

Abstract
The chemopreventive effects of three agents, rexinoid bexarotene, tyrosine kinase inhibitor gefitinib and celecoxib, were tested on mammary tumor development arising in p53 null mammary epithelium. The rexinoid bexarotene was the most efficacious inhibitor as it reduced mammary tumor development by 75% in virgin mice and significantly delayed mean tumor development by 98 days in hormone stimulated mice. The tyrosine kinase inhibitor gefitinib reduced mammary tumor incidence by 50% in virgin mice but did not significantly delay mean tumor latency in hormone stimulated mice. Celecoxib did not reduce tumor incidence or mean tumor latency in either of the two models. The high doses of the rexinoid and the tyrosine kinase inhibitor did not affect the progression of tumors arising from the premalignant mammary outgrowth line, PN8a. A comparison of these agents with tamoxifen shows the superiority of tamoxifen in preventing tumor development in p53 null mammary cells. Similarly, a comparison of the results of the p53 model with other transgenic models to the chemopreventive agents demonstrated that mammary tumors arising from different oncogenic events will respond differently to the different agents.
Keywords: Research, prevention, mammary
Introduction
The prevention of mammary tumorigenesis is a viable strategy that has been tested in conventional rodent models and recently in genetically engineered models (1). The important issues are efficacy, amount and duration of side effects, and cost. The initial strategy of continued exposure to determine efficacy and side effects has been followed by the strategy of intermittent and/or limited exposure. These latter approaches have been successfully demonstrated in breast cancer models using retinoids and selective estrogen receptor modulators (SERMs) in combination (2) and with the sole exposure of the SERM, tamoxifen (3). With genetically engineered models gradually replacing conventional models in most experimental studies, prevention studies on the different genetically engineered models are becoming more common.
There are over 100 genetically engineered models of mammary cancer (4). Only a few of these models have been characterized at the cellular and molecular levels with respect to growth control, premalignant development, hormone dependence, metastatic potential and genetic stability. The most extensively characterized models are the c-neu, c-myc, wnt, Brca-1, polyoma mT, SV40Tag and p53 null models. Prevention studies of mammary tumorigenesis have been limited to just a few of these models. Prevention agents, such as tamoxifen, 9 cis-retinoic acid, the rexinoid bexarotene (LGD1069), tyrosine kinase inhibitors (i.e., ZD1839=Iressa/gefitinib), celecoxib and soy isoflavones have been studied primarily in the c-neu and SV40 Tag models (5-13). The response of these two models to these agents varies with the genetic model and the individual agent. For instance, the rexinoid bexarotene is more effective against c-neu induced mammary tumors than against SV40Tag mammary tumors (11, 12). In MMTV-c-neu, the agents gefitinib and bexarotene are more effective than celecoxib (5, 7, 12). Both of the above models give rise to estrogen receptor-alpha negative mammary tumors.
The p53 null mammary epithelial model has been extensively characterized at the pathological, cellular and molecular levels (14-16). ER-positive DCIS is a predominant premalignant lesion and the tumors that arise are predominantly, but not exclusively, ER-negative, aneuploid and metastatic to the lung. In the only prevention study reported in this system, the SERM tamoxifen strongly inhibited mammary tumorigenesis (17), quantitatively equal or better than the inhibition observed in the c-neu and the SV40 Large T antigen models (8, 13). In the results reported herein, we continued our studies on chemoprevention agents on mammary tumorigenesis and tested the efficacy of the retinoid bexarotene, gefitinib and celecoxib in the p53 null mammary epithelial cell model. These three agents were chosen because they have been tested in other mouse models of mammary tumorigenesis. A comparison of the effects of the three agents in the p53 null mouse mammary model with the other models would provide information on the sensitivity of different oncogene driven tumorigenesis to each agent. Although two of the three agents were preventive, there was a significant difference between these two agents and between these agents and tamoxifen.
Materials and Methods
Mice
All donor and recipient mice were bred and maintained at Baylor College of Medicine. The donor mice were BALB/c p53 homozygous null, and the recipient mice were p53 wild type (15). All mice were maintained in a conventional mouse facility with food and water provided ad libitum, and the room temperature was set at 70°F. The animal facility is American Association of Laboratory Animal Care accredited.
Transplantation
The basic transplantation protocol was as described in (15). Briefly, 1 mm2 fragments of mammary duct from eight-ten week old female p53 null mice were transplanted into both cleared inguinal fat pads of three-week old female mice. The successful take rate is over 90%, thus in some groups the denominator varies by one-two because fat pads without any ductal outgrowth are not considered in the final data tabulation. The transplanted cells take 8 weeks to completely fill the fat, at which point the cells assume a steady state level of proliferation. Thus, all treatments with the chemopreventive agents started when the mice were 11 weeks of age in order to avoid any effects of the agents on the the ability of the cells to grow and fill the fat pad. In some groups of mice, at five weeks of age, a pituitary isograft was implanted under the kidney capsule to provide a continuous hormonal (prolactin/progesterone) stimulation of the mammary gland. The consequence of this hormonal stimulation is a marked decrease in mean tumor latency period and an increase in tumor incidence (15). Such mice are referred to hereafter as pituitary plus. Although the p53 deletion is the same in all donor mice, the array of secondary alterations important for neoplastic development include both common and unique events. The consequence is that the tumorigenic capabilities of mammary gland fragments vary over a small range between donor mice in the same host environment. For example, the TE50 of two untreated groups of virgin mice in two different experiments may be different (e.g., 60 weeks versus 50 weeks). Thus, each experiment always has an untreated control group to assess the effect of a particular treatment. In all the transplantation experiments described herein, there were three different donors used for each experiment with equal representation in the different groups. The doses of the three chemopreventive agents were chosen based on doses given in published experiments using transgenic mouse models.
In experiment 1, we tested the chemoprevention efficacy of the tyrosine kinase inhibitor gefitinib. There were five groups of mice. Groups 1 and 2 were virgin mice (25 transplants per group); vehicle only and treated with 100 mg/kg body weight gefitinib daily, respectively. Groups 3-5 were pituitary-isograft bearing mice (20 transplants per group); vehicle only, 10 mg/kg and 100 mg/kg gefitinib, respectively. Mice were treated with gefitinib suspended in distilled water containing 1% Tween 80 or with vehicle for 5 days/week from age 3 months until termination of the experiment. Gefitinib or vehicle was administered in 0.1 ml by gastric gavage with a 20-gauge gavage needle.
In experiment 2, we tested the chemoprevention efficacy of the rexinoid bexarotene. There were six groups of mice. Groups 1-3 were virgin mice (25 transplants per group; vehicle only, 10mg/kg and 100 mg/kg bexarotene, respectively. Groups 4-6 were pituitary-isograft bearing mice (20 transplants per group); vehicle only, 10 mg/kg and 100 mg/kg bexarotene, respectively. Mice were treated with bexarotene suspended in purified sesame oil (Croda, Inc., Mill Hall, PA) 5 days/week from age 3 months until termination of the experiment. The rexinoid or vehicle was administered by gastric gavage using a 20-gauge gavage needle in a volume of 0.1-ml.
In experiment 3, we tested the chemopreventive efficacy of the cox-2 inhibitor, celecoxib. There were 4 groups of mice. Groups 1 and 2 were virgin mice (25 transplants per group); control diet and diet containing 500ppm celecoxib. Groups 3 and 4 were pituitary-isograft bearing mice(25 transplants per group; contlo diet and diet containing 500ppm celecoxib. The diet was fed to the mice from three months of age until termination of the experiment.
In experiment 4, we tested the chemoprevention efficacy of the rexinoid bexarotene and gefitinb on the tumor-producing capability of the premalignant outgrowth line PN8a. There were two groups of virgin mice per agent (20 transplants per group; vehicle only, 100 mg/kg bexarotene and gefitinib, respectively. The agents and their vehicles were administered as described above.
Mice were weighed monthly and additionallly were examined weekly for any changes in condition of hairor motility or condition of eyes. There was little systemic side effects of the drugs except in aged mice fed the celecoxib.
Immunohistochemistry
In experiments 1-3, samples of transplants and tumors were collected and processed for BrdU and estrogen receptor immunochemistry, respectively, as previously described (16). In addition, mammary tumors in each group were processed for both H&E staining for histopathology evaluation and for immunohistochemistry.
Statistics
The tumor incidence curves were evaluated as tumor-free survival from time of transplant to first appearance of a palpable tumor. Tumor-free survival curves were estimated by the Kaplan-Meier method and compared using the Generalized Wilcoxon test. Significant differences were considered at p<.05.
Results
Figure 1 shows the results of the effect of gefitinib on the tumorigenicity of p53 null normal mammary cells transplanted into virgin and hormone stimulated BALB/c mice. In virgin mice, the high dose of gefitinib decreased the incidence of mammary tumor development (p<.05) by 50% at 56 weeks of age (8/20 vs. 4/20). However, in hormone stimulated mice where the incidence in control mice reached 90%, neither concentration of gefitinib significantly altered tumorigenesis (90% vs. 70% vs. 70%). The non-significant delay in tumorigenic rate observed between 40 and 50 weeks of age is attributed to the mild side effects of gefitinib which included eye irritation and mild hair loss. Examination of gland morphogenesis by whole mounts at 4 weeks after initiation of treatment did not show any alterations in duct morphology in virgin mice or alterations in alveolar development in the pituitary-isograft containing mice. The BrdU labeling index was examined at 2 months of gefitinib treatment. The mammary transplants in the pituitary isograft mice exhibited a significantly increased labeling index compared to those in virgin mice (31/500 vs. 6/500, respectively); however, gefitinib did not result in any significant differences in proliferation indices in either the virgin or pituitary isograft mice (Figure 2). Similarly, the distribution of ERa-positive and negative tumors was unaffected by the drug treatment (data not shown).
Figure 1.
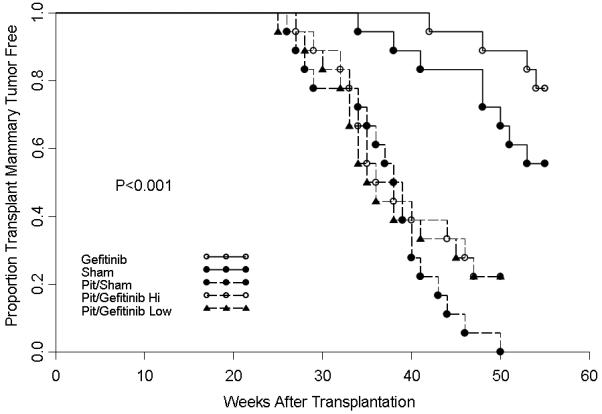
The effect of gefitinib on the tumor-producing capabilities of p53 null mammary epithelium transplanted into virgin or pituitary-isograft bearing mice. Gefitinib delayed tumorigenesis in virgin mice ( ) compared to sham controls (
) compared to sham controls ( ). Gefitinib did not have any effect on tumorigenesis in hormone-stimulated hosts.
). Gefitinib did not have any effect on tumorigenesis in hormone-stimulated hosts.
Figure 2.
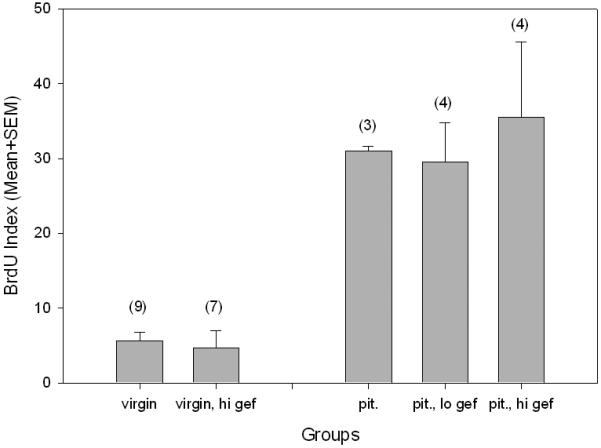
The effect of gefitinib on mammary cell proliferation as measured by BrdU incorporation. There were no differences in cell proliferation in any of the five groups of mice. Number in parentheses represents individual number of glands evaluated for BrdU. gef = gefitinib; lo = low; hi = high; pit = pituitary-isograft.
Figures 3 and 4 shows the results of the effect of bexarotene on the tumorigenicity of p53 null normal mammary cells transplanted into virgin and hormone stimulated BALB/c mice. In virgin mice, the high dose of bexarotene decreased the incidence of mammary tumor development (p<.05) by 75% at 60 weeks of age (8/25 vs. 2/25). The low dose did not affect tumor development (8/25 vs. 7/25) (p>.05) (Figure 3). In hormone stimulated mice where the incidence in control mice reached 86% (19/22) by 41 weeks after transplantation, the high concentration (16/22=73%), but not the low concentration (19/22) of bexarotene modestly but significantly delayed tumorigenesis. The TE50 was increased from 32 weeks in control to 46 weeks in the high dose retinoid treated group, although by 53 weeks after transplantation, the tumor incidence in the high dose of rexinoid attained 73% (Figure 4).
Figure 3.
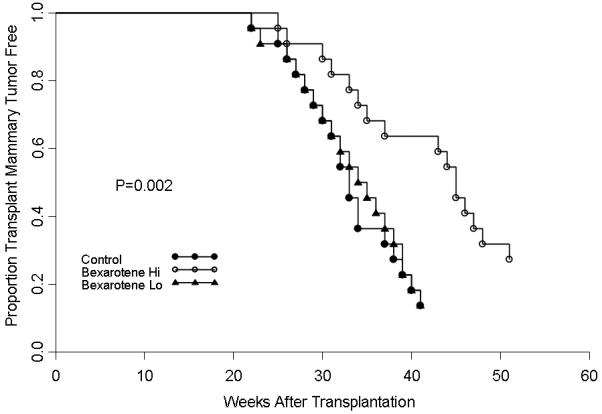
The effect of bexarotene on the tumor-producing capabilities of p53 null mammary epithelium in pituitary-isograft bearing mice. Bexarotene significantly delayed tumorigenesis at the high dose ( ) but not at the low does (
) but not at the low does ( ) compared to control mice (
) compared to control mice ( ).
).
Figure 4.
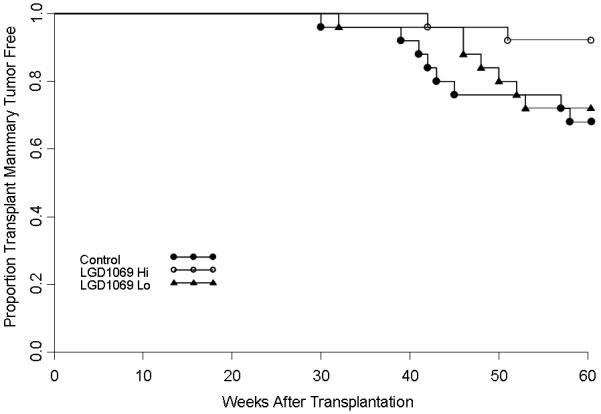
The effect of bexarotene on the tumor-producing capabilities of p53 null mammary epithelium in virgin mice. The high dose of bexarotene ( ) but not the low does (
) but not the low does ( ) delayed tumorigenesis compared to control mice (
) delayed tumorigenesis compared to control mice ( ).
).
Examination of gland morphogenesis by whole mounts at 4 weeks after initiation of treatment did not show any alterations in duct morphology in virgin mice. Alveolar morphogenesis in the pituitary-isograft containing mice was normal except the lobules were smaller in size. The BrdU labeling index in the normal gland is shown in Figure 5. In untreated virgin mice, the steady state proliferation index was low (X=3/500 cells), was slightly higher in mice exposed to the low dose of bexarotene (X=12/500 cells) but remained low in mice exposed to the high dose of bexarotene (X=1/500 cells). In pituitary-isograft bearing mice compared to virgin mice, the proliferation rate was significantly higher in mice not exposed to bexarotene (X=51/500 cells). This level was slightly lower in mice exposed to the low dose of bexarotene (X=38/500 cells) (P<.05) and much lower in mice exposed to the high dose bexarotene (X=12/500) (P<.05). Tumors from the pituitary isograft containing groups (3 untreated, 2 high bexarotene and 9 low bexarotene) were examined for BrdU-labeling index. Interestingly, there were no differences among the labeling indices for the 14 tumors with all showing greater than 10% BrdU labeled cells.
Figure 5.
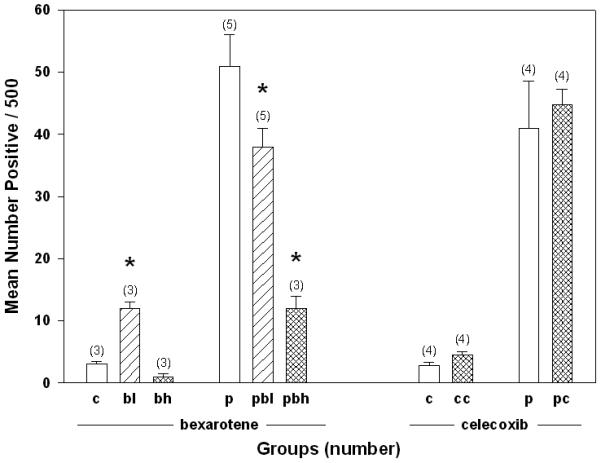
The effect of bexarotene and celecoxib on mammary cell proliferation as measured by BrdU incorporation. Asterisk marks values that are significantly different (P < 0.05). Bexarotene, but not celecoxib, altered BrdU incorporation. Numbers in parentheses represent number of individual glands evaluated for BrdU incorporation. c = control, p = pituitary, l = low dose; h = high dose; b = bexarotene; c = celecoxib.
Figure 6 shows the results of the effect of celecoxib on the tumorigenicity of p53 null normal mammary cells transplanted into virgin and hormone stimulated BALB/c mice. Celecoxib did not affect tumorigenicity in either the virgin or hormone stimulated mice. The tumor incidences in virgin mice were 48% (12/25) and 40% (10/25) at 60 weeks in control and celecoxib treated mice, respectively. The tumor incidences in hormone stimulated mice were 80% (20/25) at 50 weeks in both treated and celecoxib treated mice. Examination of gland morphogenesis by whole mounts at 4 weeks after initiation of treatment did not show any alterations in duct morphology in virgin mice or alterations in alveolar development in the pituitary-isograft containing mice. The BrdU labeling index was examined at 4 weeks after initiation of celecoxib treatment (Figure 5). The proliferation indices of the mammary transplants were the same in the control as in the celecoxib treated mice (2.8 vs. 4.5/500 in virgin mice, respectively and 41 vs. 44.8/500 in the hormone treated mice, respectively). Aged mice on celecoxib diet showed evidence of being fed celecoxib by the loss of fur as well as irritation of the eyes (red eyes, eyelids half-closed).
Figure 6.

The effect of celecoxib on the tumor-producing capabilities of p53 null mammary epithelium transplanted into virgin or pituitary-isograft bearing mice. There was no significant effect of celecoxib on tumorigenesis in either the virgin or pituitary-isograft bearing mice.
A total of 8 tumors arising in control transplants and 22 tumors arising in treated (11 each for the celecoxib and bexarotene) transplants were examined for estrogen receptor by immunohistochemistry. As seen in previous studies, the frequency of estrogen receptor positive tumors was around 25% with 2/8 from control and 5/22 from treated animals positive for ER.
The effects of bexarotene and gefitinib were tested on the tumorigenicity of p53 null premalignant mammary outgrowth line PN8a transplanted into virgin BALB/c mice. Neither the high doses of bexarotene nor of gefitinib significantly altered the tumorigenic potential or mean latency period of the premalignant outgrowth line [bexarotene: control=23/25; high dose = 20/25, (p>.05); gefitinib: control=21/25; high dose=21/25, (p>.05)]
Discussion
The experiments presented herein describe, for the first time, the effects of three different chemoprevention agents, gefitinib, rexinoid bexarotene and celecoxib on tumorigenesis in mammary cells where only p53 cell function is compromised. Several results are noteworthy. First, within the same model system and under two different experimental conditions, the results show that the rexinoid bexarotene was more efficacious than either gefitinib or celecoxib. In virgin mice, where tumors develop at a low incidence and with a long latency, both rexinoid bexarotene and gefitinib were effective (75% and 50% reduction, respectively). In hormone stimulated mice, where tumors developed at a high incidence and with a short latency, rexinoid bexarotene did not significantly reduce tumorigenesis (86% control vs. 73% treated) but did significantly delay mean tumor development by 98 days, whereas gefitinib and celecoxib did not have a significant effect on either parameter.
Both ER-negative and ER-positive tumors develop in the p53 null mammary cells (16, 18). The agent, bexarotene did not alter the distribution of ER-positive and ER-negative tumors that arose in either virgin or treated mice. This is consistent with the results of earlier reports which demonstrate that bexarotene can inhibit tumor formation in predominately ER-positive mammary tumor models as well as ER-negative mammary tumor models (12, 19).
Second, although bexarotene was effective against tumor development originating in normal p53 null mammary cells, it (as well as gefitinib) was ineffective against the progression of tumors arising in a premalignant outgrowth population. PN8a would be analogous to a high grade DCIS with respect to its potential to progress to invasive breast cancer. These results suggest that these agents might be effective against the early stages of premalignant development but are relatively ineffective against established premalignant cell populations that have significant tumorigenic potential. Perhaps an analogous situation in human breast would be low grade DCIS versus high grade DCIS. However, two studies have demonstrated recently that the SERM tamoxifen, did have a modest delaying effect on tumorigenesis in the p53 null PN8 premalignant line (17) and the PyV-m-T premalignant line 8w-B (9). More extensive studies need to be performed to assess the responsiveness of premalignant lines arising from different oncogenic events in order to determine the responsiveness of the premalignant state to chemopreventive agents. In an earlier study which examined the responsiveness of mammary premalignant lines to a standard set of cytotoxic chemotherapeutic drugs, it was impossible to predict the responsiveness of a particular premalignant line as the premalignant population responded differently from the resulting malignant population to the same cytostatic drug (20).
Third, a comparison of these three agents in the p53 null model with the c-neu model indicates that all agents were much more effective against c-neu induced mammary tumors than against p53 null mammary tumors (5, 7, 12). The most appropriate comparison is the hormone stimulated p53 null gland with the MMTV-neu gland as both models develop tumors at a high incidence and with short median latency. In the MMTV-c-neu mammary gland, bexarotene reduced tumor incidence by 75% and lengthened median tumor latency from 234 days to over 420 days. The modest prevention activity of bexarotene in the p53 null mammary gland was perhaps not too surprising as a previous study showed that bexarotene was also modestly preventive in the SV40Tag model where both p53 and Rb activity are compromised. Both these molecules exert primary functions downstream of the cyclin dependent kinases, one locus of bexarotene activity (1). The differences between the two models were even more dramatic for gefitinib as this agent reduced tumor development by 73% in c-neu mice but had no effect on tumor development in hormone stimulated p53 null mammary epithelium). Even celecoxib was more effective in the c-neu model as this agent delayed latency for 50% tumor formation from 32.3 weeks to 39.6 weeks, but had no effect in the p53 null model.
Fourth, and of most interest, is the comparison of the rexinoid (the most effective inhibitor in the experiments reported in this paper) with the effects of the SERM, tamoxifen. In a previous study, we showed that continuous tamoxifen treatment blocked mammary tumorigenesis in hormone stimulated mice by 75% and in virgin mice by 90% (18). In a second experiment reported in (3), just a three month exposure to tamoxifen in virgin mice completely blocked mammary tumor development (untreated controls= 24%). In contrast, tamoxifen was much less efficacious in the other two models discussed above, the MMTV-c-neu and the SV40LTag models (8, 13). A consideration of all these results indicates that mammary tumors derived from different primary oncogenic events will respond differently to different prevention modalities. An extension of this idea would suggest that the different subsets of human breast cancers will respond differently to the different agents. In effect, this has been demonstrated therapeutically with the results of Herceptin which targets the ErbB2 pathway (21). It is likely that using the targeted reagents as chemopreventive agents will show a similar result.
Chemoprevention, in order to be maximally effective and with no to minimal side effects, will have to utilize a different strategy than single agent exposure. One such strategy is that pioneered by Sporn and coworkers (2), who demonstrated that a combination of rexinoid and SERMs was more effective than either single agent. The experiments were performed on two very different models, chemical carcinogen-induced rat mammary tumorigenesis and MMTV-c-neu mouse mammary tumorigenesis, so the strategy shows great promise. It will be of interest if this strategy is similarly effective in other genetically engineered mouse models, including the premalignant models which have been relatively refractory to chemoprevention modalities so far. As rexinoids are currently in clinical trials, and prevention trials are lengthy and depend on surrogate markers for interpretation of effectiveness, it is important to develop optimal combinations in relevant and informative animal model systems. Such an effort will ultimately save time and money.
References
- 1.Shen Q, Brown PH. Novel agents for the prevention of breast cancer: targeting transcription factors and signal transduction pathways. Journal of mammary gland biology and neoplasia. 2003;8(1):45–73. doi: 10.1023/a:1025783221557. [DOI] [PubMed] [Google Scholar]
- 2.Liby K, Rendi M, Suh N, et al. The combination of the rexinoid, LG100268, and a selective estrogen receptor modulator, either arzoxifene or acolbifene, synergizes in the prevention and treatment of mammary tumors in an estrogen receptor-negative model of breast cancer. Clin Cancer Res. 2006;12(19):5902–9. doi: 10.1158/1078-0432.CCR-06-1119. [DOI] [PubMed] [Google Scholar]
- 3.Rajkumar L, Kittrell FS, Guzman RC, Brown PH, Nandi S, Medina D. Hormone-induced protection of mammary tumorigenesis in genetically engineered mouse models. Breast Cancer Res. 2007;9(1):R12. doi: 10.1186/bcr1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borowsky A. Special considerations in mouse models of breast cancer. Breast disease. 2007;28:29–38. doi: 10.3233/bd-2007-28104. [DOI] [PubMed] [Google Scholar]
- 5.Howe LR, Subbaramaiah K, Patel J, et al. Celecoxib, a selective cyclooxygenase 2 inhibitor, protects against human epidermal growth factor receptor 2 (HER-2)/neu-induced breast cancer. Cancer research. 2002;62(19):5405–7. [PubMed] [Google Scholar]
- 6.Li Y, Zhang Y, Hill J, et al. The rexinoid, bexarotene, prevents the development of premalignant lesions in MMTV-erbB2 mice. British journal of cancer. 2008;98(8):1380–8. doi: 10.1038/sj.bjc.6604320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu C, Speers C, Zhang Y, et al. Effect of epidermal growth factor receptor inhibitor on development of estrogen receptor-negative mammary tumors. Journal of the National Cancer Institute. 2003;95(24):1825–33. doi: 10.1093/jnci/djg117. [DOI] [PubMed] [Google Scholar]
- 8.Menard S, Aiello P, Tagliabue E, et al. Tamoxifen chemoprevention of a hormone-independent tumor in the proto-neu transgenic mice model. Cancer research. 2000;60(2):273–5. [PubMed] [Google Scholar]
- 9.Namba R, Young LJ, Maglione JE, et al. Selective estrogen receptor modulators inhibit growth and progression of premalignant lesions in a mouse model of ductal carcinoma in situ. Breast Cancer Res. 2005;7(6):R881–9. doi: 10.1186/bcr1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Piechocki MP, Dibbley SK, Lonardo F, Yoo GH. Gefitinib prevents cancer progression in mice expressing the activated rat HER2/neu. International journal of cancer. 2008;122(8):1722–9. doi: 10.1002/ijc.23231. [DOI] [PubMed] [Google Scholar]
- 11.Wu K, Kim HT, Rodriquez JL, et al. Suppression of mammary tumorigenesis in transgenic mice by the RXR-selective retinoid, LGD1069. Cancer Epidemiol Biomarkers Prev. 2002;11(5):467–74. [PubMed] [Google Scholar]
- 12.Wu K, Zhang Y, Xu XC, et al. The retinoid X receptor-selective retinoid, LGD1069, prevents the development of estrogen receptor-negative mammary tumors in transgenic mice. Cancer research. 2002;62(22):6376–80. [PubMed] [Google Scholar]
- 13.Yoshidome K, Shibata MA, Couldrey C, Korach KS, Green JE. Estrogen promotes mammary tumor development in C3(1)/SV40 large T-antigen transgenic mice: paradoxical loss of estrogen receptor alpha expression during tumor progression. Cancer research. 2000;60(24):6901–10. [PubMed] [Google Scholar]
- 14.Goepfert TM, McCarthy M, Kittrell FS, et al. Progesterone facilitates chromosome instability (aneuploidy) in p53 null normal mammary epithelial cells. Faseb J. 2000;14(14):2221–9. doi: 10.1096/fj.00-0165com. [DOI] [PubMed] [Google Scholar]
- 15.Jerry DJ, Kittrell FS, Kuperwasser C, et al. A mammary-specific model demonstrates the role of the p53 tumor suppressor gene in tumor development. Oncogene. 2000;19(8):1052–8. doi: 10.1038/sj.onc.1203270. [DOI] [PubMed] [Google Scholar]
- 16.Medina D, Kittrell FS, Shepard A, et al. Biological and genetic properties of the p53 null preneoplastic mammary epithelium. Faseb J. 2002;16(8):881–3. doi: 10.1096/fj.01-0885fje. [DOI] [PubMed] [Google Scholar]
- 17.Medina D, Kittrell FS, Hill J, Shepard A, Thordarson G, Brown P. Tamoxifen inhibition of estrogen receptor-alpha-negative mouse mammary tumorigenesis. Cancer research. 2005;65(8):3493–6. doi: 10.1158/0008.5472.CAN-04-3869. [DOI] [PubMed] [Google Scholar]
- 18.Lin SC, Lee KF, Nikitin AY, Hilsenbeck SG, Cardiff RD, Li A, Kang KW, Frank SA, Lee WH, Lee EY. Somatic mutation of p53 leads to estrogen alpha-receptor positive and -negative mouse mammary tumors with high frequency of metastasis. Cancer research. 2004;64(10):3525–3532. doi: 10.1158/0008-5472.CAN-03-3524. [DOI] [PubMed] [Google Scholar]
- 19.Gottardis MM, Bischoff ED, Shirley MA, Wagoner MA, Lamph WW, Heyman RA. Chemoprevention of mammary carcinoma by LGD1069 (Targretin): An RXR-selective ligand. Cancer research. 1996;56(12):5566–5570. [PubMed] [Google Scholar]
- 20.Medina D, Lane HW. Stage specificity of selenium-mediated inhibition of mouse mammary tumorigenesis. Biol Trace Element Res. 1983;5:297–306. doi: 10.1007/BF02987215. [DOI] [PubMed] [Google Scholar]
- 21.Finn RS, Slamon DJ. Monoclonal antibody therapy for breast cancer: herceptin. Cancer chemotherapy and biological response modifiers. 2003;21:223–33. doi: 10.1016/s0921-4410(03)21010-3. [DOI] [PubMed] [Google Scholar]