

Abstract
The discovery of the genetic causes of a rare group of immune-mediated inflammatory conditions that mimic infections and allergic conditions in their clinical presentation and the molecular understanding of the function of the mutated molecules in these diseases has led to a revolution in our understanding of the pathogenesis of systemic and local inflammation. The proteins mutated in a number of these so-called autoinflammatory diseases are part of, or regulate the activity of, intracellular molecular complexes, the inflammasomes, that sense “danger” to the body and coordinate an initial immune response. Our understanding of specific triggers of the inflammasomes, coupled with the recognition that inflammasomes are critical for activation of the proinflammatory cytokine IL-1, has provided a rational and very effective target in the treatment of a number of these rare autoinflammatory diseases. In addition, the ongoing discovery of the role of inflammasomes and IL-1 activation and secretion in a number of genetically complex disorders have fundamentally changed our view of disease pathogenesis in a growing number of disorders that were heretofore not even thought of as “immunologic” diseases.
Keywords: Autoinflammatory diseases, neonatal-onset multisystem inflammatory disease (NOMID)/chronic infantile neurologic, cutaneous and arthritis (CINCA), cryopin-associated periodic syndromes (CAPS), deficiency of the IL-1 receptor antagonist, NLRP3, IL1RN, IL-1 receptor antagonist, anakinra, neonatal disorder, genetic disease, IL-1
The concept of autoinflammation was initially developed about 10 years ago after the genetic causes of 2 hereditary fever syndromes, familial Mediterranean fever (FMF)1,2 and the former familial Hibernian fever,3 were identified by means of positional cloning. The FMF mutations occurred in a novel gene, Mediterranean fever gene (MEFV), which encodes the protein pyrin. Mutations in the p55 TNF receptor led to the renaming of familial Hibernian fever as TNF receptor–associated periodic syndrome (TRAPS). Both diseases present with episodic occurrences of fever, sterile serositis, and other more variable inflammatory manifestations but lack the clinical and laboratory markers that indicate adaptive immune dysregulation, such as autoantibodies and antigen-specific T cells. There is no evidence of infection, allergy, or immunodeficiency; disease triggers in these 2 disorders are not obvious, but nonspecific factors, such as stress and minor infections, are often reported to induce flares.
In subsequent years, the genes causing at least 10 Mendelian autoinflammatory diseases have been described and are listed in Table I. In 1999, 2 Dutch groups independently reported that mutations in the mevalonate kinase gene cause hyperimmunoglo-bulinemia D with periodic fever syndrome.4,5 In 2001, mutations in another previously unknown gene that encodes the then-novel protein cryopyrin were found to cause the autosomal dominant disorders familial cold autoinflammatory syndrome (FCAS) and Muckle-Wells syndrome (MWS).6 The following year, mutations in this same gene were identified in patients with neonatal-onset multisystem inflammatory disease (NOMID; also known as chronic infantile neurologic, cutaneous, and arthritis [CINCA] syndrome),7,8 which occurs as a sporadic disease because of the reduced reproductive fitness often associated with these severe mutations. The discovery of the genes underlying pyogenic arthritis, pyoderma gangrenosum, and acne9; Blau syndrome10; early-onset sarcoidosis11; Majeed syndrome12; cherubism13; FCAS214; and, most recently, the deficiency of the IL-1 receptor antagonist (DIRA)15,16 have opened our awareness to yet more genes and pathways, many of which have not been fully characterized.
TABLE I.
Monogenic autoinflammatory syndromes
| Disease | Year mutation published | Gene/protein | Inheritance pattern | Disease onset | Flare/fever pattern | Specific organ inflammation |
|---|---|---|---|---|---|---|
| FMF (MIM 249100) | 1997 | MEFV/pyrin | AR | 80% of the cases occur before the age of 20 y | 1–3 d | Skin, joints, peritoneum, pleura |
| TRAPS (MIM 191190) | 1999 | TNFRSF1A/ TNFRSF1A, TNFR1, p55 | AD | Median age at onset of 3 y | 1–6 wk | Skin, eyes, joints, peritoneum, pleura |
| CAPS | ||||||
| FCAS (MIM 120100) | 2001 | CIAS1 or NLRP3/cryopyrin or NLRP3 or NALP3 | AD | First 6 mo of life, cold induced | <24 h | Skin, eyes, joints |
| MWS (MIM 191900) | 2001 | CIAS1 or NLRP3/cryopyrin or NLRP3 or NALP3 | AD | Infancy to adolescence | 24–48 h | Skin, eyes, joints, inner ears, meninges (mild) |
| NOMID (MIM 607115) | 2002 | CIAS1 or NLRP3/cryopyrin or NLRP3 or NALP3 | AD/de novo | Neonatal or early infancy | Continuous with flares | Skin, eyes, joints, inner ears, meninges, bony epiphyseal hyperplasia |
| HIDS (MIM 260920) | 1999 | MVK/mevalonate kinase (MK) | AR | Median age at onset 6 mo | 3–7 d | Skin, eyes, joints, serosa, prominent lymph nodes |
| PGA (MIM 186580) | 2001 and 2005* | NOD2 or CARD15/NOD2 or CARD15 | AD/de novo | Early childhood | Continuous | Skin, eyes, joints |
| PAPA (MIM 604416) | 2002 | CD2BP1 or PSTPIP1/CD1BP1 or PSTPIP1 | AD | Early childhood | Prolonged flares | Skin, joints |
| Majeed syndrome (MIM 609628) | 2005 | LPIN2/LPIN2 | AR | Early infancy (1–19 mo) | Weeks to months | Bones, periosteum, anemia |
| Cherubism (MIM 118400) | 2001 | SH3BP2/SH3BP2 | AD | Childhood, spontaneous remission by 3rd decade | Continuous early in life | Jaws, eyes (rare) |
| FCAS2 (MIM 611762) | 2008 | NLRP12/NLRP12 or NALP12 | AD | Childhood, cold induced | 2–10 d, 1–3 × per month | Skin, hearing, joints, aphthous ulcers |
| DIRA (MIM 612852) | 2009 | IL1RN/IL-1Ra | AR | Neonatal or early infancy | Continuous with flares | Skin, bones, lungs (rare), vasculitis (rare) |
AD, Autosomal dominant; AR, autosomal recessive; CARD15, C-terminal caspase recruitment domain; DIRA, deficiency of the IL-1Ra caused by autosomal-recessive loss-of-function mutations of IL1RN; HIDS, hyperimmunoglobulinemia D with periodic fever syndrome; MIM, Mendelian inheritance in man number; MVK, mevalonate kinase gene; PAPA, pyogenic arthritis, pyoderma gangrenosum, and acne syndrome; PGA, pediatric granulomatous arthritis encompasses the familial Blau syndrome and the sporadic early-onset sarcoidosis (MIM 609464); TRAPS, TNF receptor–associated periodic syndrome.
The gene for the familial disease Blau syndrome was identified in 2001, and that for the sporadic form, sporadic early-onset sarcoidosis, was identified in 2005.
Other systemic autoinflammatory diseases with presumed more complex modes of inheritance include systemic-onset juvenile idiopathic arthritis; adult-onset Still disease; the syndrome of periodic fever with aphthous stomatitis, pharyngitis, and cervical adenitis (PFAPA); Behçet disease; and chronic recurrent multifocal osteomyelitis (CRMO). The discovery that gout is caused by increased activation of the NALP3 (NLRP3) inflammasome by uric acid crystals has illustrated the role of the NALP3 (NLRP3) inflammasome in common diseases. The concept of autoinflammation is currently evolving, and the ongoing discovery that innate immune dysregulation is also seen in patients who were thought to have classical autoimmune diseases, such as systemic lupus erythematosus and rheumatoid arthritis, indicates that in many polygenic/complex inflammatory diseases, abnormalities in the innate immune system and adaptive system jointly contribute to disease.17 A recent extensive review explores the concept of autoinflammation in diseases with mutations or immune dysregulation not only involving inflammasome components with aberrations in the IL-1 pathway but also involving nuclear factor κB activation, protein misfolding, complement regulation, cytokine signaling, and macrophage activation, reflecting the growing list of diseases that are thought to have evidence of autoinflammation.18
A major conceptual breakthrough in understanding auto-inflammatory diseases has come from Hoffman et al’s 2001 discovery6 that mutations in the cryopyrin gene cause FCAS and MWS.6 The gene that encodes cryopyrin is variously called CIAS1, NALP3, NLRP3, and, rarely, PYPAF1 and is a major component in the assembly of the NALP3 (NLRP3) inflammasome, an intracellular molecular complex that links the immune system’s ability to sense danger to a first response to such challenges by activating the crucial proinflammatory cytokine IL-1β. Despite the pivotal role of IL-1 in a number of autoinflammatory diseases, it is by no means the only cytokine pathway involved, but its exploration has certainly been aided by the availability of IL-1–inhibiting drugs for therapy. Thus this review will focus on the description of 2 autoinflammatory syndromes in which the significant contribution of IL-1 to the disease’s pathogenesis has been confirmed in clinical trials with IL-1 inhibition.
THE INFLAMMASOME AS SENSOR OF DANGER AND INSTIGATOR OF AUTOINFLAMMATION
Our immune system has evolved to protect our bodies against microbial infections and cellular waste that accumulate when cells are damaged or die. Two types of immune defense systems have emerged. The recognition of exogenous and endogenous danger by the innate immune system is mediated through pattern-recognition receptors, which we inherit and are not subject to adaptation or fine tuning through gene rearrangement or somatic mutation as we get older. Receptors of the innate immune system bind invariant microbial molecules, such as microbial cell wall components. On the other hand, the recognition of foreign antigens by the adaptive or acquired immune system is mediated by receptors on T and B cells that undergo somatic mutation, rearrangement, and specific selection and therefore allow fine tuning of the receptor specificity in response to antigen contact. This mechanism allows the development of a highly diverse T- and B-cell receptor repertoire and enables the development of immunologic memory.19
The NALP3 (NLRP3, cryopyrin) protein, which is mutated in cryopyrin-associated periodic syndromes (CAPS), and its structurally related cousin NOD2 (C-terminal caspase recruitment domain 15), which is mutated in patients with pediatric granulomatous arthritis and some patients with Crohn disease, have structural homology with plant resistance proteins20 and have established an important role of these phylogenetically conserved molecules in the recognition and response to danger in the human organism. In contrast to other pathogen-recognition receptors, such as most Toll-like receptors, the family of NOD-like receptors to which NLRP3 and NOD2 belong are intracellular sensors.21 NOD-like receptors have been shown to form active multimolecular complexes called inflammasomes that, in the case of the most often studied NALP3 inflammasome (Fig 1), result in increased caspase-1–mediated IL-1β processing and secretion.22 The NALP3 inflammasome can be triggered by a number of exogenous stimuli or “danger signals” that include conserved microbial components and large inorganic crystalline structures, such as asbestos and silica, but also endogenous danger signals that get released, for example, when cells are stressed or are dying and include uric acid, ATP, and DNA and RNA fragments. Given the diversity of stimuli that activate the NALP3 inflammasome, it is likely that it is triggered by intermediate homeostatic changes in the cell (the guard hypothesis) rather than by direct binding, as has been shown for the plant resistance proteins.
FIG 1.

The NALP3 (NLRP3) inflammasome and IL-1β–activating platform. The NALP3 (NLRP3) inflammasome is a macromolecular complex that cleaves pro–IL-1β to its biologically active form by bringing 2 molecules of pro–caspase-1 in close apposition with one another. NALP3/NLRP3, also known as cryopyrin, is comprised of an N-terminal pyrin domain (PYD), a NACHT domain, and a C-terminal leucine-rich repeat (LRR). The PYD of NALP3/NLRP3 binds the N-terminal PYD of the adaptor protein ASC (apoptosis-associated speck-like protein with a caspase recruitment domain) by homotypic interactions. The C-terminal caspase recruitment domain (CARD) of ASC binds the N-terminal CARD of pro–caspase-1, again through homotypic interactions. The NACHT domain of NALP3/NLRP3 binds the N-terminal FIIND domain (domain with function to find) of Cardinal. The C-terminal CARD of Cardinal recruits a second molecule of pro–caspase-1 to the complex. With induced proximity, the 2 pro–caspase-1 molecules undergo autocatalysis, liberating 2 catalytically active p20 and 2 catalytically active p10 domains, which form a heterotetramer capable of cleaving pro–IL-1β.
IL-1 AS A PROTOTYPIC “ALARM” CYTOKINE
IL-1 helps coordinate the immune system’s early response to exogenous and endogenous danger, serving as a prototypic alarm cytokine.23 IL-1α and IL-1β were the first members to be described in a now 11-member family of IL-1 molecules that includes at least one IL-1 receptor antagonist (IL-1Ra) molecule.24,25 The genes for 9 members of this family (all except IL-18 [IL-1F4] and IL-33 [IL-1F11]) are encoded in a cluster on the long arm of chromosome 2. In addition, the type I and II IL-1 receptors belong to a different 10-member family that also includes an accessory receptor (IL-1 receptor accessory protein [IL-1RAcP, IL-1R3]). Whereas IL-1β must be proteolytically cleaved from its 31-kd precursor form to its 17-kd fragment to be activated, the precursor form of IL-1α is already biologically active. IL-1α and IL-1β both bind to the IL-1 receptor (IL-1R) type I and to the “decoy” type II receptor, which lacks the intra-cellular Toll-like receptor domain needed for signaling.
When IL-1β or IL-1α bind to the IL-1R type I, the IL-1 receptor accessory protein (IL-1RAcP, IL-1R3) associates with the IL-1/ IL-1R1 complex, thus approximating the intracellular Toll-like receptor domains of the type I IL-1R and the IL-1RAcP into a dimer that recruits MyD88 and initiates the signaling cascade. This process is tightly regulated at a number of levels, one of which is through the IL-1 receptor antagonist (IL-1Ra), which can bind the type I IL-1R but is unable to simultaneously engage the IL-1RAcP. Thus the IL-1Ra competitively inhibits the formation of active IL-1R1/IL-1RAcP signaling complexes, thus preventing the proinflammatory signaling of IL-1α and IL-1β (Fig 2). IL-1Ra was first isolated in 1986 as a soluble factor from the urine that competed with IL-1α and IL-1β for binding to their receptor.26 In 1990, IL-1Ra was the first naturally occurring cyto-kine receptor antagonist to be purified and have its gene cloned.27 IL-1Ra has 26% to 30% homology to the gene structure of IL-1β and 19% to that of IL-1α. The IL-1 receptor antagonist gene (IL1RN) is encoded in the gene cluster on human chromosome 2q14, which also includes the genes for IL-1α and IL-1β.23 The balance between IL-1 and IL-1Ra is crucial in distinguishing proinflammatory and anti-inflammatory outcomes, and in a growing number of diseases, an imbalance of IL-1 and IL-1Ra seems to influence disease severity.28 The discovery that CAPS and DIRA are phenotypically distinct autoinflammatory diseases that are mediated by molecular lesions in 2 different proteins in the IL-1 pathway underscores the nuances and complexity of the pathway.
FIG 2.
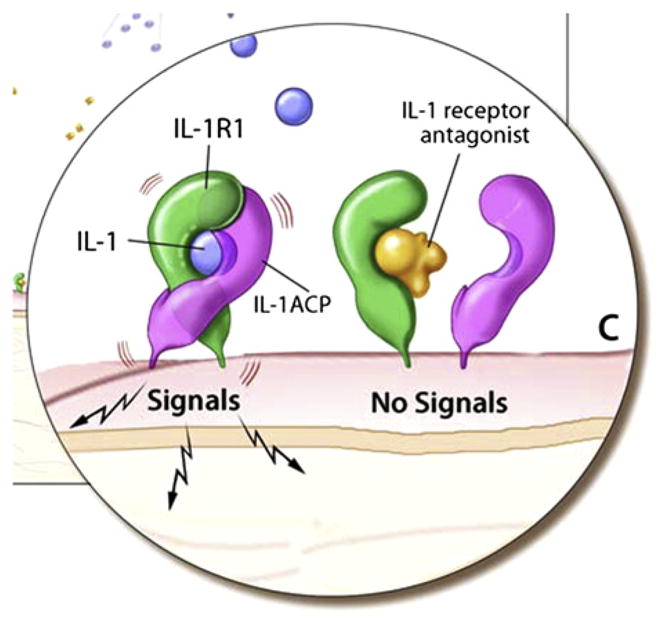
IL-1 receptor signaling. IL-1α and IL-1β can bind to the IL-1R1 receptor, which recruits the accessory receptor (IL-1ACP). This receptor complex forms a signaling unit. However, binding of the IL-1Ra to the IL-1R1 receptor inhibits IL-1 binding and does not allow for association with the accessory receptor, and therefore no signaling through the receptor occurs.
As noted above, IL-1β is activated when it is cleaved by the inflammasome. Because it is the most powerful endogenous fever-inducing molecule (pyrogen) known, there are multiple checkpoints to control its production and effects, including both the regulation of inflammasome activation and the control of its end-organ activity through IL-1Ra. Although the major sources of IL-1β are blood monocytes, tissue macrophages, and dendritic cells,24 it should be noted that leukocytes producing this cytokine are found in immunologically privileged organs, such as the kidney, heart, skeletal muscle, and brain.29 Release of IL-1β leads to induction of IL-1β itself, TNF, inducible nitric oxide synthase, COX-2, prostaglandin E2, nitric oxide, and type 2 phospholipase A, depending on the target cell type. In contrast to IL-1β, the biologic activity of IL-1α is not dependent on the inflammasome, and the biologically active IL-1α precursor is present intracellularly (and might be released with cell death) and on the cell membrane.24 IL-1α is expressed at high levels in epithelial cells, such as in the intestine and lungs, as well as lymphoreticular organs, such as the spleen and liver. Because IL-1 plays such a pivotal role in the initiation of inflammation, it has been an important therapeutic target, although the most impressive results of targeting IL-1 have occurred only recently as this strategy has been used in the treatment of autoinflammatory diseases.
EARLY CLINICAL STUDIES WITH IL-1RA IN SEPSIS AND RHEUMATOID ARTHRITIS
Based on the observation that IL-1β blockade had attenuated the severity of disease and mortality in experimental models of shock and sepsis,30 clinical trials of the recombinant IL-1Ra anakinra were conducted in the early 1990s to asses its utility in these 2 conditions. The results of a phase II study suggested a possible benefit, with anakinra reducing 28-day all-cause mortality in a dose-dependent manner.31 However, based on a subsequent phase III trial that failed to demonstrate a reduction in 28-day mortality,32 it was not approved for this indication. Several years later, anakinra was US Food and Drug Administration (FDA) approved for the treatment in rheumatoid arthritis, an autoimmune disease characterized by chronic inflammation of the joint lining (synovial membrane). Although anakinra is well tolerated despite its requisite daily dosing and common injection site reactions, anti-TNF agents appear to have superior clinical efficacy in patients with rheumatoid arthritis.33 To date, there have been no trials published indicating that other IL-1–blocking agents are more effective than anakinra in patients with rheumatoid arthritis.
Two disorders that are caused by dysregulated IL-1 responses with remarkable clinical responses to IL-1 blockade will be discussed in detail in this article. These include the spectrum of CAPS that is caused by mutations in NLRP3 (NALP3, CIAS1)6–8 and DIRA that is caused by homozygous mutations in the IL-1 receptor antagonist gene (IL1RN).15,16 Selected clinical trials data with IL-1–blocking agents in other autoinflammatory diseases will also be briefly discussed.
CAPS
Clinical presentation
Historically, CAPS was described not as a single entity but as 3 different disorders that were not recognized as related to one another until their common genetic cause was established. These diseases are familial cold urticaria (now known as FCAS), MWS, and NOMID, which was first named CINCA syndrome. These conditions were initially not recognized as related because of striking differences in severity, multiorgan disease manifestations, and long-term morbidity and mortality, although they do share manifestation of episodic fever and urticaria-like rash (Fig 3, A) and increases in acute-phase reactants. Although patients with MWS or NOMID typically present with an urticaria-like rash at birth, patients with FCAS might present later in life. Patients with FCAS present with a classical history of “cold-induced” episodes of inflammation that manifest with fever, urticaria-like rash, joint pain, conjunctivitis, and headaches. Episodes can last for 12 to 48 hours and then subside. Although patients suffer from these attacks, in most cases long-term outcome is favorable, and amyloidosis is rare.34
FIG 3.

Clinical manifestations of NOMID/CINCA and DIRA. A through D, Clinical images of NOMID. Fig 3, A, shows an urticaria-like rash in a patient with NOMID. Fig 3, B, demonstrates the characteristic bony proliferation within the growth plate, as indicated by red arrows. Fig 3, C, indicates cochlear enhancement in a patient with NOMID pretreatment. Fig 3, D, shows resolution of cochlear enhancement (cochleitis) in the same patient after 3 months’ treatment with anakinra. E through G, Clinical images of DIRA. Fig 3, E, shows a pustular rash on the neck, arm, and trunk. Fig 3, F, shows heterotopic bone formation and periosteal elevation on the proximal femurs bilaterally (red arrows). Fig 3, G, shows pathognomonic widening of multiple anterior ribs in a neonate with DIRA (red arrowheads).
In patients with MWS, episodes of fever, urticarial rash, and arthritis can be continuous and in most instances are not provoked by cold. Conjunctivitis, episcleritis, and optic disc edema are also seen, and progressive perceptive hearing loss develops in the second to fourth decade; in a European cohort amyloidosis was reported in up to 25% of patients.35,36
NOMID was first described in 2 siblings with a continuous rash from birth, lymphadenopathy, uveitis, and mental retardation.37 Anne Marie Prieur coined the acronym CINCA syndrome and asserted that it is distinct from systemic-onset juvenile idiopathic arthritis (or pediatric Still disease). Dr Prieur observed that abnormal bony overgrowth of the knees and chronic meningitis were seen in children with CINCA (NOMID) that were absent in the patients with Still disease.38,39 The characteristic arthropathy seen in patients with NOMID is caused by uncontrolled overgrowth of the patella and epiphyses of the long bones (Fig 3, B) and presents in up to 60% of patients.40 Joint contractures are an important cause of disability, but the most serious manifestations of NOMID are the consequence of chronic aseptic meningitis. Resulting central nervous system (CNS) manifestations include increased intracranial pressure, ventriculomegaly, cerebral atrophy, seizures, sensorineural hearing loss that begins in childhood and can lead to deafness, progressive vision loss caused by optic nerve atrophy from chronically increased intracranial pressures, and mental retardation.42,43 Other findings include short stature, frontal bossing of the skull, and, rarely, flattening of the nasal bridge. If untreated, the reported mortality in patients with NOMID is reported to be 20% before adulthood.43
FCAS, MWS, and NOMID/CINCA are all transmitted in an autosomal dominant fashion. However, although a history of other affected family members can usually be elicited from patients with FCAS and MWS, NOMID/CINCA often occurs as a de novo mutation, with no family history of CAPS. It should be noted that this is often due to the fact that, in the past, many patients with NOMID never had children. With effective treatment, this picture is likely to change. Laboratory research and clinical investigations conducted in parallel revealed the pivotal role of IL-1β in causing the clinical disease phenotype of CAPS. The discovery that genetic mutations in exon 3 of the gene NLRP3 (NALP3, CIAS1) cause all 3 disease phenotypes explains the IL-1β overproduction seen in all 3. These illnesses comprise a disease spectrum, with FCAS being the mildest and NOMID the most severe disease on the continuum. Nevertheless, up to 40% of patients with clinical NOMID have no demonstrable mutations in NLRP3, suggesting the likelihood that mutations will eventually be found in other inflammasomerelated genes.
Patients with FCAS, MWS, and NOMID often receive diagnoses late in life, when IL-1–mediated organ damage has already occurred. Early on, patients often receive misdiagnoses of allergies or viral infections, and the correct diagnosis is often only made when the patients are much older and might have permanent hearing loss. An early clue to the correct diagnosis can be obtained from a careful history. A nonpruritic rash that might burn or even sting but rarely itches and the presence of a neutrophilic dermal infiltrate on a skin biopsy specimen can help to make an early diagnosis. Microscopic examination of lesional skin reveals a predominant perivascular neutrophilic infiltrate, dermal edema, and dilated blood vessels, without the presence of vasculitis, mast cells, or mast cell degranulation. The latter are indicative of histamine release and are typically seen in allergic urticarial skin rashes.44–46 The characteristic neutrophilic infiltrate on skin histology should raise an early suspicion of an autoinflammatory disease, including CAPS, which would allow for timely treatment.
Clinical responses to treatment with IL-1–blocking agents in patients with NOMID
The clinical results for IL-1 blockade are striking; patients with CAPS respond well to treatment with anakinra and, more recently, the newer, long-acting IL-1 inhibitors. Clinical studies have shown significant improvement in the clinical symptoms of CAPS, including rash, headaches, fevers, and joint pain, and marked improvement in inflammatory markers, with remission in many patients, even in 60% of patients with NOMID. IL-1 blockade with anakinra in patients with NOMID can reverse organ inflammation imaged on magnetic resonance imaging, including CNS leptomeningitis and cochlear inflammation (Fig 3, C and D), which is the cause of progressive hearing loss.41 Preliminary data in very young children suggest that disability might be prevented if therapy can be initiated early in life, which requires early diagnosis (our own unpublished data). Clinical features that lead to permanent IL-1–mediated organ damage and those that are responsive to treatment with IL-1 blockade are listed in Table II. The dose of anakinra needed to suppress inflammation in patients with CAPS depends on disease severity and clinical phenotype and is lowest in patients with FCAS (0.5–1.5 mg/kg/d in most patients) and up to 3.5 mg/kg/d in patients with MWS and up to 6 mg/kg/d in patients with NOMID/CINCA. Despite multiple open-label studies showing the remarkable benefit of anakinra in patients with CAPS, this drug has not been FDA approved for the treatment of these conditions. However, recent successful drug development programs with the long-acting IL-1 inhibitor rilonacept (IL-1 Trap; Regeneron Pharmaceuticals, Inc, Tarry-town, NY), led to the first FDA-approved therapy for CAPS.47,48 A second long-acting IL-1 inhibitor, canakinumab (Novartis AG, Basel, Switzerland), also showed efficacy in patients with CAPS and was recently approved by the FDA for the treatment of CAPS.49
TABLE II.
Clinical features of NOMID and their response to IL-1–blocking therapy
| Inflammation | Symptoms/signs | End-organ damage | Preventable Partially reversible if damage not permanent Further progression is halted |
|---|---|---|---|
| Systemic inflammation | Fever, headaches | Amyloidosis in 30% | Yes* |
| Conjunctivitis, anterior and posterior uveitis, subcorneal infiltrates | Eye pain, cloudy vision | Retinal scarring, corneal clouding | Yes* |
| Papilledema | Headache | Progressive optic nerve atrophy | Yes* |
| Cochleitis | Progressive reversible hearing loss | Progressive permanent hearing loss | Yes* |
| Leptomeningitis | Headache, CSF pleocytosis | Hydrocephalus, brain atrophy, cognitive impairment | Leptomeningitis is fully reversible, hydrocephalus only when treated early, evidence of halting further progression is emerging |
| Bony enlargement | Bone and joint pain | Deformities, contractures | Maybe preventable with early treatment, but once a lesions is formed, progression is not halted with treatment. |
CSF, Cerebrospinal fluid.
Applies to all categories.
DIRA
We recently reported a series of 9 patients with a severe autoinflammatory condition with some similarity to NOMID who had homozygous mutations in IL1RN.15 Eight of the 9 patients had inactivating point mutations in IL1RN, whereas the remaining patient, a child from Puerto Rico, harbored a 175-kb genomic deletion that encompassed IL1RN and 5 other genes in the IL1 family (IL1F9, IL1F6, IL1F8, IL1F5, IL1F10, and IL1RN, from centromere to telomere). In an accompanying case report in the New England Journal of Medicine, another group reported an unrelated patient, also of Puerto Rican ancestry, with the same large genomic deletion and similar clinical findings.16 The mortality of our early reported cases included 3 of 9 identified patients during early childhood. In recognition of the distinct clinical and genetic features of this illness, we have proposed the name “deficiency of the IL-1 receptor antagonist (DIRA)” to denote these patients.
Mutations in IL1RN lead to complete absence of IL-1Ra protein and thus unopposed action of IL-1 on the IL-1 receptor because the competitive antagonist is lacking. Affected children present with their first symptoms within the first 2.5 weeks of life. Fetal distress, pustular rash, joint swelling, oral mucosal lesions, and pain with movement were common presenting features. Over time, all children had cutaneous pustulosis, ranging from discrete crops of pustules to generalized severe pustulosis or ichthyosiform lesions (Fig 3, E). Similar to patients with NOMID, lesional skin biopsy specimens show extensive neutrophilic infiltration in the dermis, but in contrast to NOMID, the neutrophils are also present in the epidermis, and pustule formation along the hair follicles, acanthosis, and hyperkeratosis are seen on biopsy. Histopathologic evidence of vasculitis was observed in the connective and fat tissue adjacent to bone in 1 patient,15 and extensive thrombosis was found in another child.16 Pain and joint swelling are early indicators of bone involvement, and extensive epiphyseal ballooning, similar to the bony deformities seen in patients with NOMID, were observed in one of 3 children from Puerto Rico. Radiographic characteristics of the bony lesions include balloon-like widening of the anterior rib ends, which was seen in all affected children; periosteal elevation along multiple long bones and heterotopic ossification around the hip; and multifocal osteolytic lesions (Fig 3, F and G). Signs of cord compression were seen in one of several children who had vertebral osteolytic lesions that led to vertebral collapse. Bone biopsy specimens are sterile, with variable histology showing purulent osteomyelitis, fibrosis, and sclerosis. Cerebral vasculitis/ vasculopathy was present in 1 patient on magnetic resonance imaging, and pulmonary hemosiderosis with progressive interstitial fibrosis was seen in 1 patient who had died. Interestingly, most children do not have high-grade fever despite a marked increase in the erythrocyte sedimentation rate and C-reactive protein levels. Therapy with DMARDs and high doses of corticosteroids only partially controlled clinical symptoms and acute-phase reactants.
In contrast to patients with NOMID (Table III), patients with DIRA do not have CNS or inner ear inflammation. Patients with point mutations respond dramatically to treatment with anakinra, which is the recombinant form of the very protein these children are missing. Patients with the genomic deletion exhibit a gratifying but less complete response to anakinra. The IL1RN mutations are present in founder populations in Newfoundland The Netherlands, Puerto Rico, and possibly Lebanon.15,16 Heterozygous carriers are asymptomatic and have no detectable cyto-kine abnormalities in vitro. The clinical presentation early in life can resemble that of neonatal sepsis and osteomyelitis. Failure to recognize and treat with anakinra can lead to the development of a severe inflammatory response syndrome and death from multiorgan failure. The dramatic effect of treatment with anakinra that can be lifesaving, makes the early recognition of this disease very important.
TABLE III.
A comparison of NOMID/CINCA with DIRA
| NOMID/CINCA | DIRA | |
|---|---|---|
| Gene | CIAS1/NLRP3 | IL1RN |
| Functional consequence | Increased inflammasome activation (IL-1β, IL-18, IL-33) | Decreased inhibition of IL-1 (α and β) |
| Skin | Urticarial rash | Pustulosis |
| Bone | Epiphyseal overgrowth | Multifocal osteomyelitis |
| CNS involvement | Aseptic meningitis, blindness, hearing loss | None known |
IL-1 BLOCKADE IN OTHER AUTOINFLAMMATORY DISEASES
Anakinra has also been used to prevent attacks and reduce systemic inflammation in patients with colchicine-resistant FMF,50–52 hyperimmunoglobulinemia D with periodic fever syndrome,53,54 and TNF receptor–associated periodic syndrome,55–57 and good responses were also reported in patients with Blau syndrome58 and some with pyogenic arthritis, pyoderma gangrenosum, and acne.59 In addition to the effect in monogenic diseases, a number of presumed polygenic autoinflammatory diseases have successfully been treated with IL-1 inhibition. Some likely polygenic autoinflammatory diseases that share clinical similarities with monogenic autoinflammatory diseases also show impressive responses to IL-1 blockade. These include acute and chronic gout,60,61 pseudogout, and Schnitzler syndrome,62 a rare acquired urticarial disease with clinical similarities to MWS that is also associated with a monoclonal IgM gammopathy. A subset of patients with pediatric63–68 and adult Morbus Still disease67,69,70 is also responsive to IL-1 blockade. A case report of a patient with Behçet disease71 and the improvement in glucose tolerance in type II diabetes treated with IL-1 blockade72 suggest that IL-1 involvement and IL-1–mediated organ damage are not only limited to a small group of rare diseases but that autoinflammation might be a concept that could be useful in thinking about the pathogenesis and treatment of some common diseases that are not traditionally thought to be immunologic diseases.
SUMMARY
The systemic autoinflammatory diseases are a group of illnesses characterized by episodic or sometimes chronic inflammation without evidence of high-titer autoantibodies or antigen-specific T lymphocytes. Monogenic autoinflammatory diseases are inborn errors of the phylogenetically ancient innate immune system, with its predominance of myeloid cells and germline receptors, whereas monogenic autoimmune diseases affect primarily acquired or adaptive immunity, with its predominance of lymphocytes and receptors that somatically rearrange and mutate. There is accumulating evidence that some more common polygenic immune disorders lie at the interface between auto-inflammatory and autoimmune disease, combining elements of innate and adaptive immunopathology. IL-1 is one of the key mediators of the innate immune system, and several monogenic autoinflammatory diseases show evidence of dysregulation of IL-1 signaling. A macromolecular complex denoted the inflammasome cleaves IL-1β from its biologically inactive precursor form to its 17-kd fragment, which is a major mediator of fever and inflammation. The best studied of the various molecular variants of the inflammasome is the NLRP3 (NALP3) inflammasome. In CAPS (FCAS, MWS, and NOMID/CINCA) there is constitutive activation of the NLRP3 inflammasome caused by autosomal dominant mutations in the NLRP3 gene. This leads to excessive IL-1β activation and varying degrees of inflammation, with the most severe manifestations seen in patients with NOMID/ CINCA. The activation and effects of IL-1β are highly regulated. The IL-Ra protein, which is encoded by the IL1RN gene, blocks IL-1 signaling by competitively inhibiting binding of IL-1β to its receptor. In a newly described autoinflammatory disease denoted DIRA, patients have autosomal recessive, inactivating mutations in the gene encoding IL-1Ra, leading to a profound inflammatory phenotype. Although CAPS and DIRA exhibit similarities both phenotypically and immunologically, there are important clinical differences that likely teach us about the differences between inflammasome and IL-1Ra expression in local tissues. Regardless of these differences, the recombinant IL-1Ra anakinra shows marked efficacy in patients with both CAPS and DIRA. With the recognition of the expanding spectrum of autoinflammatory disease, and the corresponding involvement of IL-1β in some of these other more genetically complex conditions, IL-1 is an attractive therapeutic target in a growing family of illnesses that only recently were not suspected to be immunologically mediated.
INFORMATION FOR CATEGORY 1 CME CREDIT.
Credit can now be obtained, free for a limited time, by reading the review articles in this issue. Please note the following instructions.
Method of Physician Participation in Learning Process: The core material for these activities can be read in this issue of the Journal or online at the JACI Web site: www.jacionline.org. The accompanying tests may only be submitted online at www.jacionline.org. Fax or other copies will not be accepted.
Date of Original Release: December 2009. Credit may be obtained for these courses until November 30, 2011.
Copyright Statement: Copyright © 2009-2011. All rights reserved.
Overall Purpose/Goal: To provide excellent reviews on key aspects of allergic disease to those who research, treat, or manage allergic disease.
Target Audience: Physicians and researchers within the field of allergic disease.
Accreditation/Provider Statements and Credit Designation: The American Academy of Allergy, Asthma & Immunology (AAAAI) is accredited by the Accreditation Council for Continuing Medical Education (ACCME) to provide continuing medical education for physicians. The AAAAI designates these educational activities for a maximum of 1 AMA PRA Category 1 Credit™. Physicians should only claim credit commensurate with the extent of their participation in the activity.
List of Design Committee Members: Authors: Raphaela Goldbach-Mansky MD, MHS, and Daniel L. Kastner MD, PhD
Activity Objectives
To define monogenic and polygenic autoinflammatory disorders.
To describe the role of the NALP3/NLRP3 inflammasome.
To define the role of IL-1 and its receptor antagonist in autoinflammatory processes.
To compare the molecular and clinical features of the autoinflammatory diseases neonatal-onset multisystem inflammatory disease (NOMID/CINCA) and deficiency of the IL-1 receptor antagonist (DIRA).
Recognition of Commercial Support: This CME activity is supported by an educational grant from Merck & Co., Inc.
Disclosure of Significant Relationships with Relevant
Commercial Companies/Organizations: The authors are employees of the National Institutes of Health.
Acknowledgments
Supported by an educational grant from Merck & Co., Inc.
Abbreviations
- CAPS
Cryopyrin-associated periodic syndromes
- CNS
Central nervous system
- DIRA
Deficiency of the IL-1 receptor antagonist
- FCAS
Familial cold autoinflammatory syndrome
- FDA
US Food and Drug Administration
- FMF
Familial Mediterranean fever
- IL-1R
IL-1 receptor
- IL-1Ra
IL-1 receptor antagonist
- IL-1RAcP
IL-1 receptor accessory protein
- IL1RN
IL-1 receptor antagonist gene
- MWS
Muckle-Wells syndrome
References
- 1.The International FMF Consortium. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell. 1997;90:797–807. doi: 10.1016/s0092-8674(00)80539-5. [DOI] [PubMed] [Google Scholar]
- 2.The French FMF Consortium. A candidate gene for familial Mediterranean fever. Nat Genet. 1997;17:25–31. doi: 10.1038/ng0997-25. [DOI] [PubMed] [Google Scholar]
- 3.McDermott MF, Aksentijevich I, Galon J, McDermott EM, Ogunkolade BW, Centola M, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell. 1999;97:133–44. doi: 10.1016/s0092-8674(00)80721-7. [DOI] [PubMed] [Google Scholar]
- 4.Drenth JP, Cuisset L, Grateau G, Vasseur C, van de Velde-Visser SD, de Jong JG, et al. Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. International Hyper-IgD Study Group. Nat Genet. 1999;22:178–81. doi: 10.1038/9696. [DOI] [PubMed] [Google Scholar]
- 5.Houten SM, Kuis W, Duran M, de Koning TJ, Royen-Kerkhof A, Romeijn GJ, et al. Mutations in MVK, encoding mevalonate kinase, cause hyperimmunoglobu-linaemia D and periodic fever syndrome. Nat Genet. 1999;22:175–7. doi: 10.1038/9691. [DOI] [PubMed] [Google Scholar]
- 6.Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet. 2001;29:301–5. doi: 10.1038/ng756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feldmann J, Prieur AM, Quartier P, Berquin P, Certain S, Cortis E, et al. Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. Am J Hum Genet. 2002;71:198–203. doi: 10.1086/341357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aksentijevich I, Nowak M, Mallah M, Chae JJ, Watford WT, Hofmann SR, et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum. 2002;46:3340–8. doi: 10.1002/art.10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wise CA, Gillum JD, Seidman CE, Lindor NM, Veile R, Bashiardes S, et al. Mutations in CD2BP1 disrupt binding to PTP PEST and are responsible for PAPA syndrome, an autoinflammatory disorder. Hum Mol Genet. 2002;11:961–9. doi: 10.1093/hmg/11.8.961. [DOI] [PubMed] [Google Scholar]
- 10.Miceli-Richard C, Lesage S, Rybojad M, Prieur AM, Manouvrier-Hanu S, Hafner R, et al. CARD15 mutations in Blau syndrome. Nat Genet. 2001;29:19–20. doi: 10.1038/ng720. [DOI] [PubMed] [Google Scholar]
- 11.Kanazawa N, Okafuji I, Kambe N, Nishikomori R, Nakata-Hizume M, Nagai S, et al. Early-onset sarcoidosis and CARD15 mutations with constitutive nuclear factor-kappaB activation: common genetic etiology with Blau syndrome. Blood. 2005;105:1195–7. doi: 10.1182/blood-2004-07-2972. [DOI] [PubMed] [Google Scholar]
- 12.Ferguson PJ, Chen S, Tayeh MK, Ochoa L, Leal SM, Pelet A, et al. Homozygous mutations in LPIN2 are responsible for the syndrome of chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anaemia (Majeed syndrome) J Med Genet. 2005;42:551–7. doi: 10.1136/jmg.2005.030759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ueki Y, Tiziani V, Santanna C, Fukai N, Maulik C, Garfinkle J, et al. Mutations in the gene encoding c-Abl-binding protein SH3BP2 cause cherubism. Nat Genet. 2001;28:125–6. doi: 10.1038/88832. [DOI] [PubMed] [Google Scholar]
- 14.Jeru I, Duquesnoy P, Fernandes-Alnemri T, Cochet E, Yu JW, Lackmy-Port-Lis M, et al. Mutations in NALP12 cause hereditary periodic fever syndromes. Proc Natl Acad Sci U S A. 2008;105:1614–9. doi: 10.1073/pnas.0708616105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aksentijevich I, Masters SL, Ferguson PJ, Dancey P, Frenkel J, Royen-Kerkhoff A, et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Engl J Med. 2009;360:2426–37. doi: 10.1056/NEJMoa0807865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reddy S, Jia S, Geoffrey R, Lorier R, Suchi M, Broeckel U, et al. An autoinflammatory disease due to homozygous deletion of the IL1RN locus. N Engl J Med. 2009;360:2438–44. doi: 10.1056/NEJMoa0809568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McGonagle D, McDermott MF. A proposed classification of the immunological diseases. PLoS Med. 2006;3:e297. doi: 10.1371/journal.pmed.0030297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease (*) Annu Rev Immunol. 2009;27:621–68. doi: 10.1146/annurev.immunol.25.022106.141627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819–26. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- 20.Schneider DS. Plant immunity and film noir: what gumshoe detectives can teach us about plant-pathogen interactions. Cell. 2002;109:537–40. doi: 10.1016/s0092-8674(02)00764-x. [DOI] [PubMed] [Google Scholar]
- 21.Ye Z, Ting JP. NLR, the nucleotide-binding domain leucine-rich repeat containing gene family. Curr Opin Immunol. 2008;20:3–9. doi: 10.1016/j.coi.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 22.Petrilli V, Dostert C, Muruve DA, Tschopp J. The inflammasome: a danger sensing complex triggering innate immunity. Curr Opin Immunol. 2007;19:615–22. doi: 10.1016/j.coi.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 23.Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. 1996;87:2095–147. [PubMed] [Google Scholar]
- 24.Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–50. doi: 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- 25.Towne JE, Garka KE, Renshaw BR, Virca GD, Sims JE. Interleukin (IL)-1F6, IL-1F8, and IL-1F9 signal through IL-1Rrp2 and IL-1RAcP to activate the pathway leading to NF-kappaB and MAPKs. J Biol Chem. 2004;279:13677–88. doi: 10.1074/jbc.M400117200. [DOI] [PubMed] [Google Scholar]
- 26.Balavoine JF, de Rochemonteix B, Williamson K, Seckinger P, Cruchaud A, Dayer JM. Prostaglandin E2 and collagenase production by fibroblasts and synovial cells is regulated by urine-derived human interleukin 1 and inhibitor(s) J Clin Invest. 1986;78:1120–4. doi: 10.1172/JCI112669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hannum CH, Wilcox CJ, Arend WP, Joslin FG, Dripps DJ, Heimdal PL, et al. Interleukin-1 receptor antagonist activity of a human interleukin-1 inhibitor. Nature. 1990;343:336–40. doi: 10.1038/343336a0. [DOI] [PubMed] [Google Scholar]
- 28.Arend WP. The balance between IL-1 and IL-1Ra in disease. Cytokine Growth Factor Rev. 2002;13:323–40. doi: 10.1016/s1359-6101(02)00020-5. [DOI] [PubMed] [Google Scholar]
- 29.Hacham M, Argov S, White RM, Segal S, Apte RN. Distinct patterns of IL-1 alpha and IL-1 beta organ distribution—a possible basis for organ mechanisms of innate immunity. Adv Exp Med Biol. 2000;479:185–202. doi: 10.1007/0-306-46831-x_16. [DOI] [PubMed] [Google Scholar]
- 30.Cain BS, Meldrum DR, Harken AH, McIntyre RC., Jr The physiologic basis for anticytokine clinical trials in the treatment of sepsis. J Am Coll Surg. 1998;186:337–50. doi: 10.1016/s1072-7515(98)00036-2. [DOI] [PubMed] [Google Scholar]
- 31.Fisher CJ, Jr, Slotman GJ, Opal SM, Pribble JP, Bone RC, Emmanuel G, et al. Initial evaluation of human recombinant interleukin-1 receptor antagonist in the treatment of sepsis syndrome: a randomized, open-label, placebo-controlled multicenter trial. Crit Care Med. 1994;22:12–21. doi: 10.1097/00003246-199401000-00008. [DOI] [PubMed] [Google Scholar]
- 32.Fisher CJ, Jr, Dhainaut JF, Opal SM, Pribble JP, Balk RA, Slotman GJ, et al. Recombinant human interleukin 1 receptor antagonist in the treatment of patients with sepsis syndrome. Results from a randomized, double-blind, placebo-controlled trial. Phase III rhIL-1ra Sepsis Syndrome Study Group. JAMA. 1994;271:1836–43. [PubMed] [Google Scholar]
- 33.O’Dell JR. Therapeutic strategies for rheumatoid arthritis. N Engl J Med. 2004;350:2591–602. doi: 10.1056/NEJMra040226. [DOI] [PubMed] [Google Scholar]
- 34.Thornton BD, Hoffman HM, Bhat A, Don BR. Successful treatment of renal amyloidosis due to familial cold autoinflammatory syndrome using an interleukin 1 receptor antagonist. Am J Kidney Dis. 2007;49:477–81. doi: 10.1053/j.ajkd.2006.10.026. [DOI] [PubMed] [Google Scholar]
- 35.Muckel TJ, Wells M. Urticaria, deafness, and amyloidosis: a new heredo-familial syndrome. QJM. 1962;31:235–48. [PubMed] [Google Scholar]
- 36.Muckle TJ. The “Muckle-Wells” syndrome. Br J Dermatol. 1979;100:87–92. doi: 10.1111/j.1365-2133.1979.tb03572.x. [DOI] [PubMed] [Google Scholar]
- 37.Ansell MB, Bywaters EG, Elderkin FM. Familial arthropathy with rash, uveitis and mental retardation. Proc R Soc Med. 1975;68:584–5. doi: 10.1177/003591577506800922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prieur AM, Griscelli C. Chronic meningo-cutaneo-articular syndrome in children. Rev Rhum Mal Osteoartic. 1980;47:645–9. [PubMed] [Google Scholar]
- 39.Prieur AM, Griscelli C. Arthropathy with rash, chronic meningitis, eye lesions, and mental retardation. J Pediatr. 1981;99:79–83. doi: 10.1016/s0022-3476(81)80961-4. [DOI] [PubMed] [Google Scholar]
- 40.Hill SC, Namde M, Dwyer A, Poznanski A, Canna S, Goldbach-Mansky R. Arthropathy of neonatal onset multisystem inflammatory disease (NOMID/CINCA) Pediatr Radiol. 2007;37:145–52. doi: 10.1007/s00247-006-0358-0. [DOI] [PubMed] [Google Scholar]
- 41.Goldbach-Mansky R, Dailey NJ, Canna SW, Gelabert A, Jones J, Rubin BI, et al. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N Engl J Med. 2006;355:581–92. doi: 10.1056/NEJMoa055137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aksentijevich I, Putnam D, Remmers EF, Mueller JL, Le J, Kolodner RD, et al. The clinical continuum of cryopyrinopathies: novel CIAS1 mutations in North American patients and a new cryopyrin model. Arthritis Rheum. 2007;56:1273–85. doi: 10.1002/art.22491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Prieur AM, Griscelli C, Lampert F, Truckenbrodt H, Guggenheim MA, Lovell DJ, et al. A chronic, infantile, neurological, cutaneous and articular (CINCA) syndrome. A specific entity analysed in 30 patients. Scand J Rheumatol Suppl. 1987;66:57–68. doi: 10.3109/03009748709102523. [DOI] [PubMed] [Google Scholar]
- 44.Hoffman HM, Wanderer AA, Broide DH. Familial cold autoinflammatory syndrome: phenotype and genotype of an autosomal dominant periodic fever. J Allergy Clin Immunol. 2001;108:615–20. doi: 10.1067/mai.2001.118790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lieberman A, Grossman ME, Silvers DN. Muckle-Wells syndrome: case report and review of cutaneous pathology. J Am Acad Dermatol. 1998;39:290–1. doi: 10.1016/s0190-9622(98)70094-5. [DOI] [PubMed] [Google Scholar]
- 46.Shinkai K, McCalmont TH, Leslie KS. Cryopyrin-associated periodic syndromes and autoinflammation. Clin Exp Dermatol. 2008;33:1–9. doi: 10.1111/j.1365-2230.2007.02540.x. [DOI] [PubMed] [Google Scholar]
- 47.Goldbach-Mansky R, Shroff SD, Wilson M, Snyder C, Plehn S, Barham B, et al. A pilot study to evaluate the safety and efficacy of the long-acting interleukin-1 inhibitor rilonacept (interleukin-1 Trap) in patients with familial cold autoinflammatory syndrome. Arthritis Rheum. 2008;58:2432–42. doi: 10.1002/art.23620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hoffman HM, Throne ML, Amar NJ, Sebai M, Kivitz AJ, Kavanaugh A, et al. Efficacy and safety of rilonacept (interleukin-1 Trap) in patients with cryopyrin-associated periodic syndromes: results from two sequential placebo-controlled studies. Arthritis Rheum. 2008;58:2443–52. doi: 10.1002/art.23687. [DOI] [PubMed] [Google Scholar]
- 49.Lachmann HJ, Kone-Paut I, Kuemmerle-Deschner JB, Leslie KS, Hachulla E, Quartier P, et al. Use of canakinumab in the cryopyrin-associated periodic syndrome. N Engl J Med. 2009;360:2416–25. doi: 10.1056/NEJMoa0810787. [DOI] [PubMed] [Google Scholar]
- 50.Gattringer R, Lagler H, Gattringer KB, Knapp S, Burgmann H, Winkler S, et al. Anakinra in two adolescent female patients suffering from colchicine-resistant familial Mediterranean fever: effective but risky. Eur J Clin Invest. 2007;37:912–4. doi: 10.1111/j.1365-2362.2007.01868.x. [DOI] [PubMed] [Google Scholar]
- 51.Kuijk LM, Govers AM, Frenkel J, Hofhuis WJ. Effective treatment of a colchicine-resistant familial Mediterranean fever patient with anakinra. Ann Rheum Dis. 2007;66:1545–6. doi: 10.1136/ard.2007.071498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roldan R, Ruiz AM, Miranda MD, Collantes E. Anakinra: new therapeutic approach in children with familial Mediterranean fever resistant to colchicine. Joint Bone Spine. 2008;75:504–5. doi: 10.1016/j.jbspin.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 53.Bodar EJ, van der Hilst JC, Drenth JP, van der Meer JW, Simon A. Effect of etanercept and anakinra on inflammatory attacks in the hyper-IgD syndrome: introducing a vaccination provocation model. Neth J Med. 2005;63:260–4. [PubMed] [Google Scholar]
- 54.Cailliez M, Garaix F, Rousset-Rouviere C, Bruno D, Kone-Paut I, Sarles J, et al. Anakinra is safe and effective in controlling hyperimmunoglobulinaemia D syndrome-associated febrile crisis. J Inherit Metab Dis. 2006;29:763. doi: 10.1007/s10545-006-0408-7. [DOI] [PubMed] [Google Scholar]
- 55.Simon A, Bodar EJ, van der Hilst JC, van der Meer JW, Fiselier TJ, Cuppen MP, et al. Beneficial response to interleukin 1 receptor antagonist in traps. Am J Med. 2004;117:208–10. doi: 10.1016/j.amjmed.2004.02.039. [DOI] [PubMed] [Google Scholar]
- 56.Gattorno M, Pelagatti MA, Meini A, Obici L, Barcellona R, Federici S, et al. Persistent efficacy of anakinra in patients with tumor necrosis factor receptor-associated periodic syndrome. Arthritis Rheum. 2008;58:1516–20. doi: 10.1002/art.23475. [DOI] [PubMed] [Google Scholar]
- 57.Sacre K, Brihaye B, Lidove O, Papo T, Pocidalo MA, Cuisset L, et al. Dramatic improvement following interleukin 1beta blockade in tumor necrosis factor receptor-1-associated syndrome (TRAPS) resistant to anti-TNF-alpha therapy. J Rheumatol. 2008;35:357–8. [PubMed] [Google Scholar]
- 58.Arostegui JI, Arnal C, Merino R, Modesto C, Antonia CM, Moreno P, et al. NOD2 gene-associated pediatric granulomatous arthritis: clinical diversity, novel and recurrent mutations, and evidence of clinical improvement with interleukin-1 blockade in a Spanish cohort. Arthritis Rheum. 2007;56:3805–13. doi: 10.1002/art.22966. [DOI] [PubMed] [Google Scholar]
- 59.Dierselhuis MP, Frenkel J, Wulffraat NM, Boelens JJ. Anakinra for flares of pyogenic arthritis in PAPA syndrome. Rheumatology (Oxford) 2005;44:406–8. doi: 10.1093/rheumatology/keh479. [DOI] [PubMed] [Google Scholar]
- 60.So A, De Smedt T, Revaz S, Tschopp J. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res Ther. 2007;9:R28. doi: 10.1186/ar2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McGonagle D, Tan AL, Shankaranarayana S, Madden J, Emery P, McDermott MF. Management of treatment resistant inflammation of acute on chronic tophaceous gout with anakinra. Ann Rheum Dis. 2007;66:1683–4. doi: 10.1136/ard.2007.073759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ryan J, de Koning HD, Beck LA, Booty MG, Kastner DL, Simon A. IL-1 blockade in Schnitzler syndrome: ex vivo findings correlate with clinical remission. J Allergy Clin Immunol. 2008;121:260–2. doi: 10.1016/j.jaci.2007.09.021. [DOI] [PubMed] [Google Scholar]
- 63.Reiff A. The use of anakinra in juvenile arthritis. Curr Rheumatol Rep. 2005;7:434–40. doi: 10.1007/s11926-005-0047-2. [DOI] [PubMed] [Google Scholar]
- 64.Pascual V, Allantaz F, Arce E, Punaro M, Banchereau J. Role of interleukin- 1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J Exp Med. 2005;201:1479–86. doi: 10.1084/jem.20050473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gattorno M, Piccini A, Lasiglie D, Tassi S, Brisca G, Carta S, et al. The pattern of response to anti-interleukin-1 treatment distinguishes two subsets of patients with systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2008;58:1505–15. doi: 10.1002/art.23437. [DOI] [PubMed] [Google Scholar]
- 66.Lequerre T, Quartier P, Rosellini D, Alaoui F, De Bandt M, Mejjad O, et al. Inter-leukin-1 receptor antagonist (anakinra) treatment in patients with systemic-onset juvenile idiopathic arthritis or adult onset Still disease: preliminary experience in France. Ann Rheum Dis. 2008;67:302–8. doi: 10.1136/ard.2007.076034. [DOI] [PubMed] [Google Scholar]
- 67.Quartier P. Still’s disease (systemic-onset juvenile idiopathic arthritis) Arch Pediatr. 2008;15:865–6. doi: 10.1016/S0929-693X(08)71944-4. [DOI] [PubMed] [Google Scholar]
- 68.Lovell DJ, Ruperto N, Goodman S, Reiff A, Jung L, Jarosova K, et al. Adalimumab with or without methotrexate in juvenile rheumatoid arthritis. N Engl J Med. 2008;359:810–20. doi: 10.1056/NEJMoa0706290. [DOI] [PubMed] [Google Scholar]
- 69.Fitzgerald AA, Leclercq SA, Yan A, Homik JE, Dinarello CA. Rapid responses to anakinra in patients with refractory adult-onset Still’s disease. Arthritis Rheum. 2005;52:1794–803. doi: 10.1002/art.21061. [DOI] [PubMed] [Google Scholar]
- 70.Vasques Godinho FM, Parreira Santos MJ, Canas dS. Refractory adult onset Still’s disease successfully treated with anakinra. Ann Rheum Dis. 2005;64:647–8. doi: 10.1136/ard.2004.026617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Botsios C, Sfriso P, Furlan A, Punzi L, Dinarello CA. Resistant Behcet disease responsive to anakinra. Ann Intern Med. 2008;149:284–6. doi: 10.7326/0003-4819-149-4-200808190-00018. [DOI] [PubMed] [Google Scholar]
- 72.Larsen CM, Faulenbach M, Vaag A, Volund A, Ehses JA, Seifert B, et al. Interleu-kin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med. 2007;356:1517–26. doi: 10.1056/NEJMoa065213. [DOI] [PubMed] [Google Scholar]