenomenon similar to the well-known observation for voltage-gated cation channels (Armstrong, 1971, 1974; Armstrong and Bezanilla, 1977). Thermodynamic constraints then dictate that inactivation promotes opening of the activation gate.
Scheme 3 also explains reopening of the locked-open channels in the absence of ATP upon washout of the inhibitor. This observation is analogous to the gating mechanism of some potassium channels where depolarization-induced, inactivated channels reopen transiently upon hyperpolarization, as the inactivation gate opens before the activation gate closes (Ruppersberg et al., 1991). If the activation gate is allowed to close and indeed closes much faster than recovery from the inactivated state (Iinh), one will not see reopening. Scheme 3 predicts just that: this reopening will only be seen with channels with a slow closing rate (e.g., locked-open channels). When the closing rate of the inactivated channels (Iinh→I’inh) is much faster than the rate of reopening from the inactivated open state (Iinh→Oinh), the recovery from inactivation will take place predominantly through the so-called “invisible pathway” (i.e., Iinh→I’inh→Cinh) upon removal of the inhibitor. Indeed, for WT-CFTR gated by ATP, removal of the inhibitor in the absence of ATP did not elicit any transient current response (unpublished data). On the other hand, for locked-open channels, the slow closing of the activation gate allows the inactivated open state to travel to the inhibitor-bound open state to become visible (i.e., Iinh→Oinh in Scheme 3).
In addition to the aforementioned differences between WT and E1371S channels regarding the mechanism of recovery from inactivation, Scheme 3 also predicts differences in inactivation per se. For E1371S-CFTR, because the closing rate is much slower than the rate of inactivation (i.e., Oinh→Cinh << Oinh→Iinh), the application of ATP to channels preexposed to CFTRinh-172 (Fig. 8) will generate a biphasic current response as a result of Cinh→Oinh→Iinh. However, the same protocol, when used for WT-CFTR, will produce different results because the transition Oinh→Iinh is slow compared with other transitions that dissipate the Oinh state. Indeed, as seen in Fig. 10, the application of ATP to WT-CFTR preexposed to CFTRinh-172 yields current rise in two phases without the appearance of inactivation. It should be noted that this apparent inactivation can be seen with WT-CFTR if we slightly modify the protocol. Also seen in Fig. 10, when ATP is applied in the continuous presence of CFTRinh-172, an initial increase of the current is followed by inactivation. This is simply because the presence of ATP and the inhibitor together shifts the reaction to favor the dominance of the Oinh state, which then dissipates into the Iinh state (see supplemental text for simulations).
Structural implications
As an ion channel, CFTR must possess a gate that controls the ion permeation pathway. Numerous functional studies have provided compelling evidence that this activation gate is controlled by ATP binding to CFTR’s two NBDs and subsequent dimerization of the ATP-bound NBDs (for review see Chen and Hwang, 2008; Hwang and Sheppard, 2009). However, being a member of the ABC transporter family, CFTR likely evolves from an active transporter. Phylogenic analysis has classified CFTR into the ABCC subfamily, in which many members function to export organic anions (Dean and Annilo, 2005). If we accept the idea that the ancestor of CFTR is an exporter, one can assume that this ancestor protein likely possesses two gates as a transporter requires. This primordial protein will have its substrate-binding site accessible to the cytoplasmic side before ATP binds, and dimerization of its two ATP-bound NBDs will trigger conformational changes that render the binding site exposed to the extracellular side. This classical alternating access model for a transporter has gained support from crystallographic studies that demonstrate outward-facing and inward-facing configurations of the TMDs of ABC proteins (Locher et al., 2002; Dawson and Locher, 2006, 2007; Hollenstein et al., 2007; Hvorup et al., 2007; Pinkett et al., 2007). Based on this idea, we have proposed that the dimerization-opened activation gate of CFTR is located at the extracellular end of the TMD (Tsai et al., 2009; also see Csanády, 2010). Thus, during evolution, the other gate located at the intracellular end of the TMD for this ancestor protein is degenerated so that CFTR can function as an ion channel (Chen and Hwang, 2008).
Although our work does not provide direct evidence pointing to a definitive structural mechanism for the action of CFTRinh-172, here we will discuss several possibilities. First, because binding of CFTRinh-172 to the channel leads to complete shutdown of the single-channel current, one has to conclude that the pore is obstructed. This can be a result of a direct blockade of the pore by CFTRinh-172. As explained above, the inhibitor may dock to the superficial position of the pore before it travels to a deeper position to block chloride ion movement through the permeation pathway. The observation that mutations of the R347 residue in the sixth transmembrane segment alter the apparent affinity for CFTRinh-172 appears to support this idea (Caci et al., 2008), but these mutational effects could be secondary to changes in gating kinetics. Furthermore, the residue R347, mutations of which produce most changes in the apparent affinity, is not a pore-lining residue (Dawson et al., 1999; McCarty and Zhang, 2001; Ge et al., 2004; Bai et al., 2010). Furthermore, the idea that CFTRinh-172 inhibits CFTR by plugging the pore is difficult to explain why ATP, which keeps the gate open, in fact slows down current recovery upon removal of the inhibitor.
Second, CFTRinh-172 may work by decoupling NBDs from the activation gate controlled by ATP-induced NBD dimerization. This decoupling hypothesis is not only difficult to substantiate (or disapprove) because of the nonspecific nature of this hypothesis, but it is also difficult to explain how this decoupling preferentially occurs in the open state versus the closed state. Perhaps more importantly, the decoupling hypothesis does not provide clear insights for further examining this idea through experimentation.
Third, part of the CFTR protein itself, like in the case of plugging the K+ channel pore by the inactivation ball (Hoshi et al., 1990; Zagotta et al., 1990), blocks the permeation pathway upon the binding of CFTRinh-172. In light of the evolutionary relationship between CFTR channels and ABC transporters, and the fact that many of our data can be explained by the action of an inactivation gate, this interesting possibility is worth consideration. One can imagine that opening the activation gate by ATP will cause constriction of the intracellular end of TMDs, where the inhibitor blocks the pore. Regardless, this hypothesis predicts that the opening of the activation gate should facilitate the closing of the inactivation gate because during a transport cycle, these two gates are coupled. Our data indeed show that inhibitor-bound open channels are inactivated more readily than the inhibitor-bound closed channels (see above).
No doubt, the two-gate hypothesis proposed here needs to be further tested. Because this idea is borrowed from the voltage-gated ion channel field, perhaps many of the strategies used previously to understand how voltage-gated cation channels activate and inactivate can be used to further examine this hypothesis.
Acknowledgments
We thank Wen-Chi Wu, Cindy Chu, and Shenghui Hu for technical assistance. We are also grateful to Dr. Robert Bridges, Rosalind Franklin University of Medicine and Science, and Cystic Fibrosis Foundation Therapeutics for the supply of CFTRinh-172.
This work was supported by grants NIHR01DK55835 and NIHR01HL53455 (to T.-C. Hwang), a Postdoctoral Fellowship (KOPEIK09F0) from Cystic Fibrosis Foundation (to Z. Kopeikin), and a research grant (KAKENHI22590212) from Japan Society for the Promotion of Science (to Y. Sohma). This investigation was conducted in a facility constructed with support from Research Facilities Improvement Program (grant C06 RR-016489-01) from the National Center for Research Resources, National Institutes of Health.
Christopher Miller served as editor.
Footnotes
Abbreviations used in this paper:
- ABC
- ATP-binding cassette
- CHO
- Chinese hamster ovary
- NBD
- nucleotide-binding domain
- P-ATP
- N6-(2-phenylethyl)-ATP
- TMD
- transmembrane domain
- WT
- wild type
References
- Aleksandrov A.A., Aleksandrov L., Riordan J.R. 2002. Nucleoside triphosphate pentose ring impact on CFTR gating and hydrolysis. FEBS Lett. 518:183–188 10.1016/S0014-5793(02)02698-4 [DOI] [PubMed] [Google Scholar]
- Armstrong C.M. 1971. Interaction of tetraethylammonium ion derivatives with the potassium channels of giant axons. J. Gen. Physiol. 58:413–437 10.1085/jgp.58.4.413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong C.M. 1974. Ionic pores, gates, and gating currents. Q. Rev. Biophys. 7:179–210 10.1017/S0033583500001402 [DOI] [PubMed] [Google Scholar]
- Armstrong C.M., Bezanilla F. 1977. Inactivation of the sodium channel. II. Gating current experiments. J. Gen. Physiol. 70:567–590 10.1085/jgp.70.5.567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y., Li M., Hwang T.C. 2010. Dual roles of the sixth transmembrane segment of the CFTR chloride channel in gating and permeation. J. Gen. Physiol. 136:293–309 10.1085/jgp.201010480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett K.E., Keely S.J. 2000. Chloride secretion by the intestinal epithelium: molecular basis and regulatory aspects. Annu. Rev. Physiol. 62:535–572 10.1146/annurev.physiol.62.1.535 [DOI] [PubMed] [Google Scholar]
- Bhattacharya S.K. 1995. Cholera outbreaks: role of oral rehydration therapy. J. Indian Med. Assoc. 93:237–238 [PubMed] [Google Scholar]
- Bompadre S.G., Cho J.H., Wang X., Zou X., Sohma Y., Li M., Hwang T.C. 2005. CFTR gating II: effects of nucleotide binding on the stability of open states. J. Gen. Physiol. 125:377–394 10.1085/jgp.200409228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bompadre S.G., Sohma Y., Li M., Hwang T.C. 2007. G551D and G1349D, two CF-associated mutations in the signature sequences of CFTR, exhibit distinct gating defects. J. Gen. Physiol. 129:285–298 10.1085/jgp.200609667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caci E., Caputo A., Hinzpeter A., Arous N., Fanen P., Sonawane N., Verkman A.S., Ravazzolo R., Zegarra-Moran O., Galietta L.J.V. 2008. Evidence for direct CFTR inhibition by CFTR(inh)-172 based on Arg347 mutagenesis. Biochem. J. 413:135–142 10.1042/BJ20080029 [DOI] [PubMed] [Google Scholar]
- Cai Z., Taddei A., Sheppard D.N. 2006. Differential sensitivity of the cystic fibrosis (CF)-associated mutants G551D and G1349D to potentiators of the cystic fibrosis transmembrane conductance regulator (CFTR) Cl− channel. J. Biol. Chem. 281:1970–1977 10.1074/jbc.M510576200 [DOI] [PubMed] [Google Scholar]
- Chen T.Y., Hwang T.C. 2008. CLC-0 and CFTR: chloride channels evolved from transporters. Physiol. Rev. 88:351–387 10.1152/physrev.00058.2006 [DOI] [PubMed] [Google Scholar]
- Chen T.Y., Tsai M.F., Hwang T.C. 2010. Structure and mechanisms in chloride channels. The Comprehensive Biophysics. Egelman E.H., editor Elsevier, New York: In press [Google Scholar]
- Cotten J.F., Welsh M.J. 1999. Cystic fibrosis-associated mutations at arginine 347 alter the pore architecture of CFTR. Evidence for disruption of a salt bridge. J. Biol. Chem. 274:5429–5435 10.1074/jbc.274.9.5429 [DOI] [PubMed] [Google Scholar]
- Csanády L. 2000. Rapid kinetic analysis of multichannel records by a simultaneous fit to all dwell-time histograms. Biophys. J. 78:785–799 10.1016/S0006-3495(00)76636-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csanády L. 2010. Degenerate ABC composite site is stably glued together by trapped ATP. J. Gen. Physiol. 135:395–398 10.1085/jgp.201010443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson R.J., Locher K.P. 2006. Structure of a bacterial multidrug ABC transporter. Nature. 443:180–185 10.1038/nature05155 [DOI] [PubMed] [Google Scholar]
- Dawson R.J., Locher K.P. 2007. Structure of the multidrug ABC transporter Sav1866 from Staphylococcus aureus in complex with AMP-PNP. FEBS Lett. 581:935–938 10.1016/j.febslet.2007.01.073 [DOI] [PubMed] [Google Scholar]
- Dawson D.C., Smith S.S., Mansoura M.K. 1999. CFTR: mechanism of anion conduction. Physiol. Rev. 79:S47–S75 [DOI] [PubMed] [Google Scholar]
- Dean M., Annilo T. 2005. Evolution of the ATP-binding cassette (ABC) transporter superfamily in vertebrates. Annu. Rev. Genomics Hum. Genet. 6:123–142 10.1146/annurev.genom.6.080604.162122 [DOI] [PubMed] [Google Scholar]
- Ge N., Muise C.N., Gong X., Linsdell P. 2004. Direct comparison of the functional roles played by different transmembrane regions in the cystic fibrosis transmembrane conductance regulator chloride channel pore. J. Biol. Chem. 279:55283–55289 10.1074/jbc.M411935200 [DOI] [PubMed] [Google Scholar]
- Hollenstein K., Frei D.C., Locher K.P. 2007. Structure of an ABC transporter in complex with its binding protein. Nature. 446:213–216 10.1038/nature05626 [DOI] [PubMed] [Google Scholar]
- Hoshi T., Zagotta W.N., Aldrich R.W. 1990. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 250:533–538 10.1126/science.2122519 [DOI] [PubMed] [Google Scholar]
- Hvorup R.N., Goetz B.A., Niederer M., Hollenstein K., Perozo E., Locher K.P. 2007. Asymmetry in the structure of the ABC transporter-binding protein complex BtuCD-BtuF. Science. 317:1387–1390 10.1126/science.1145950 [DOI] [PubMed] [Google Scholar]
- Hwang T.C., Sheppard D.N. 1999. Molecular pharmacology of the CFTR Cl− channel. Trends Pharmacol. Sci. 20:448–453 10.1016/S0165-6147(99)01386-3 [DOI] [PubMed] [Google Scholar]
- Hwang T.C., Sheppard D.N. 2009. Gating of the CFTR Cl− channel by ATP-driven nucleotide-binding domain dimerisation. J. Physiol. 587:2151–2161 10.1113/jphysiol.2009.171595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locher K.P., Lee A.T., Rees D.C. 2002. The E. coli BtuCD structure: a framework for ABC transporter architecture and mechanism. Science. 296:1091–1098 10.1126/science.1071142 [DOI] [PubMed] [Google Scholar]
- Ma T., Thiagarajah J.R., Yang H., Sonawane N.D., Folli C., Galietta L.J.V., Verkman A.S. 2002. Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J. Clin. Invest. 110:1651–1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarty N.A., Zhang Z.R. 2001. Identification of a region of strong discrimination in the pore of CFTR. Am. J. Physiol. Lung Cell. Mol. Physiol. 281:L852–L867 [DOI] [PubMed] [Google Scholar]
- Muanprasat C., Sonawane N.D., Salinas D., Taddei A., Galietta L.J.V., Verkman A.S. 2004. Discovery of glycine hydrazide pore-occluding CFTR inhibitors: mechanism, structure–activity analysis, and in vivo efficacy. J. Gen. Physiol. 124:125–137 10.1085/jgp.200409059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinkett H.W., Lee A.T., Lum P., Locher K.P., Rees D.C. 2007. An inward-facing conformation of a putative metal-chelate-type ABC transporter. Science. 315:373–377 10.1126/science.1133488 [DOI] [PubMed] [Google Scholar]
- Riordan J.R., Rommens J.M., Kerem B., Alon N., Rozmahel R., Grzelczak Z., Zielenski J., Lok S., Plavsic N., Chou J.L., et al. 1989. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 245:1066–1073 10.1126/science.2475911 [DOI] [PubMed] [Google Scholar]
- Ruppersberg J.P., Frank R., Pongs O., Stocker M. 1991. Cloned neuronal IK(A) channels reopen during recovery from inactivation. Nature. 353:657–660 10.1038/353657a0 [DOI] [PubMed] [Google Scholar]
- Schultz B.D., Singh A.K., Devor D.C., Bridges R.J. 1999. Pharmacology of CFTR chloride channel activity. Physiol. Rev. 79:S109–S144 [DOI] [PubMed] [Google Scholar]
- Taddei A., Folli C., Zegarra-Moran O., Fanen P., Verkman A.S., Galietta L.J.V. 2004. Altered channel gating mechanism for CFTR inhibition by a high-affinity thiazolidinone blocker. FEBS Lett. 558:52–56 10.1016/S0014-5793(04)00011-0 [DOI] [PubMed] [Google Scholar]
- Thiagarajah J.R., Verkman A.S. 2003. CFTR pharmacology and its role in intestinal fluid secretion. Curr. Opin. Pharmacol. 3:594–599 10.1016/j.coph.2003.06.012 [DOI] [PubMed] [Google Scholar]
- Tsai M.-F., Shimizu H., Sohma Y., Li M., Hwang T.-C. 2009. State-dependent modulation of CFTR gating by pyrophosphate. J. Gen. Physiol. 133:405–419 10.1085/jgp.200810186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Goor F., Hadida S., Grootenhuis P.D.J. 2008. Pharmacological rescue of mutant CFTR function for the treatment of cystic fibrosis. Top. Med. Chem. 3:91–120 10.1007/7355_2008_022 [DOI] [Google Scholar]
- Van Goor F., Hadida S., Grootenhuis P.D., Burton B., Cao D., Neuberger T., Turnbull A., Singh A., Joubran J., Hazlewood A., et al. 2009. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. USA. 106:18825–18830 10.1073/pnas.0904709106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergani P., Nairn A.C., Gadsby D.C. 2003. On the mechanism of MgATP-dependent gating of CFTR Cl− channels. J. Gen. Physiol. 121:17–36 10.1085/jgp.20028673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergani P., Lockless S.W., Nairn A.C., Gadsby D.C. 2005. CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains. Nature. 433:876–880 10.1038/nature03313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreich F., Riordan J.R., Nagel G. 1999. Dual effects of ADP and adenylylimidodiphosphate on CFTR channel kinetics show binding to two different nucleotide binding sites. J. Gen. Physiol. 114:55–70 10.1085/jgp.114.1.55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh M.J., Smith A.E. 1993. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell. 73:1251–1254 10.1016/0092-8674(93)90353-R [DOI] [PubMed] [Google Scholar]
- Welsh M.J., Ramsey B.W., Accurso F., Cutting G.R. 2001. Cystic fibrosis. The Metabolic and Molecular Basis of Inherited Disease. Scriver C.R., Beaudet A.L., Sly W.S., Valle D., McGraw-Hill Inc, New York: 5121–5188 [Google Scholar]
- Zagotta W.N., Hoshi T., Aldrich R.W. 1990. Restoration of inactivation in mutants of Shaker potassium channels by a peptide derived from ShB. Science. 250:568–571 10.1126/science.2122520 [DOI] [PubMed] [Google Scholar]
- Zhang X.D., Tseng P.Y., Yu W.P., Chen T.Y. 2009. Blocking pore-open mutants of CLC-0 by amphiphilic blockers. J. Gen. Physiol. 133:43–58 10.1085/jgp.200810004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M., Morais-Cabral J.H., Mann S., MacKinnon R. 2001. Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature. 411:657–661 10.1038/35079500 [DOI] [PubMed] [Google Scholar]
- Zhou Z., Hu S., Hwang T.-C. 2002. Probing an open CFTR pore with organic anion blockers. J. Gen. Physiol. 120:647–662 10.1085/jgp.20028685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z., Wang X., Li M., Sohma Y., Zou X., Hwang T.-C. 2005. High affinity ATP/ADP analogues as new tools for studying CFTR gating. J. Physiol. 569:447–457 10.1113/jphysiol.2005.095083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z., Wang X., Liu H.Y., Zou X., Li M., Hwang T.-C. 2006. The two ATP binding sites of cystic fibrosis transmembrane conductance regulator (CFTR) play distinct roles in gating kinetics and energetics. J. Gen. Physiol. 128:413–422 10.1085/jgp.200609622 [DOI] [PMC free article] [PubMed] [Google Scholar]