

Abstract
The relationships between immune and neural function are an increasingly important area of study for neuropsychiatric disorders, in particular depression. This is exemplified by the growing number of publications on cytokines and depression during the last 10 years, as compared to earlier decades. This review summarizes the current theories and novel treatment strategies for depression, with a focus on cytokine-induced depression. Neuroimmune mechanisms are now viewed as central to the development of depressive symptoms and emerging evidence is beginning to identify the neural circuits involved in cytokine-induced depression. The current diagnostic categories for depression, as defined by the Diagnostic and Statistical Manual of Mental Disorders, however, are not etiologically or biologically derived, and it has been proposed that “depression”, likely reflects multiple pathogeneses leading to varying symptom constellations. As we move toward a better biological understanding of depression-related symptom constellations or syndromes, the term “depression” may prove inadequately broad, and an integration of interdisciplinary literatures will increase in importance. Future research should aim to characterize these depression-related symptom constellations or syndromes better with the goal of optimizing treatment strategies.
Keywords: Depression, Cytokines, Inflammation, Antidepressants, Animal models, Sickness behavior, Biomarkers
Introduction
Cytokines [e.g., interferons (IFN) and interleukins (IL)] are pleitropic, immunomodulatory signaling molecules that have been increasingly implicated in the development of neuropsychiatric disorders, especially major depressive disorder. In 1927 Julius Wagner-Jauregg won the Nobel Prize for the seminal observation that activation of the immune system by an infectious agent (i.e., malaria inoculation) can affect psychiatric functioning. On the basis of this and related discoveries, it was concluded that cytokines signal the brain and can serve as mediators between the immune and central nervous systems. Subsequently, Maes et al., investigated plasma concentrations and in vitro production of several cytokines, including IL-1 and IL-6 which led to the additional conclusion that there is an increase in pro-inflammatory cytokines in patients with major depression that seems to correlate with severity of illness and measures of hypothalamic–pituitary–adrenal (HPA) axis hyperactivity (Maes et al., 1993, 1995). Since the early 1990s there has been a dramatic increase in the number of papers published on the topic of cytokines and major depression (Fig. 1).
Fig. 1.

The dramatic increase in research on the topic of cytokine-induced depression is illustrated by the significantly higher number of original and review articles, rapid communications, letters to the editor and case reports published in the last 9 years, as compared to earlier decades. PubMed was searched from 1980 to present with “cytokines and major depression” used as the search terms. It is noteworthy that from 2000 to present, of the 616 articles published, 29% were review articles, highlighting the importance of this topic yet the need for more original research.
It is debated whether or not cytokine-induced depression is analogous to major depressive disorder or a major depressive episode, as defined by the DSM-IV-TR. A recent study seeking an answer to this question investigated the similarities and differences between cytokine-induced depression and idiopathic major depression in otherwise healthy participants. The authors reported considerable overlap in the depressive symptom profiles between the two groups, with the exception that, compared to medically healthy patients with major depression, patients with IFN-α-induced depression had significantly greater psychomotor retardation and weight loss and significantly less severe feelings of guilt (Capuron et al., 2009). Thus, rather than differentiating between cytokine-induced depression and idiopathic depression, this review focuses on (1) the neuroimmune mechanisms that are central to the development or progression of depressive symptoms across diagnostic categories, and (2) the need for improved diagnostic definitions that are supported by biological as well as psychological data.
Depression (aka sickness, conservation-withdrawal and passive stress-coping behaviors in animal models)
The Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM-IV-TR) defines a depressive episode as a period of greater than 2 weeks that is characterized by five or more symptoms ranging from depressed mood (sadness, emptiness, tearfulness), anhedonia, feelings of worthlessness or guilt, and suicidal ideation, to changes in weight, appetite, sleep, energy, and psychomotor function, to reduced ability to think or concentrate (DSM-IV-TR, 2000). These symptoms must cause clinically significant stress or impairment in social, occupational or other important areas of functioning, and they must be present for most of the day, nearly every day during a depressive episode. DSM-IV-TR diagnoses, however, are not etiologically or biologically derived, and it has been proposed that various psychiatric disorders, including depression, likely reflect multiple pathogeneses leading to assorted depressive symptom constellations. In turn, research variously operationalizes depression in humans using clinical rating scales (e.g., Beck Depression Inventory, Hamilton Depression Rating Scale, and Depression Anxiety Stress Scale) or psychiatric interviews and in animals using models of sickness behavior, social or behavioral withdrawal, or anhedonia. Table 1 provides a comparison between symptoms of depression in humans with “depressive-like” behaviors in rodents. Collectively, these studies show how behavioral alterations modeling various aspects of depressive illness can be used to study specific depressive symptoms and to test mechanistic hypotheses and novel treatment interventions. However, it is important to note that sickness behavior modeled in an animal is not analogous to a major depressive episode experienced by a human. Although there are a considerable number of overlapping features (Table 1), feelings of worthlessness and recurrent thoughts of death cannot be assessed in animals. Similarly, pyrexia, a common symptom of sickness behavior, is not a symptom typically associated with major depressive episodes, suggesting that these conditions have some common as well as distinct biological mechanisms. The similarities between sickness behavior and depression are thought to result, in part, from an overproduction of endogenous proinflammatory cytokines and a dysregulation of the HPA axis.
Table 1.
Comparison of DSM-IV-TR diagnostic criteria with features of “depressive-like” behavior in rodents.
| DSM-IV-TR symptoms of depressiona | Cytokine-induced sickness or “depressive-like” behavior in rodents | Reference |
|---|---|---|
| Anhedonia | Decrease in sucrose consumption and drinking rate; decrease in responding for rewarding brain stimulation | Anisman et al., 1998; Merali et al., 2003; Sammut et al., 2002, 2001 |
| Significant loss of weight or appetite | Weight loss and decreased food consumption | Makino et al., 2000; Plata-Salaman et al., 1988; Swiergiel et al., 1997 |
| Insomnia or hypersomnia | Increased sleeping | Krueger and Majde, 2003 |
| Psychomotor agitation or retardation | Decreased locomotor activity, voluntary wheel-running behavior, and social exploration | Dantzer et al., 1991; Wood et al., 2006; Pitychoutis et al., 2009 |
| Fatigue or loss of energy | Decreased locomotor activity and voluntary wheel-running behavior; increased conservation-withdrawal behavior | Dantzer et al., 1991; Wood et al., 2006; Minor et al., 2006 |
| Diminished ability to think or concentrate, or indecisiveness | Decreased spatial memory on Morris water maze; increased latency of escape to a foot shock using a shuttle box; inhibition of long-term potentiationb | Bonaccorso et al., 2003; Kelly et al., 2003; Song et al., 2006; reviewed in Khairova et al., 2009 |
Additional symptoms of depression listed in the DSM-IV-TR (and not shown in Table 1) include: feelings of worthlessness or excessive or inappropriate guilt, and recurrent thoughts of death (DSM-IV-TR, 2000).
Decreased capacity for long-term potentiation is associated with stress exposure and this change in synaptic plasticity can be restored by treatment with various types of antidepressants, such as tianeptine and fluoxetine (Dupin et al., 2006; Yaka et al., 2007).
In humans, exposure to endotoxins (e.g., LPS) or proinflammatory cytokines induces a number of neuropsychiatric symptoms. When modeled in animals this constellation of symptoms is often referred to as “sickness behavior”. Within the depression literature, it is at times implied that “sickness behavior” refers to the transient and therefore adaptive response to immune activation, while “depression” may be a chronic and therefore maladaptive response to immune activation, especially chronic inflammation or stress. However, this distinction is not universal across studies, and different studies utilize varying definitions as well as paradigms to study depressive symptoms. For example, the literature on cytokine-induced depression is frequently separate from the research on cytokine-induced cognitive impairment. Yet, reduced ability to concentrate is a DSM-IV-TR symptom of depression (Table 1). Moreover, inflammation, depression, and cognitive impairment co-occur across many conditions including aging and dementia; medical conditions such as infectious diseases [e.g., hepatitis C viral infection (HCV), HIV, malaria)], cardiovascular disease and risk factors (e.g., stroke, ischemia, high cholesterol, diabetes, bypass surgery), inflammatory conditions (e.g., arthritis), and autoimmune disorders (e.g., lupus, multiple sclerosis); cytokine-based therapies (e.g., IFN-α for HCV); and substance use disorders (see also co-morbid depression is common in patients with medical conditions). Given the high burden of depression in medical conditions (and vice versa), it is important that psychiatric assessments include questions to additionally evaluate general health status. As we move toward a better biological understanding of etiologically related depressive symptom constellations or syndromes, the term “depression” may prove inadequately broad, and an integration of what now appears to be arbitrarily distinct literatures will increase in importance. Identification of reliable biological measures of depressive symptoms will be instrumental in this process. Ideally, a diagnosis of depression should be supported not only by psychiatric rating scales but also by biological factors, as is the case with other medical conditions and disorders (e.g., diabetes, hypertension, thyroid disease).
Cytokines in the central nervous system
Cytokines in the central nervous system: 1) are constitutively expressed, 2) can have functions such as neuroprotection or neurodegeneration, and 3) can be regulated by nonimmune factors, such as neurotransmitters and hormones. Peripheral cytokines can also access the brain and affect function via vagal nerve activation, a leaky or compromised blood–brain barrier, and active transport across the blood–brain barrier, or binding to cell-surface proteins on brain endothelial cells. The most recent findings in this area support and extend these concepts by showing that during an immune and inflammatory response, acute activation of tumor necrosis factor-alpha (TNF-α) leads to chronic increases in brain levels of proinflammatory cytokines (Qin et al., 2007); administration of IFN activates expression of several IFN-stimulated genes in brain as well as in peripheral organs (Wang et al., 2008); exposure to a psychosocial stressor, greatly augments the effects of immune activation on sickness, plasma corticosterone and hippocampal norepinephrine, as well as on the levels of circulating IL-6, TNF-α and IL-10 (Anisman et al., 2007a,b; Gibb et al., 2008); IFN-γ participates in the death of dopaminergic neurons by regulating microglial activity (Mount et al., 2007)—thus, IFN-γ induced activation of microglia and consequent neuronal loss may contribute to the modulatory effects of cytokines on depressive symptoms. Collectively, this body of research has led investigators in the field to appreciate that hyperactivation of the immune system and associated signaling cascades results in increased levels of proinflammatory cytokines accompanied by glucocorticoid and immune system dysregulation and deleterious neuropsychiatric effects (e.g., depression). In addition to studying the up-regulation of inflammatory molecules, such as TNF-α or IL-1β, further research to identify and investigate the role of proteins not increased or downregulated during an immune challenge may contribute towards a better understanding of the pathways and molecular targets involved in cytokine-induced depression.
Cytokines and other immune molecules impact neuropsychiatric functions such as mood and cognition in part through their modulation of neuronal anatomy and function. Neuronal plasticity is critical for normal regulation of mood, cognition, and behavior across the lifespan. Cytokines and other immune factors play a key role in modulating early brain development as well as adult neuronal plasticity; however, prolonged exposure to proinflammatory cytokines can impair neuronal plasticity, thereby contributing to cognitive and mood disorders (McAfoose and Baune, 2009). Although peripheral cytokines can enter the CNS or impact central cytokine expression, cytokines are also constitutively expressed in the healthy brain. Brain regions with the highest concentrations of proinflammatory cytokine receptors, specifically receptors for IL-1β, IL-6, and TNF-α, include the hypothalamus, hippocampus, and cortex (Boka et al., 1994; Khairova et al., 2009; Parnet et al., 1994; Schobitz et al., 1993), regions critical for antidepressant response and cognitive functioning. At pathophysiologically elevated levels, TNF-α and IL-1β have both been shown to impair normal neuronal plasticity and to inhibit long-term potentiation (LTP) (Butler et al., 2004; Cunningham et al., 1996). In short, a large and growing literature indicates that central cytokines are critical to the regulation of neuronal plasticity and survival, and that chronic disruption of the balance of these cytokines due to stress, disease (e.g., Alzheimer's disease, infectious diseases, cardiovascular conditions, autoimmune disorders), or medication/substance use (e.g., IFN therapy, substance use disorders) can lead to long-lasting changes in brain anatomy and function, and therefore long-term impairments in mood, cognition, and behavior (see also review on cytokines and Alzheimer's disease by Landreth et al. in this issue of Neurobiology of Disease).
Co-morbid depression is common in patients with medical conditions
There is abundant evidence that depression involves alterations in multiple aspects of immunity that may contribute to the development or exacerbation of a number of medical disorders and also may play a role in the etiology of depressive symptoms. The relationship between medical illness and depression has been particularly well-described in cardiovascular disease (Wirtz et al., 2009; Grippo and Johnson, 2009; Halaris, 2009; Davidson et al., 2009), multiple sclerosis (Gold and Irwin, 2006), cancer (Wood et al., 2006; Orre et al., 2009), bone metabolism (Williams et al., 2009), rheumatoid arthritis (Malemud and Miller, 2008; Uguz et al., 2009), irritable bowel syndrome (Lee et al., 2008; Tache and Brunnhuber, 2008), Huntington's disease (Björkqvist et al., 2008), asthma (Van Lieshout et al., 2009), fibromyalgia and other chronic pain conditions (Wang et al., 2009; Marchand et al., 2005; Edwards et al., 2008), viral and bacterial infections (Loftis et al., 2008; Huckans et al., 2009; Leslie et al., 2008), and substance use disorders (Friedman and Eisenstein, 2004; Huckans et al., 2009). The role of the immune system in depression and co-morbid medical conditions is also supported in animal models which provide additional insights into the mechanisms associated with cytokine-induced depression (Varghese et al., 2006; Kiank et al, 2006; O'Mahony et al., 2009; Ghia et al., 2009) (Table 2). Collectively, these studies illustrate the prevalence of depressive symptoms observed across diverse medical populations and highlight the importance of interdisciplinary research efforts.
Table 2.
Animal models of depression—based on stimulation of the peripheral immune system to induce exaggerated neuroinflammatory response and prolonged sickness.
| Animal modela | Depressive symptoms investigated | Associated biomarkers measured | Main outcomes | References |
|---|---|---|---|---|
| IFN-α, rats | Changes in neurogenesis in dentate gyrus, as a correlate of depression | Hippocampal IL-1β protein levels Number of bromo-deoxyuridine-labeled cells |
Increased IL-1β in hippocampus Decreased number of proliferating cells in the dentate gyrus |
Kaneko et al., 2006 |
| IFN-α, mice | Anxiety, as measured by the elevated plus maze (EPM) Behavioral despair or learned helplessness, as measured by forced swim (FST) and tail suspension tests (TST) |
Expression of adhesion molecules on leukocytes and the recruitment of lymphocyte subsets into the brain | Increased immobility of mice in the late phase of FST, without significant effects in TST and EPM The percentages of CD4+ and CD8+ lymphocytes as well as the percentages of lymphocyte function-associated antigen 1 (LFA-1)-expressing CD4+ and CD8+ lymphocytes were increased in brain |
Orsal et al., 2008 |
| IFN-α, rats | Somatic and psychomotor symptoms (e.g., changes in temperature, body weight, food intake, and locomotor behavior) Anhedonia, as measured by brain stimulation reward (BSR) thresholds elicited from the ventral tegmental area |
N/A | Increased sickness behavior No significant changes in the hedonic status of BSR |
Kentner et al., 2007 |
| IFN-α, rats | Anhedonia, as measured by sucrose consumption | N/A | Decreased sucrose consumption and drinking rate | Sammut et al., 2001, 2002 |
| IFN-α, rats | Somatic symptoms (e.g., food consumption, body weight changes) Behavioral despair, as measured by FST Anhedonia, as measured by sucrose pellet self-administration |
Monoamine turnover in several brain regions Corticosterone release Plasma TNF-α and IL-6 and release IL-1β and IL-6 mRNA expression in hippocampus |
No significant changes in sucrose consumption No significant changes in corticosterone release, plasma TNF-α or IL-6 release No significant differences in IL-1β or IL-6 mRNA expression in hippocampus |
De La Garza et al., 2005 |
| IFN-α, rats | Somatic and psychomotor symptoms (e.g., body weight changes and locomotor behavior) Behavioral despair, as measured by FST |
Signal transducer and activator of transcription 1 (STAT1) Phospho-STAT1 |
No significant changes in depressive-like behaviors No changes in the expression or phosphorylation of STAT1 |
Loftis et al., 2006a,b |
| LPS, rats | Somatic and psychomotor symptoms (e.g., changes in food intake, locomotor behavior, and social exploration) Behavioral despair, as measured by FST Anhedonia, as measured by sucrose consumption |
Serotonergic and dopaminergic activity in hypothalamus, hippocampus, prefrontal cortex, amygdala and striatum | Enhanced serotonergic function and increased swimming duration in FST for female rats Increased suppression of locomotor activity for male rats |
Pitychoutis et al., 2009 |
| LPS, rats | Behavioral despair, as measured by FST Psychomotor retardation, as measured by social exploration |
N/A | No effect on FST Reduced social interaction |
Deak et al., 2005 |
| LPS, rats | Somatic and psychomotor symptoms (e.g., changes in body temperature and social behavior) | c-fos in brain IL-1β in periphery and circumventricular organs and choroid plexus |
Reduced social interaction, induction of fever and body weight loss Increased IL-1β production in systemic circulation as well as in brain circumventricular organs and the choroid plexus. Induced c-fos expression in the amygdala and bed nucleus of the stria terminalis |
Konsman et al., 2008 |
| LPS and IL-1β, mice | Anxiety and psychomotor function, as measured by the open field and elevated plus maze (EPM) tests | N/A | Dose-dependent decreases in open arm entries and the time spent on the open arms of the EPM IL-1β and LPS decreased the number of line crossings in the center of the open field The doses of IL-1β and LPS necessary to induce these effects also decreased locomotor activity, indicating a reduction in general activity |
Swiergiel and Dunn, 2007 |
| LPS and stress, mice | Somatic and psychomotor symptoms, as measured by locomotor and exploratory activities, curled body posture, ptosis (drooping eyelids), and the presence of ruffled and greasy fur | Plasma corticosterone, IL-6, TNF-α, IL-10, IL-1β, IFN-γ TNF-α, IL-10, IL-1β, IL-4, IFN-γ mRNA in hippocampus, prefrontal cortex, nucleus tractus solitaries Norepinephrine utilization in brain |
Enhanced NE utilization within the prefrontal cortex and hippocampus The LPS-induced elevation of IL-1β, IL-6 and TNF-α mRNA expression in the hippocampus, prefrontal cortex and nucleus tractus solitaries was diminished in animals that experienced stress |
Gibb et al., 2008 |
| LPS and aging, mice | Somatic and psychomotor symptoms, as measured by feeding behavior and locomotor, feeding activities Behavioral despair, as measured by FST and TST |
Peripheral and brain IDO Serotonin utilization in brain |
Increased duration of immobility in aged mice Induction of peripheral and brain IDO and an increased turnover of brain serotonin |
Godbout et al., 2008 |
| BCG inoculation | Somatic and psychomotor symptoms (e.g., changes in body weight and voluntary wheel running) Behavioral despair, as measured by FST and TST Anhedonia, as measured by sucrose preference test |
Plasma IFN-γ, TNF-α Peripheral IDO activation |
Transient body weight loss, reduced locomotor activity and induction of fever; these sickness behaviors were followed by… Increased duration of immobility in FST and TST Reduced voluntary wheel running and decreased preference for sucrose Increased plasma IFN-γ and TNF-α concentrations and IDO activation |
Moreau et al., 2008 |
Abbreviations: EPM, elevated plus maze; FST, forced swim test; IDO, indoleamine 2,3-dioxygenase; LPS, lipopolysaccharide; TST, tail suspension tests
The route of administration, dose, duration, type and formulation of IFN-α (e.g., recombinant human, mouse or pegylated IFN) vary across studies.
Mechanisms of cytokine-induced depression
Cytokines play an active role in the molecular events influencing synaptic transmission, neuronal plasticity, and depressive behaviors. However, although cytokine-induced depression is well-established (Raison et al., 2009; Anisman, 2009), the specific mechanisms by which activation of the innate immune system and expression of specific depressive symptoms are related remain poorly understood, in part, because of the complex and diverse processes involved. The potential mechanisms leading to cytokine-induced depression are numerous and were recently reviewed by Miller et al. (2009b), Maes et al. (2009), Dantzer et al. (2008), Dantzer (2009), and others. Included among these mechanisms are several lines of evidence demonstrating how cytokines can contribute to HPA axis hyperactivity (Maes et al., 1993) as well as affect the serotonergic and dopaminergic systems (Bonaccorso et al., 2002; Capuron et al., 2002a,b) and subsequently lead to depressive symptomatology. There is, however, a need for a more integrated view of depression. Given that in humans as well as in animal models of depression (Tables 1 and 2), treatment with cytokines produces depressive symptoms and that depression occurs more frequently in those with medical conditions, cytokines and other inflammatory molecules may function as mediators of depressive symptoms. Currently, there is not a consensus in the literature regarding the role of the immune system and inflammatory pathways on the etiology of depression. In fact, it is still debated whether or not inflammatory responses occur in brain (Galea et al., 2007; Dantzer et al., 2008). Fig. 2 summarizes the putative psychoneuroimmunological factors and biochemical correlates associated with depression.
Fig. 2.
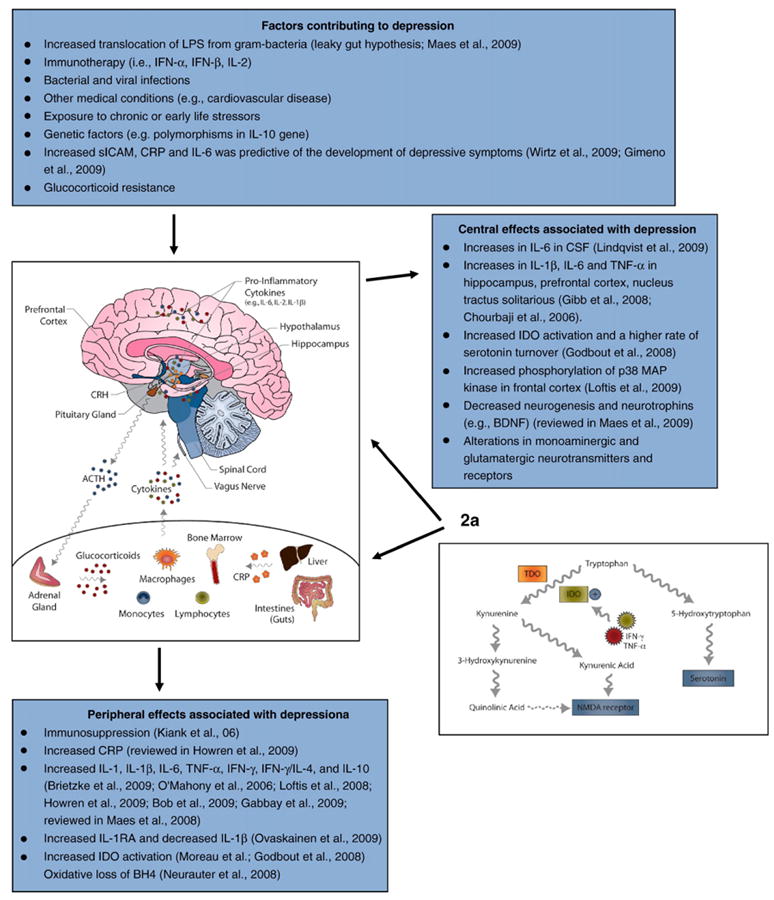
Immune and central nervous systems interact and contribute to the etiology and pathophysiology of cytokine-induced depression. Text boxes summarize the putative factors contributing to the development of depressive symptoms as well as the accompanying alterations in peripheral and central neuroimmune modulators. Current theory proposes that years of cumulative risk factors (e.g., genetic polymorphisms, repeated brain injuries or vascular insults, exposure to stress and toxins, or oxidative stress) combine to activate danger associated molecular pattern detectors (DAMPs) within the innate immune system, in turn modifying microglial receptors [e.g., toll-like receptors (TLRs), receptors for highly glycosylated end products (RAGEs)], resulting in the chronic overproduction of proinflammatory cytokines such as TNF-α, IL-1β, and IL-6 (Maccioni et al., 2009). Related immune mechanisms activating pathogen associated molecular pattern molecules (PAMPs) are also speculated to contribute to the etiology of depressive symptoms and may be especially relevant for depression accompanying specific medical conditions. 2a. IDO pathway and its role in mediating cytokine-induced depression. Cytokines, such as IFN-γ, can induce the enzyme indoleamine 2,3-dioxygenase (IDO) or tryptophan 2,3-dioxygenase (TDO; an analogous enzyme located in the liver), which induces the metabolism of tryptophan. Activation of IDO decreases the amount of tryptophan for conversion to serotonin and increases the possibility of oxidative stress. Specifically, kynurenine, a metabolite of tryptophan, can be transported across the blood–brain barrier into the brain where it is further metabolized in macrophages, microglia and astrocytes leading to the generation of potentially neurotoxic compounds: 3-hydroxykynurenine (3-HK) and quinolinic acid (QA). Taken together, these studies provide support for the hypothesis that alterations in the regulation of certain cytokines such as IL-1, IL-6, and TNF-α released during an immune response activate the hepatic synthesis of CRP and components of the HPA axis, alter neurotransmitter networks activity, and induce fatigue, loss of appetite, anhedonia, and other depressive symptoms.
Human biomarkers associated with cytokine-induced depression
Increasing evidence implicates proinflammatory cytokines (e.g., IL-1β, TNF-α, and IL-6) and a dysregulation of immune mediators [e.g., acute phase response protein, C-reactive protein (CRP), nitric oxide and glucocorticoids] in the etiology of depression and related symptoms of anorexia, sleep disturbance, fatigue, and cognitive impairment (Fig. 2). A systematic review of articles published between January 1967 and January 2008 was performed in order to assess the size and direction of the relationships among depression and IL-1, IL-6 and CRP (Howren et al., 2009). Effect sizes were calculated and meta-analyzed, and each inflammatory marker was positively associated with depression, with the strongest associations found in clinically depressed patient groups (Howren et al., 2009). Maes et al. (2009) similarly reviewed research showing that depression is associated with an increased production of proinflammatory cytokines, IL-1β, IL-6, TNF-α, and IFN-γ.
In addition to changes in peripheral levels of circulating or stimulated cytokine production, cytokine alterations in the CNS accompanying depressive symptoms have also been observed. Specifically, IL-1β, IL-6, IL-8 and TNF-α were measured in the cerebrospinal fluid of suicide attempters and healthy control participants. IL-6 was significantly higher in suicide attempters than in healthy controls, with patients who performed more violent suicide attempts showing the highest IL-6 levels. There was also a significant positive correlation between depression rating scale scores and cerebrospinal fluid IL-6 levels in all patients (Lindqvist et al., 2009). To further characterize and localize the central mechanisms underlying mood disturbances and immune activation, a double-blind, randomized crossover study was conducted which compared the effects of typhoid vaccination or saline injection on changes in mood, cytokine levels and brain activity. Typhoid but not saline injections produced an increase in circulating IL-6 accompanied by a significant increase in depressive symptoms. Interestingly, typhoid-induced changes in mood correlated with enhanced activity within subgenual anterior cingulate cortex (sACC) (a region implicated in the etiology of depression) and reduced connectivity of sACC to amygdala, medial prefrontal cortex, nucleus accumbens, and superior temporal sulcus, which was modulated by peripheral IL-6 (Harrison et al., 2009). Collectively, these studies support a role for proinflammatory cytokines and a dysregulation of immune mediators in the etiology of depression and begin to suggest possible neural circuits involved in cytokine-induced depression.
However, conflicting results regarding the relationship between elevated proinflammatory cytokines and depressive symptoms have also been described (Brambilla et al., 2004; Rothermundt et al., 2001; Ovaskainen et al., 2009; Wirtz et al., 2009). The discrepant results exist, in part, because of methodological differences in the following:
1) The assessment and definition of depression
Depression rating scales are known to differ in terms of the instruments' conceptualizations of depression. Clinical studies vary in regard to the use of self-report (e.g., Beck Depression Inventory) or clinician-administered (e.g., Hamilton Depression Rating Scale) depression rating scales, and this can affect the depressive variables that are being measured. For example, compared to the Hamilton Depression Rating Scale, a greater number of items on the Beck Depression Inventory are related to depressive cognitive attitudes, as opposed to items that measure functional impairments or somatic symptoms (Lambert et al., 1986). In addition, studies based in clinical care as opposed to research settings may use medical chart diagnoses rather than criterion-based symptom rating scales to assess depression. Interestingly, Gabbay et al. (2009a) recently found that adolescents with major depressive disorder and suicidal symptomatology had significantly lower TNF-α levels, as compared to nonsuicidal adolescents with major depressive disorder. These results highlight the potential variability in inflammatory markers associated with differing depressive symptom profiles.
2) The use of medication and relevant demographic controls
Given the putative anti-inflammatory effects of many antidepressant medications (Hashioka et al., 2007; Carvalho and Pariante, 2008; Lim et al., 2009), controlling for the use of antidepressants (as well as other medications known to affect inflammation, such as is steroids) is an important factor to consider when assessing the expression of cytokines and related proteins. Differences in (a) body mass, (b) age, (c) sex, (d) the presence of medical conditions, chronic or acute infections, and (e) substance use can also impact the expression of depressive symptoms and inflammatory factors (Stewart et al., 2009; Karakelides et al., 2009; Loftis et al., 2008).
3) Cytokine sampling
The variability in inflammatory markers associated with depressive symptoms may also be due to the timing of the assessments and source of cytokines measured. There is increasing evidence for a circadian influence on immune function (Coogan and Wyse, 2008). Consequently, the timing of blood or tissue sampling may affect the concentration of cytokines or other immune markers under investigation. In addition for clinical studies, changes in cytokine expression can vary depending on whether measurements are obtained from plasma, serum, cerebrospinal fluid (CSF) or stimulated peripheral blood mononuclear samples. For example in plasma samples, TNF-α levels were not significantly different in patients with major depressive disorder as compared to controls, but TNF-α levels were significantly lower than normal in patients with dysthymia (Brambilla et al., 2004). However, Boufidou et al. (2009) found that serum and CSF TNF-α levels were positively associated with depressive mood.
4) The failure to consider the role of genetic and environmental risk factors
Genetic polymorphisms in genes [e.g., for IL-10 (Traks et al., 2008) and serotoninergic proteins (Kraus et al., 2007; Lotrich et al., 2009)] may be involved in an increased risk for major depressive disorder. Further, adverse childhood experiences have been described as major environmental risk factors for depressive disorder. Aguilera et al. (2009) extended previous reports and found that stressful childhood experiences, in particular childhood sexual abuse, predicted higher levels of adult depressive symptoms and that polymorphisms in the genes for brain-derived neurotrophic factor (BDNF) and the serotonin transporter seemed to moderate the effects of childhood sexual abuse on adult depressive symptoms. Thus, future studies may benefit from considering the role of genetic factors as well as childhood history to determine the extent of prior stress exposure. Knowledge of genotypes and prior stressors may further help identify protective or resiliency factors that could prevent the development of neuropsychiatric disorders in patients at risk.
The lack of agreement in the literature regarding the role of cytokine pathology in depressive symptoms described also shows that the psychoneuroimmunological processes contributing to depression are complex and that the interrelationships among systems and circuits are not completely understood.
The diagnostic category of major depressive disorder is composed of varied symptoms, impacting mood, somatic, as well as cognitive functioning (Table 1). Thus, it is likely that the biochemical markers associated with depression depend not only on the etiology of the depressive disorder but also on its specific symptom profile. For example, patients experiencing impaired thinking and concentration as the prominent depressive symptom may show a different pattern of cytokine alterations, as compared to patients experiencing somatic symptoms as the primary depressive concern. It has been suggested that by inducing long-term activation of microglia to release TNF-α and IL-1 in a positive feedback manner, immune-mediated glutamatergic alterations may be associated with cognitive impairments (reviewed in McNally et al., 2008 and Khairova et al., 2009). Fatigue and other somatic symptoms of depression; however, may be associated with a different pattern of immune dysregulation (Loftis et al., 2008; Gold and Irwin, 2009).
The time course for cytokine-induced depression onset is different across symptom dimensions. Capuron et al. (2002a,b) found that in patients with HCV, the neurovegetative and somatic symptoms including weight loss, fatigue and pain appeared within 2 weeks of IFN-α therapy, but symptoms of depressed mood, anxiety and cognitive dysfunction appeared later during HCV treatment. A prospective study by Gimeno et al. (2009) further demonstrated that inflammation precedes the cognitive symptoms of depression, as baseline CRP and IL-6 levels were predictive of the development of cognitive symptoms of depression (approximately 12 years later). More research is needed to better characterize the temporal relationships among inflammatory mediators and the etiology of depressive symptoms. However, these data suggest that different pathogenic mechanisms may mediate the various behavioral manifestations of depression—an observation with direct clinical relevance, as symptoms of fatigue and weight loss are less responsive to typical antidepressant treatment (Capuron et al., 2002a,b) and improved treatment approaches are needed (see Novel treatment strategies).
One strategy that has been used to describe the pattern of immune dysregulation associated with cytokine-induced depression considers the balance of T helper lymphocyte 1 (Th1) proinflammatory cytokines and Th2 anti-inflammatory cytokines, such that depression is characterized by a shift in the pro/anti-inflammatory or Th1/Th2 cytokine ratio. Three studies recently tested the hypothesis that patients with depression would show a dysregulation of the immune system, as measured by a Th1/Th2 cytokine imbalance or shift. Sixty-one bipolar patients were recruited for assessment of serum cytokine levels. Of these, 14 were in euthymic state, 23 and 24 were in manic and depressive episodes, respectively. Cytokines involved in Th1/Th2 balance, such as TNF-α, IL-2, IL-4, IL-6, IL-10, and IFN-γ were measured. Patients in depressive episodes showed increased IL-6 levels but no significant differences in Th2 cytokine expression (Brietzke et al., 2009). Gabbay et al. (2009b) extended this work to adolescents to examine a similar hypothesis of immune system dysregulation in adolescents with MDD. Thirty adolescents with MDD and 15 healthy comparisons, group-matched for age, were enrolled. Plasma cytokines (IFN-γ, TNF-α, IL-6, IL-1β, and IL-4) were measured. Adolescents with MDD had significantly elevated plasma IFN-γ levels and IFN-γ/IL-4 ratios; however, IL-4 levels were not significantly different across the groups. A trend for IL-6 to be elevated in the MDD group was also observed (Gabbay et al., 2009b). In another study examining the role of pro- and anti-inflammatory cytokines in depression, depressed participants (n=12) showed significantly lower IL-10 levels and higher IL-6/IL-10 ratios, as compared to controls (n=11). IL-6 levels were higher in the depressed group, but this difference did not reach statistical significance (Dhabhar et al., 2009). Taken together, these studies provide support for the hypothesis that alterations in certain cytokines such as IL-1, IL-6, TNF-α, and IL-10 are associated fatigue, loss of appetite, anhedonia, and other depressive symptoms. However, to clarify the role of Th2 responses in depression and to provide support for the hypothesis that depression is characterized by a shift in the pro/anti-inflammatory or Th1/Th2 cytokine ratio, more research is needed.
Proinflammatory cytokines, neurotransmission and oxidative stress
How does the cytokine hypothesis of depression fit with what is known about the role of serotonin in depression? Proinflammatory cytokines such as IFN affect serotonin metabolism by stimulating indoleamine-pyrrole 2,3-dioxygenase (IDO or INDO EC 1.13.11.52) which leads to a peripheral reduction of tryptophan and serotonin (Taylor and Feng, 1991; Curreli et al., 2001; Bonaccorso et al., 2002). A number of papers have identified IDO as an enzyme involved in depressogenesis, not only because of its affects on serotonin biosynthesis (Turner et al., 2006) but also because of its putative contributions to excitotoxicity and oxidative stress. IDO is highly inducible by proinflammatory cytokines (e.g., IFN-γ and TNF-α) and is secreted by activated macrophages and other immunoregulatory cells, which catalyzes the degradation of tryptophan (serotonin precursor) to kynurenine. As is shown in Fig. 2, kynurenine degradation leads to the formation of 3-hydroxykynurenine (3-HK, generates free-radical species that can cause oxidative stress), quinolinic acid (QA, a glutamate receptor agonist), and kynurenic acid (KA, an NMDA receptor antagonist hypothesized to be neuroprotective). Given the evidence supporting a role for increased glutamate receptor activity in depression, this IDO-mediated imbalance of kynurenine pathway metabolites might contribute to cytokine-induced depression (Muller and Schwarz, 2007). Thus, cytokine- and IDO-mediated degradation of tryptophan through the kynurenine pathway is hypothesized to influence serotonergic biosynthesis and neurotransmission in the brain resulting in significant neuropsychiatric consequences (Fig. 2).
Proinflammatory cytokines, such as IFN-γ, stimulate not only IDO but also the biosynthesis of 5,6,7,8-tetrahydrobiopterin (BH4), which is a cofactor for several aromatic amino acid monooxygenases and thus is involved in the biosynthesis of the neurotransmitter serotonin and the catecholamines dopamine, epinephrine, and norepinephrine. In macrophages, IFN-γ also causes the generation of reactive oxygen species, which can reduce BH4 levels. Recent data suggest that oxidative loss of BH4 in chronic inflammatory conditions can lower the biosynthesis of catecholamines, which may relate to altered neurotransmission in patients with depression (Neurauter et al., 2008). Maes et al. (2009) further theorized that inflammatory, oxidative, and nitrosative pathways and an increased translocation of lipopolysaccharide from gram-negative bacteria are causally related to depression (i.e., “leaky gut” hypothesis). Maes et al. (2009) suggested that depression is associated with an autoimmune response directed against disrupted lipid membrane components, such as phosphatidyl-inositol, by-products of lipid peroxidation, and nitric oxide-modified amino acids, which have become immunogenic (Maes et al., 2009).
The complexity of this pathophysiological framework is enriched by the growing evidence showing that these inflammatory processes related to depression may be influenced by psychological stress as well as by other environmental and genetic factors. In a study of 103 healthy women, participants reporting high levels of chronic interpersonal stress at baseline displayed greater increases in their leukocyte messenger ribonucleic acid (mRNA) for the transcription factor nuclear factor-kappaB (NF-kappaB) over the next 6 months. Chronic interpersonal stress at baseline was also associated with increasingly pronounced IL-6 responses to lipopolysaccharide, suggesting that chronic interpersonal difficulties contribute to the dysregulation of pro- and anti-inflammatory signaling molecules (Miller et al., 2009a).
The relationships among inflammation, aging, and depression
In the absence of neurological disease or other medical conditions, aging is associated with weakened peripheral immunity and increased neuroinflammation. Older adults evidence weakened innate and humoral responses to novel antigens (in part due to reduced numbers of naive T cells), an elevated peripheral and central inflammatory profile (e.g., reduced expression of anti-inflammatory cytokines and increased expression of proinflammatory cytokines), and an increased population of reactive or “primed” glial cells [e.g., increased microglial expression of major histocompatibility complexes (MHC), scavenger receptors, and complement receptors, and increased astroglial expression of glial fibrillary protein (GFAP)] (Godbout and Johnson, 2009; Parachikova et al., 2007). These factors increase the potential for exaggerated neuroinflammatory responses to peripheral immune stimulation (e.g., due to pathogens, injury, or stress) and contribute to increased incidence of cognitive and psychiatric disorders among older adults, particularly following illness (Penninx et al., 1999).
Approximately 15–30% of older adults suffer from depression associated with a medical condition (Mulsant and Ganguli, 1999), and older adults are at increased risk for delirium and dementia following peripheral infections (Jackson et al., 2004). Depression is frequently a prodrome of dementia, and the incidence of depression among patients with Alzheimer's disease is estimated to be greater than 40%. Several recent reviews summarize how neuroinflammation contributes to the cognitive and brain changes associated with Alzheimer's disease (Maccioni et al., 2009), dementia and depression (Leonard, 2007), and aging (Godbout and Johnson, 2009; Dilger and Johnson, 2008; Giunta, 2008). Overall, this literature demonstrates that, similar to major depression without an associated medical condition, Alzheimer's disease and dementia are associated with increased expression of peripheral and central proinflammatory cytokines, increased microglial activation, increased activation of TDO and IDO, reduced serotonin synthesis, and increased production of neurotoxins including quinolinic acid and 3-hydroxykynurenine (Leonard, 2007). However, the question of whether or not there is greater susceptibility to developing depression in otherwise healthy individuals with no underlying medical conditions still needs to be answered. Further investigation into the role of genetic (see Contribution of genetic factors to the etiology of cytokine-induced depression) and environmental (e.g., history of prior stress exposure) factors will help to answer this important question.
Contribution of genetic factors to the etiology of cytokine-induced depression
It is generally accepted that depressive symptoms are associated with increased expression of inflammatory markers. Thus, some of the variability across studies with regard to biomarker profiles in cytokine-induced depression may be due to differences in genetic risk factors. Several studies have considered the possibility that there are common genes associated with depressive symptoms and inflammation. Specifically, polymorphisms in the IL-10 gene cluster may be involved in the increased risk for major depressive disorder (Traks et al., 2008). In addition, gene expression microarray studies have shown that several receptors for immune genes, such as the IFN α/β and IL-8 receptors were differentially regulated in the frontal cortex of patients with bipolar disorder (Bezchlibnyk et al. 2001; Iwamoto et al. 2004). Recently, another study found that, compared to healthy controls, the expression of inflammatory genes (i.e., TNF-α, IL-1, and IL-6) was increased in the monocytes of patients with bipolar disorder (62% euthymic, 17% depressed, 17% manic, and 5% of unknown mood state) as well as in the offspring of bipolar patients (Padmos et al., 2008). A subsequent study conducted in twins found further support for the heritability of cytokine-induced depression, as a significant association between severity of current depressive symptoms and increased levels of inflammatory markers (e.g., CRP and IL-6) was observed. The heritability of IL-6, CRP, and depressive symptoms was estimated at 0.37, 0.65, and 0.48, respectively (Su et al., 2009).
In addition to differences in the expression of immune genes, a study of 42 participants with and without major depression also investigated the role of serotonergic genes and found that the mRNA expression of the serotonin transporter, as well as IL-1β, IL-6, IFN-γ and TNF-α were higher in the patients with depression as compared with the healthy controls (Tsao et al., 2006). Although Tsao et al. (2006) did not distinguish SERT mRNA expression in depressed patients with and without immune activation, the investigators used strict inclusion/exclusion criteria for their depressed participants and specifically excluded individuals with major physical disorders as well as participants experiencing a common cold, influenza, or any other inflammation over the 3 months prior to study entry. The hypothesis that genetic differences in the serotonin reuptake transporter promoter (5-HTTLPR) may interact with the immune system and influence depression risk in patients undergoing IFN therapy for HCV was recently supported by Lotrich et al. (2009). In a sample of 71 patients with HCV, the SS allele was associated with greater susceptibility to depression and a higher rate of IFN-induced depression. In contrast, the LA allele was associated with a lower rate of depression during IFN therapy, and analyses indicated that genotype LA/LA was significantly different from S/S but not from S/LA (Lotrich et al., 2009). These findings are consistent with an earlier study of 139 HCV patients treated with IFN therapy designed to investigate the impact of serotonergic gene variants which found that homozygosity for the HTR1A-1019G variant significantly increased both incidence and severity of IFN-induced depression (Kraus et al., 2007).
Variability in cytokine and neurotransmitter genes may additionally be accompanied by differences in neuroendocrine gene expression, as glucocorticoid (GC) receptor gene polymorphisms could also explain the differences observed in depressive symptom profiles (Kumsta et al., 2008). It is generally accepted that depression is accompanied by reduced responses to GC on the dexamethasone suppression test (reviewed in Pariante, 2004). In a sample of 206 healthy research participants, Kumsta et al. (2008) investigated the effects of four GC receptor gene polymorphisms on GC responsiveness. No significant differences in the ability of dexamethasone to suppress IL-6 production were detected across the genotype groups. However, individuals with the intronic BclI polymorphism had lower GC sensitivity of subdermal blood vessels, as determined by the least degree of skin blanching when compared to individuals with the other genotypes. Authors concluded that GC responsiveness is highly variable not only between individuals but also between tissues in the same individual, thus supporting the relevance of genetic variability in explaining differential GC sensitivities. Taken together, the gene expression changes identified and summarized herein are consistent with and extend the observations found in the clinical biomarker literature. The following sections link this body of evidence to various animal models of depression, thereby reinforcing the use of pre-clinical models to better understand the pathogeneses of depressive symptoms and identify novel therapeutic targets.
Animal models and hypothesized mechanisms of cytokine-induced depression
As in clinical studies, inflammatory factors and cytokine-specific alterations of monoamine and neuroendocrine function are similarly observed in animal models of depression. Newly developed animal models of depression based on induced inflammation are summarized in Table 2; these animal models accompany and contribute to the studies using previously established models of depression–which remain highly relevant, as these models not only result in the expression of depressive behaviors but also impact and alter immune function [e.g., neonatal separation/early life stress models (Varghese et al., 2006; O'Mahony et al., 2009); chronic/repeated stress models (Kiank et al., 2006; Detanico et al., 2009; O'Mahony et al., 2006; Anisman et al., 2007a,b); separation from group housing models (Martin and Brown, 2010), olfactory bulbectomy models (Song et al., 2009a,b; Myint et al., 2007); and social dominance/subordination models (Kroes et al., 2006)]. The long list of animal models (including genetically altered animals–see Genetically engineered rodent models used to study depression) and behavioral measures designed to investigate depression illustrate the complexities associated with studying neuropsychiatric disorders.
The literature shows that a number of the behavioral responses accompanying immune activation (e.g., increased immobility on the forced swim or tail suspension tests), are not consistently observed across studies. For example, Deak et al. (2005) found that behavioral responses during the forced swim test are not affected by systemic LPS injections, even using doses of LPS sufficient to cause significant reductions in social behavior. Orsal et al. (2008) similarly found that IFN-α had no effect on the tail suspension test (i.e., another measure of behavioral despair or learned helplessness) and only modest effects on swimming behavior during the forced swim test. Interestingly, Neveu et al. (1998) found that lateralization in mice can affect IL-1-induced behavior on the forced swim test, such that IL-1-induced immobility is less pronounced in left- as compared to right-pawed mice. Taken together, these findings suggest that “depression” or sickness associated with immune activation may not consistently affect behavioral despair, as measured using the forced swim or tail suspension tests. Alternative measures of behavioral depression, such as sucrose preference testing (Sammut et al., 2001) or social interaction assessments (Konsman et al.,2008) may be more sensitive to the depressive-like effects of proinflammatory cytokines.
Of the behavioral measures described in Table 2, changes in sucrose preference appear to be one of the more consistently observed behavioral responses. This measure of anhedonia has been used in models of depression associated with and without direct immune activation (Sammut et al., 2001; Moreau et al., 2008; Navarre et al., 2009) and is also responsive to antidepressant treatment (Sammut et al., 2002; Pandey et al., 2009). Given that a diagnosis of depression currently requires the presence of at least one of the two core symptoms, depressed mood and anhedonia (DSM-IV-TR), including this measure in behavioral batteries to assess depression seem critical.
Animals can theoretically be used to model all but two of the DSM-IV-TR criteria for a major depressive episode—recurrent thoughts of death and feelings of worthlessness or excessive guilt. Thus, the ideal animal study would use a combination of behavioral assessments to most comprehensively model clinical depression using a rodent. There are, however, a limited number of published studies that have used such a design to study depression. This paucity results in part because of technical limitations, as the depressive-like symptoms induced in some models can be transient and do not permit repeated behavioral testing in the same animal.
Given the high percentage of patients who develop depressive symptoms during IFN therapy (Loftis and Hauser, 2004), an animal model to investigate the etiology of these neuropsychiatric side effects would be highly beneficial; however, there is considerable debate in the literature regarding the utility of IFN to induce “depressive-like” symptoms in rodents. Increasingly, investigators are appreciating the species specificity of IFN (Langer and Pestka, 1988; De La Garza et al., 2005; Loftis et al., 2006a). Our data suggest that recombinant human pegylated IFN-α does not bind type I IFN receptors in the rat CNS or periphery, as immunoblotting of frontal cortex and liver tissue revealed that STAT1 was not phosphorylated following IFN-α administration, an indicator of IFN receptor binding (Tanabe et al., 2005; Loftis et al., 2006a). Although our data do not rule out the possibility that pegylated IFN-α may not have crossed the blood–brain barrier, our findings are in agreement with De La Garza et al. (2005) who found that neither recombinant human pegylated IFN-α nor non-pegylated IFN-α induced neuroendocrine or neuroimmune activation in Wistar rats. To further address the issue of species specificity, we evaluated sequence similarity between: 1) human and rat type I IFN receptors and 2) human and mouse type I IFN receptors. Results indicated that there is approximately 40.9% identity between rat and human receptors and 45.7% identity between mouse and human receptors (Loftis et al., 2006b). While lack of IFN receptor species specificity is a plausible explanation, Makino et al. (2000) found that when given to rats, human IFN-α but not rat IFN-α, -β or -γ changed the immobility time during the forced swim test (Makino et al., 2000). In addition, IFN-α can bind to other neurotransmitter systems, such as opioid receptors (Wang et al., 2006), suggesting that the neuropsychiatric side effects of IFN-α might not be mediated through type I IFN receptors. Future studies using IFN to behaviorally and biologically model depression in rodents should consider these factors.
Alternatively, investigators have used treatment with other proinflammatory cytokines (e.g., IL-1β, IL-2, IL-6) or endotoxin exposure [e.g., lippopolysaccharide (LPS)] to induced depressive symptoms and associated biochemical changes, with varying effects (Table 2). For example, IL-1β, produced by activated innate immune cells (i.e., macrophages and monocytes) upon injury or infection, is an important cytokine not only for the induction of sickness behavior (Dantzer, 2001) but also likely plays a significant role in depression (Maes et al., 1993; Owen et al., 2001; van den Biggelaar et al., 2007).
Similar to the debate regarding cytokine-induced depression and idiopathic depression in humans (Capuron et al., 2009), it has been argued that sickness behavior is not depression. A recent study aimed to develop a novel model to identify the neurobiological basis of depressive-like behavior induced by chronic inflammation, independent of sickness behavior. Investigators measured the behavioral consequences of chronic inoculation of mice with the bacteria, Bacillus Calmette-Guerin (BCG), which is used against tuberculosis and activates both lung and brain IDO (Moreau et al., 2008). BCG treatment induced an acute period of sickness (e.g., body weight loss, reduction of motor activity, and increased body temperature) and delayed depressive-like behaviors (e.g., increased duration of immobility in both forced swim and tail suspension tests, reduced voluntary wheel running, and decreased preference for sucrose) lasting over several weeks. Mice treated with BCG reportedly exhibited normal locomotor activity at the time of the forced swim test; however, they evidenced significantly decreased swimming and climbing behaviors on the forced swim test as well as significantly reduced wheel running 1 week after BCG treatment, thus complicating the interpretation of these delayed depressive-like behaviors. Moreau et al. (2008) additionally observed a temporally distinct pattern of IDO activation, as increased lung IDO activation was associated with the delayed depressive-like but not with the sickness behaviors. The authors suggest that IDO activation likely plays a role in the development of depressive-like behavior during chronic immune activation. It would be interesting to know if BCG-induced increases in IDO activation are also accompanied by decreases in serotonin, as activation of IDO decreases the amount of tryptophan for conversion to serotonin and is one hypothesized mechanism of cytokine-induced depression (Fig. 2a).
Because it is known that preexisting vulnerability to depression may affect outcomes of an immune challenge, a number of studies combine the use immune activation with other depression risk factors, such as aging or exposure to stress to more comprehensively study the disorder. Collectively, these studies generally report a more pronounced induction of depressive-like behavior as well as greater peripheral and central changes in neurotransmitter and cytokine expression. Thus, in view of the combined and often synergistic effects of immune activation and accompanying risk factors, it is suggested that models of depression based on immune activation should consider the “stressor backdrop” upon which acute or chronic immune activation occurs (Gibb et al., 2008).
Questions regarding the effects of chronic immune activation on neuropsychiatric functioning are further complicated; however, by the observation that depressing the immune system can also lead to depressive-like symptoms. Recently, experimentally induced immunodepression was shown to reduce performance on cognitive behavioral tasks in mice (Barnard et al., 2009), illustrating that the role of immune dysregulation in the etiology of depressive symptoms is more than increased expression of proinflammatory cytokines. Further, patients with alcohol use disorders show decreased immune responses (Lau et al., 2009), yet these individual have an increased prevalence of mood disorders (Grant et al., 2004). It appears that a delicate balance of cytokines is required for normal regulation of neuropsychiatric functioning. A better understanding of the bidirectional communication scheme which has been proposed to explain cytokine-induced depression may be gained by considering the role not only of the innate but also of the adaptive immune system, including processes such as immunological memory and tolerance. It was recently suggested that immunity to certain self-antigens (or a lack thereof) when exposed to increased levels of risk factors (e.g., environmental or genetic) may be an important underlying factor in the etiology or pathophysiology of neurodegenerative processes and neuropsychiatric impairments (Schwartz and Ziv, 2008).
Genetically engineered rodent models used to study depression
Cytokines and cytokine-mediated signaling appears to play a critical role in the etiology and pathophysiology of depression, yet there exist a number of theories regarding their specific effects on behavior. One strategy that has been used to test such hypotheses is to alter the expression of genes that are involved in the central nervous and immune systems and to study their respective roles in animal behavior and effects on neuroimmune and neuroendocrine factors. Accordingly, chronic overexpression of brain IL-1β caused chronic leukocyte recruitment, axonal injury and prolonged depression of spontaneous behavior (i.e., reduced food pellet burrowing) in rats. Interestingly, IL-1β was not detected in circulating blood, despite the extended production of hepatic and circulating chemokines, liver damage and weight loss observed (Campbell et al., 2007). IL-6, another proinflammatory cytokine with a putative role in the pathogenesis of depression was investigated using a knockout model. Specifically, IL-6-deficient mice [IL-6(−/−)] were evaluated with number of depression-related tests (e.g., forced swim, tail suspension, and sucrose preference test). IL-6(−/−) mice showed reduced behavioral despair in the forced swim, and tail suspension test, and enhanced hedonic behavior in the sucrose preference test (Chourbaji et al., 2006). A similar panel of behavioral tests (e.g., forced swim and sucrose consumption tests) was used to assess TNF receptor deficient [TNFR1(−/−) and TNFR2(−/−)] mice for depressive-like behaviors. Deletion of either TNFR1 or TNFR2 leads to an antidepressant-like response in the forced swim test and mice lacking TNFR2 demonstrated a hedonic response in the sucrose drinking test compared with controls (Simen et al., 2006). Taken together, these results are consistent with the hypothesis that IL-1β, IL-6 and TNF-α can induce depressive-like symptoms and show that both TNF receptor subtypes can be involved in this response, with somewhat differing roles in regard to the expression of depressive symptoms (e.g., behavioral despair vs. anhedonia).
Given the hypothesized influence of stress on the development of depressive symptoms, it is interesting to note that several knockout mouse models [e.g., μ-opioid receptor knockout mice (Ide et al., 2010); norepinephrine transporter knockout mice (Haenisch et al., 2009); IL-1 receptor null mice (Koo and Duman, 2009)] show reduced emotional and biochemical responses following certain types of stress exposure.
In addition to genetically altering the expression of cytokines to better understand depression, investigators have also manipulated genes associated with the expression or signaling functions of cytokines. For example, given that activation of the purinergic P2X (7) receptors results in the rapid release of IL-1β, a study was conducted to behaviorally profile P2X(7) receptor knockout mice using a panel of tests and measurements to assess depressive-like behavior, including the forced swim and tail suspension tests and spontaneous locomotor activity and food intake assessments. Mice lacking the P2X(7) receptor exhibited antidepressant-like behavior in the tail suspension and forced swim tests (Basso et al., 2009). The investigators found no significant effects of genotype on locomotor activity or on novelty suppressed feeding, as measured by latency to start eating and time spent eating. However, knockout mice consumed more food than wild-type mice in a familiar environment, and the authors suggested that this difference may be attributed to the role of IL-1 in the regulation of physiological functions such as food intake and body weight (Basso et al., 2009). Manipulating the signaling functions of cytokines and their receptors by altering downstream effector molecules may also provide information about relevant inflammatory pathways involved in depression. Recently, we reported that treatment with IL-1β increased the phosphorylation of p38 MAP kinase in rat frontal cortex (Loftis et al., 2009a,b), a kinase that activates specific intracellular signaling pathways and transcription factors leading to the increased production of proinflammatory modulators. Thus, use of a conditional p38 MAP kinase knockout mouse model may also be useful in testing novel therapeutic strategies for depression (Kim et al., 2008). Taken together, these data support the relevance of proinflammatory cytokines in the expression of depressive-like behavior, and suggest alternative targets that could be of potential interest for the treatment of cytokine-induced depression.
Novel treatment strategies
The use of anti-inflammatory approaches for the treatment of depression is being examined at both pre-clinical and clinical levels. Many antidepressant medications have specific anti-inflammatory effects (Lim et al., 2009; Carvalho and Pariante, 2008) and significant immunoregulatory activities, such as reducing the number of Th1 cells secreting IFN-γ (Bengtsson et al., 1992; Zhu et al., 1998), increasing the production of IL-6 and IL-10 (Kubera et al., 2000), and inhibiting IFN-γ-induced microglial production of IL-6 and nitric oxide (Hashioka et al., 2007). Increasing evidence suggests that antidepressants operate in part to repair neurotransmission by enhancing neurogenesis (Malberg et al., 2000) as well as axonal and dendritic sprouting (Fujioka et al., 2004; Vaidya et al., 1999). However, current pharmacologic approaches are effective in less than 50% of patients and immune activation in patients with depression is also associated with resistance to treatment with traditional antidepressant medications. For example, (1) depressed patients non-responsive to drug treatment have increased immunity shown by elevated CD4+ T-cell activity and proinflammatory cytokine expression (Maes et al., 1997; O'Brien et al., 2007), (2) suppression of IL-6 and TNF-α does not occur in depressed patients who fail to respond to antidepressants (O'Brien et al., 2007), and (3) higher levels of TNF-α in patients with depression might predict a non-response to treatment with selective serotonin reuptake inhibitors (SSRIs) (Eller et al., 2008).
Targeted anti-inflammatory compounds to replace or augment the existing therapies are needed. The mechanism of action for these new treatments may involve modifying proinflammatory cytokine signaling pathways in the brain to better ameliorate the inflammation in depression and to improve the clinical efficacy of antidepressants (Malemud and Miller, 2008; Maes et al., 2009). Pre-clinical studies comparing the effects of typical and novel antidepressant treatments show that a number of different pharmacotherapeutic strategies have anti-inflammatory effects. For example, Pandey et al. (2009) used an olfactory bulbectomized model of depression in rats to evaluate the antidepressant effects of 5-((4-benzo [alpha] isothiazol-3-yl) piperazin-1-yl) methyl)-6-chloroindolin-2-one (BIP-1), a compound with affinity for serotonin type 2A receptors. Fourteen days of treatment with BIP-1 or the tricyclic antidepressant amitriptyline reduced the behavioral changes induced by olfactory bulbectomy, as shown using open field exploration, social interaction, hyperemotionality (i.e., behavioral responses to a variety of physical stimuli), and sucrose preference testing. Further, the combination of BIP-1 and amitryptyline administration resulted in a shorter course (7 days) of antidepressant treatment to achieve similar behavioral outcomes; however inflammatory markers were not assessed in this study (Pandey et al., 2009). In an earlier study, Hashioka et al. (2007) examined various types of antidepressants (i.e., imipramine, fluvoxamine, reboxetine), as well as lithium chloride on IFN-γ activation in murine microglial cells. Pretreatment with imipramine, fluvoxamine or reboxetine (but not lithium chloride) resulted in significant and dose-dependent suppression of IL-6 production. However, it remains unknown how the reduction in microglial IL-6 production specifically affects depressive-like behaviors. More research is clearly needed to better evaluate the effects of different classes of antidepressants on inflammatory pathways in relation to alterations in depressive-like symptoms.
Review of the clinical literature revealed a lack of studies directly comparing the efficacy of different classes of antidepressants in treating immune-associated depression. There have been a few clinical trials comparing the effects of placebo vs. SSRI antidepressants in patients with HCV undergoing interferon therapy (e.g., Morasco et al., 2007; Kraus et al., 2008). Recently Song et al. (2009a,b) compared the effects of electroacupuncture, fluoxetine or placebo treatment in 95 patients with major depressive disorder. These investigators found that both electroacupuncture and fluoxetine treatments had anti-inflammatory effects, as indicated by a reduction in IL-1β levels. Electroacupuncture additionally increased TNF-α and decreased IL-4 levels, and these changes were interpreted as a treatment-induced restoration of the balance between Th1 and Th2 cytokines (Song et al., 2009a,b).
In the development of new compounds as well as in the use of novel adjunctives to current treatments, it will also be important to appreciate the existence of depressive subtypes which could require different therapeutic approaches (Gold and Irwin, 2009; Charlton, 2009). Ultimately, treatment guidelines may need to be adjusted to include subpopulations of patients potentially resistant to typical antidepressant medications (e.g., SSRIs). The development of biologically based screening tests and assessment tools to identify these patient populations will help clinicians better identify such patients and subsequently target their depressive symptom profile. Table 3 lists some of the more promising pharmaceutical, dietary and psychosocial interventions currently under consideration for the treatment of cytokine-induced and other depressive disorders.
Table 3.
Novel antidepressant treatment targets and strategies.
| Intervention | Antidepressant efficacy | Referencea |
|---|---|---|
| Cyclo-oxygenase-2 (COX-2) inhibition (e.g., celecoxib) | In a 6-week double-blind, placebo-controlled trial, the combination of fluoxetine + celecoxib (n = 20) showed superiority over fluoxetine + placebo (n = 20), as measured using the Hamilton Depression Rating Scale. | Myint et al., 2007; Muller and Schwarz, 2008; Nery et al., 2008; Song et al., 2009; Akhondzadeh et al., 2009 |
| Suppression of SAPK/MAPK and/or JAK/STAT signaling components | Administration of a p38 MAP kinase inhibitor resulted in an attenuation of proinflammatory cytokines in the hippocampus and improved spatial memory on the Y-maze. | Malemud and Miller, 2008; Munoz et al., 2007; Loftis et al., 2009 |
| TNF-α inhibition/antagonism (e.g., adalimumab, etanercept, and infliximab) | A limited number of clinical trials and open-label studies suggest that TNF-α antagonists may reduce depressive symptoms, in particular fatigue, and improve quality of life. | Jiang et al., 2008; Soczynska et al., 2009 |
| Melatonin | Pre-clinical and clinical data show that melatonin reduces adhesion molecules and proinflammatory cytokines (e.g., IL-6, IL-8 and TNF-α). Using an unpredictable chronic stress model in mice, melatonin counteracted the stress-induced degradation of coat and decrease in grooming behavior in the splash test Melatonin treatment also decreased corticosterone levels, similar to the decreases observed following imipramine treatment. | Detanico et al., 2009; Maldonado et al., 2009 |
| Probiotics (e.g., Bifidobacteria) | Rats receiving probiotic treatment showed no significant differences on the FST, as compared to controls. However, probiotic treatment resulted in a reduction of IFN-γ, TNF-α, and IL-6 following mitogen stimulation of whole blood cultures. | Desbonnet et al., 2008 |
| Curcumin | In chronically stressed rats, curcumin treatment increased hippocampal neurogenesis (similar to imipramine treatment) and prevented stress-induced decreases in hippocampal serotonin receptor mRNA and BDNF protein levels. | Xu et al., 2007 |
| Ethyl-eicosapentaenoate (EPA) | In a rat olfactory bulbectomy model of depression, EPA significantly attenuated behavioral changes in the open field test and improved spatial memory, as compared to control rats. EPA treatment additionally resulted in a reduction of IL-1β and corticosterone levels. | Song et al., 2009 |
| Folic acid | A clinical study comparing fluoxetine+folic acid (n=14) with fluoxetine+placebo (n=13) found that patients receiving folate supplementation had significantly lower depression rating scale scores after 6 weeks of treatment. | Resler et al., 2008 |
| Psychotherapy (e.g., cognitive behavioral therapy and mindfulness interventions) | In a study comparing mindfulness meditation, cognitive behavioral therapy and education, patients with recurrent depression benefited from mindfulness meditation; in a separate group, IL-6 production was decreased following cognitive behavioral therapy in patients with depressive symptoms. | Fazzino et al., 2009; Zautra et al., 2008 |
| Vaccination (e.g., immunization with CNS-related antigens as a therapeutic means for treating depression) | In rats, immunization with a modified peptide [i.e., a segment of myelin basic protein (MBP)] resulted in the amelioration of anhedonia but no treatment effect was seen on the FST. Vaccination also resulted in significantly higher BDNF expression in hippocampus and increased cell proliferation. | Lewitus et al., 2009 |
Additional supporting references are provided for studies not specifically summarized under “Antidepressant efficacy”.
Conclusions and future perspectives
This review summarized current theories regarding the relationships between immune activation and neural function, as they relate to the development and expression of depressive symptoms. Collectively, this body of clinical and pre-clinical research supports the theory that inflammation and immune dysregulation can influence neurotransmitter metabolism, neuroendocrine function, synaptic plasticity and growth factor production, thus altering neural circuitry and contributing to depressive symptomatology. The current diagnostic categories for depression, as defined by the Diagnostic and Statistical Manual of Mental Disorders, however, are not etiologically or biologically derived, and it suggested that “depression” reflects multiple pathogeneses leading to varying symptom constellations.
Thus, there is a need for: 1) better tools (e.g., biological markers/neuroimmune disease signatures in addition to self-report or clinician-administered rating scales) to diagnose and treat depression, 2) a new definition of depression, one that can better differentiate among the various symptom profiles and etiologies (e.g., depression in medically ill populations vs. depression due to prolonged grief reactions)–this is important because treatment strategies may need to be different depending on the pathophysiological mechanisms contributing to the depressive symptoms, 3) more longitudinal studies in order to determine the direction and time course associated with circulating levels of inflammatory markers and symptoms of depression, and 4) improved animal models that allow for better a differentiation between the neuroimmune and pathophysiologic mechanisms associated with sickness versus other depressive-like behaviors.
Further research into the role of specific cytokines, inflammatory molecules and associated signaling cascades in the etiology of depressive symptoms will be important, as indicated by the promising results from recent treatment studies. Although there are some conflicting reports in the literature, due in part to methodological differences, studies using novel treatment strategies highlight the therapeutic potential of anti-inflammatory-based approaches for treating patients with depressive disorders.
Acknowledgments
We wish to thank the following individuals for their respective contributions to this paper: Lynsey Gebelin and Starr DeGennaro (data compilation for Fig. 1), Gray Whelan (graphic design and illustrations for Fig. 2), and Max Strater (editorial support).
References
- Aguilera M, Arias B, Wichers M, Barrantes-Vidal N, Moya J, Villa H, van Os J, Ibáñez MI, Ruipérez MA, Ortet G, Fañanás L. Early adversity and 5-HTT/BDNF genes: new evidence of gene–environment interactions on depressive symptoms in a general population. Psychol Med. 2009;39:1425–1432. doi: 10.1017/S0033291709005248. [DOI] [PubMed] [Google Scholar]
- Akhondzadeh S, Jafari S, Raisi F, Nasehi AA, Ghoreishi A, Salehi B, Mohebbi-Rasa S, Raznahan M, Kamalipour A. Clinical trial of adjunctive celecoxib treatment in patients with major depression: a double blind and placebo controlled trial. Depress Anxiety. 2009;26(7):607–611. doi: 10.1002/da.20589. [DOI] [PubMed] [Google Scholar]
- Anisman H, Kokkinidis L, Borowski T, Merali Z. Differential effects of interleukin (IL)-1beta, IL-2 and IL-6 on responding for rewarding lateral hypothalamic stimulation. Brain Res. 1998;779:177–187. doi: 10.1016/s0006-8993(97)01114-1. [DOI] [PubMed] [Google Scholar]
- Anisman H, Poulter MO, Gandhi R, Merali Z, Hayley S. Interferon-alpha effects are exaggerated when administered on a psychosocial stressor backdrop: cytokine, corticosterone and brain monoamine variations. J Neuroimmunol. 2007a;186:45–53. doi: 10.1016/j.jneuroim.2007.02.008. [DOI] [PubMed] [Google Scholar]
- Anisman H, Prakash P, Merali Z, Poulter MO. Corticotropin releasing hormone receptor alterations elicited by acute and chronic unpredictable stressor challenges in stressor-susceptible and resilient strains of mice. Behav Brain Res. 2007b;181:180–190. doi: 10.1016/j.bbr.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Anisman H. Cascading effects of stressors and inflammatory immune system activation: implications for major depressive disorder. J Psychiatry Neurosci. 2009;34:4–20. [PMC free article] [PubMed] [Google Scholar]
- Barnard CJ, Collins SA, Daisley JN, Behnke JM. Immunodepression reduces learning performance in male laboratory mice (Mus musculus) Physiol Behav. 2009;96:362–369. doi: 10.1016/j.physbeh.2008.10.021. [DOI] [PubMed] [Google Scholar]
- Basso AM, Bratcher NA, Harris RR, Jarvis MF, Decker MW, Rueter LE. Behavioral profile of P2X7 receptor knockout mice in animal models of depression and anxiety: relevance for neuropsychiatric disorders. Behav Brain Res. 2009;198:83–90. doi: 10.1016/j.bbr.2008.10.018. [DOI] [PubMed] [Google Scholar]
- Bengtsson BO, Zhu J, Thorell LH, Olsson T, Link H, Walinder J. Effects of zimeldine and its metabolites, clomipramine, imipramine and maprotiline in experimental allergic neuritis in Lewis rats. J Neuroimmunol. 1992;39:109–122. doi: 10.1016/0165-5728(92)90180-s. [DOI] [PubMed] [Google Scholar]
- Bezchlibnyk YB, Wang JF, McQueen GM, Young LT. Gene expression differences in bipolar disorder revealed by cDNA array analysis of post-mortem frontal cortex. J Neurochem. 2001;79:826–834. doi: 10.1046/j.1471-4159.2001.00628.x. [DOI] [PubMed] [Google Scholar]
- Björkqvist M, Wild EJ, Thiele J, Silvestroni A, Andre R, Lahiri N, Raibon E, Lee RV, Benn CL, Soulet D, Magnusson A, Woodman B, Landles C, Pouladi MA, Hayden MR, Khalili-Shirazi A, Lowdell MW, Brundin P, Bates GP, Leavitt BR, Möller T, Tabrizi SJ. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington's disease. J Exp Med. 2008;205(8):1869–1877. doi: 10.1084/jem.20080178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boka G, Anglade P, Wallach D, Javoy-Agid F, Agid Y, Hirsch EC. Immunocytochemical analysis of tumor necrosis factor and its receptors in Parkinson's disease. Neurosci Lett. 1994;172:151–154. doi: 10.1016/0304-3940(94)90684-x. [DOI] [PubMed] [Google Scholar]
- Bonaccorso S, Marino V, Puzella A, Pasquini M, Biondi M, Artini M, Almerighi C, Verkerk R, Meltzer H, Maes M. Increased depressive ratings in patients with hepatitis C receiving interferon-alpha-based immunotherapy are related to interferon-alpha-induced changes in the serotonergic system. J Clin Psychopharmacol. 2002;22:86–90. doi: 10.1097/00004714-200202000-00014. [DOI] [PubMed] [Google Scholar]
- Bonaccorso S, Maier SF, Meltzer HY, Maes M. Behavioral changes in rats after acute, chronic and repeated administration of interleukin-1beta: relevance for affective disorders. J Affect Disord. 2003;77:143–148. doi: 10.1016/s0165-0327(02)00118-0. [DOI] [PubMed] [Google Scholar]
- Boufidou F, Lambrinoudaki I, Argeitis J, Zervas IM, Pliatsika P, Leonardou AA, Petropoulos G, Hasiakos D, Papadias K, Nikolaou C. CSF and plasma cytokines at delivery and postpartum mood disturbances. J Affect Disord. 2009;115:287–292. doi: 10.1016/j.jad.2008.07.008. [DOI] [PubMed] [Google Scholar]
- Brambilla F, Monteleone P, Maj M. Interleukin-1beta and tumor necrosis factor-alpha in children with major depressive disorder or dysthymia. J Affect Disord. 2004;78:273–277. doi: 10.1016/S0165-0327(02)00315-4. [DOI] [PubMed] [Google Scholar]
- Brietzke E, Stertz L, Fernandes BS, Kauer-Sant'anna M, Mascarenhas M, Escosteguy Vargas A, Chies JA, Kapczinski F. Comparison of cytokine levels in depressed, manic and euthymic patients with bipolar disorder. J Affect Disord. 2009;116(3):214–217. doi: 10.1016/j.jad.2008.12.001. [DOI] [PubMed] [Google Scholar]
- Butler MP, O'Connor JJ, Moynagh PN. Dissection of tumor-necrosis factor-alpha inhibition of long-term potentiation (LTP) reveals a p38 mitogen-activated protein kinase-dependent mechanism which maps to early-but not late-phase LTP. Neuroscience. 2004;124:319–326. doi: 10.1016/j.neuroscience.2003.11.040. [DOI] [PubMed] [Google Scholar]
- Campbell SJ, Deacon RM, Jiang Y, Ferrari C, Pitossi FJ, Anthony DC. Overexpression of IL-1beta by adenoviral-mediated gene transfer in the rat brain causes a prolonged hepatic chemokine response, axonal injury and the suppression of spontaneous behaviour. Neurobiol Dis. 2007;27:151–163. doi: 10.1016/j.nbd.2007.04.013. [DOI] [PubMed] [Google Scholar]
- Capuron L, Gumnick JF, Musselman DL, Lawson DH, Reemsnyder A, Nemeroff CB, Miller AH. Neurobehavioral effects of interferon-alpha in cancer patients: phenomenology and paroxetine responsiveness of symptom dimensions. Neuropsychopharmacology. 2002a;26:643–652. doi: 10.1016/S0893-133X(01)00407-9. [DOI] [PubMed] [Google Scholar]
- Capuron L, Ravaud A, Neveu PJ, Miller AH, Maes M, Dantzer R. Association between decreased serum tryptophan concentrations and depressive symptoms in cancer patients undergoing cytokine therapy. Mol Psychiatry. 2002b;7:468–473. doi: 10.1038/sj.mp.4000995. [DOI] [PubMed] [Google Scholar]
- Capuron L, Fornwalt FB, Knight BT, Harvey PD, Ninan PT, Miller AH. Does cytokine-induced depression differ from idiopathic major depression in medically healthy individuals? J Affect Disord. 2009;119(1–3):181–185. doi: 10.1016/j.jad.2009.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho LA, Pariante CM. In vitro modulation of the glucocorticoid receptor by antidepressants. Stress. 2008;11:411–424. doi: 10.1080/10253890701850759. [DOI] [PubMed] [Google Scholar]
- Charlton BG. A model for self-treatment of four sub-types of symptomatic ‘depression’ using non-prescription agents: neuroticism (anxiety and emotional instability); malaise (fatigue and painful symptoms); demotivation (anhedonia) and seasonal affective disorder ‘SAD’. Med Hypotheses. 2009;72:1–7. doi: 10.1016/j.mehy.2008.09.021. [DOI] [PubMed] [Google Scholar]
- Chourbaji S, Urani A, Inta I, Sanchis-Segura C, Brandwein C, Zink M, Schwaninger M, Gass P. IL-6 knockout mice exhibit resistance to stress-induced development of depression-like behaviors. Neurobiol Dis. 2006;23:587–594. doi: 10.1016/j.nbd.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Coogan AN, Wyse CA. Neuroimmunology of the circadian clock. Brain Res. 2008;1232:104–112. doi: 10.1016/j.brainres.2008.07.087. [DOI] [PubMed] [Google Scholar]
- Cunningham AJ, Murray CA, O'Neill LA, Lynch MA, O'Connor JJ. Interleukin-1 beta (IL-1 beta) and tumour necrosis factor (TNF) inhibit long-term potentiation in the rat dentate gyrus in vitro. Neurosci Lett. 1996;203:17–20. doi: 10.1016/0304-3940(95)12252-4. [DOI] [PubMed] [Google Scholar]
- Curreli S, Romerio F, Mirandola P, Barion P, Bemis K, Zella D. Human primary CD4+ T cells activated in the presence of IFN-alpha 2b express functional indoleamine 2,3-dioxygenase. J Interferon Cytokine Res. 2001;21:431–437. doi: 10.1089/107999001750277916. [DOI] [PubMed] [Google Scholar]
- Dantzer R. Cytokine-induced sickness behavior: mechanisms and implications. Ann NY Acad Sci. 2001;933:222–234. doi: 10.1111/j.1749-6632.2001.tb05827.x. [DOI] [PubMed] [Google Scholar]
- Dantzer R. Cytokine, sickness behavior, and depression. Immunol Allergy Clin North Am. 2009;29:247–264. doi: 10.1016/j.iac.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer R, Bluthe RM, Kelley KW. Androgen-dependent vasopressinergic neurotransmission attenuates interleukin-1-induced sickness behavior. Brain Res. 1991;557:115–120. doi: 10.1016/0006-8993(91)90123-d. [DOI] [PubMed] [Google Scholar]
- Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson KW, Schwartz JE, Kirkland SA, Mostofsky E, Fink D, Guernsey D, Shimbo D. Relation of inflammation to depression and incident coronary heart disease (from the Canadian Nova Scotia Health Survey [NSHS95] Prospective Population Study) Am J Cardiol. 2009;103:755–761. doi: 10.1016/j.amjcard.2008.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Garza R, 2nd, Asnis GM, Pedrosa E, Stearns C, Migdal AL, Reinus JF, Paladugu R, Vemulapalli S. Recombinant human interferon-alpha does not alter reward behavior, or neuroimmune and neuroendocrine activation in rats. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29:781–792. doi: 10.1016/j.pnpbp.2005.03.008. [DOI] [PubMed] [Google Scholar]
- Deak T, Bellamy C, D'Agostino LG, Rosanoff M, McElderry NK, Bordner KA. Behavioral responses during the forced swim test are not affected by anti-inflammatory agents or acute illness induced by lipopolysaccharide. Behav Brain Res. 2005;160:125–134. doi: 10.1016/j.bbr.2004.11.024. [DOI] [PubMed] [Google Scholar]
- Desbonnet L, Garrett L, Clarke G, Bienenstock J, Dinan TG. The probiotic Bifidobacteria infantis: an assessment of potential antidepressant properties in the rat. J Psychiatr Res. 2008;43:164–174. doi: 10.1016/j.jpsychires.2008.03.009. [DOI] [PubMed] [Google Scholar]
- Detanico BC, Piato AL, Freitas JJ, Lhullier FL, Hidalgo MP, Caumo W, Elisabetsky E. Antidepressant-like effects of melatonin in the mouse chronic mild stress model. Eur J Pharmacol. 2009;607:121–125. doi: 10.1016/j.ejphar.2009.02.037. [DOI] [PubMed] [Google Scholar]
- Dhabhar FS, Burke HM, Epel ES, Mellon SH, Rosser R, Reus VI, Wolkowitz OM. Low serum IL-10 concentrations and loss of regulatory association between IL-6 and IL-10 in adults with major depression. J Psychiatr Res. 2009;43(11):962–969. doi: 10.1016/j.jpsychires.2009.05.010. [DOI] [PubMed] [Google Scholar]
- Dilger RN, Johnson RW. Aging, microglial cell priming, and the discordant central inflammatory response to signals from the peripheral immune system. J Leukoc Biol. 2008;84:932–939. doi: 10.1189/jlb.0208108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupin N, Mailliet F, Rocher C, Kessal K, Spedding M, Jay TM. Common efficacy of psychotropic drugs in restoring stress-induced impairment of prefrontal plasticity. Neurotox Res. 2006;10:193–198. doi: 10.1007/BF03033356. [DOI] [PubMed] [Google Scholar]
- Edwards RR, Kronfli T, Haythornthwaite JA, Smith MT, McGuire L, Page GG. Association of catastrophizing with interleukin-6 responses to acute pain. Pain. 2008;140:135–144. doi: 10.1016/j.pain.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eller T, Vasar V, Shlik J, Maron E. Pro-inflammatory cytokines and treatment response to escitalopram in major depressive disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32:445–450. doi: 10.1016/j.pnpbp.2007.09.015. [DOI] [PubMed] [Google Scholar]
- Fazzino F, Obregon F, Morles M, Rojas A, Arocha L, Mata S, Lima L. Taurine transporter in lymphocytes of patients with major depression treated with venlafaxine plus psychotherapy. Adv Exp Med Biol. 2009;643:217–224. doi: 10.1007/978-0-387-75681-3_22. [DOI] [PubMed] [Google Scholar]
- Friedman H, Eisenstein TK. Neurological basis of drug dependence and its effects on the immune system. J Neuroimmunol. 2004;147:106–108. doi: 10.1016/j.jneuroim.2003.10.022. [DOI] [PubMed] [Google Scholar]
- Fujioka T, Fujioka A, Duman RS. Activation of cAMP signaling facilitates the morphological maturation of newborn neurons in adult hippocampus. J Neurosci. 2004;24:319–328. doi: 10.1523/JNEUROSCI.1065.03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbay V, Klein RG, Guttman LE, Babb JS, Alonso CM, Nishawala M, Katz Y, Gaite MR, Gonzalez CJ. A preliminary study of cytokines in suicidal and nonsuicidal adolescents with major depression. J Child Adolesc Psychopharmacol. 2009a;19:423–430. doi: 10.1089/cap.2008.0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbay V, Klein RG, Alonso CM, Babb JS, Nishawala M, De Jesus G, Hirsch GS, Hottinger-Blanc PM, Gonzalez CJ. Immune system dysregulation in adolescent major depressive disorder. J Affect Disord. 2009b;115:177–182. doi: 10.1016/j.jad.2008.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galea I, Bechmann I, Perry VH. What is immune privilege (not)? Trends Immunol. 2007;28:12–18. doi: 10.1016/j.it.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Ghia JE, Blennerhassett P, Deng Y, Verdu EF, Khan WI, Collins SM. Reactivation of inflammatory bowel disease in a mouse model of depression. Gastroenterology. 2009;136:2280–2288. doi: 10.1053/j.gastro.2009.02.069. [DOI] [PubMed] [Google Scholar]
- Gibb J, Hayley S, Gandhi R, Poulter MO, Anisman H. Synergistic and additive actions of a psychosocial stressor and endotoxin challenge: circulating and brain cytokines, plasma corticosterone and behavioral changes in mice. Brain Behav Immun. 2008;22:573–589. doi: 10.1016/j.bbi.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Gimeno D, Kivimaki M, Brunner EJ, Elovainio M, De Vogli R, Steptoe A, Kumari M, Lowe GD, Rumley A, Marmot MG, Ferrie JE. Associations of C-reactive protein and interleukin-6 with cognitive symptoms of depression: 12-year follow-up of the Whitehall II study. Psychol Med. 2009;39:413–423. doi: 10.1017/S0033291708003723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giunta S. Exploring the complex relations between inflammation and aging (inflamm-aging): anti-inflamm-aging remodelling of inflamm-aging, from robustness to frailty. Inflamm Res. 2008;57:558–563. doi: 10.1007/s00011-008-7243-2. [DOI] [PubMed] [Google Scholar]
- Godbout JP, Johnson RW. Age and neuroinflammation: a lifetime of psychoneuroimmune consequences. Immunol Allergy Clin North Am. 2009;29:321–337. doi: 10.1016/j.iac.2009.02.007. [DOI] [PubMed] [Google Scholar]
- Godbout JP, Moreau M, Lestage J, Chen J, Sparkman NL, O' Connor J, Castanon N, Kelley KW, Dantzer R, Johnson RW. Aging exacerbates depressive-like behavior in mice in response to activation of the peripheral innate immune system. Neuropsychopharmacology. 2008;33:2341–2351. doi: 10.1038/sj.npp.1301649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold SM, Irwin MR. Depression and immunity: inflammation and depressive symptoms in multiple sclerosis. Neurol Clin. 2006;24:507–519. doi: 10.1016/j.ncl.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Gold SM, Irwin MR. Depression and immunity: inflammation and depressive symptoms in multiple sclerosis. Immunol Allergy Clin North Am. 2009;29:309–320. doi: 10.1016/j.iac.2009.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant BF, Stinson FS, Dawson DA, Chou SP, Dufour MC, Compton W, Pickering RP, Kaplan K. Prevalence and co-occurrence of substance use disorders and independent mood and anxiety disorders: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Arch Gen Psychiatry. 2004;61:807–816. doi: 10.1001/archpsyc.61.8.807. [DOI] [PubMed] [Google Scholar]
- Grippo AJ, Johnson AK. Stress, depression and cardiovascular dysregulation: a review of neurobiological mechanisms and the integration of research from preclinical disease models. Stress. 2009;12:1–21. doi: 10.1080/10253890802046281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haenisch B, Bilkei-Gorzo A, Caron MG, Bönisch H. Knockout of the norepinephrine transporter and pharmacologically diverse antidepressants prevent behavioral and brain neurotrophin alterations in two chronic stress models of depression. J Neurochem. 2009;111:403–416. doi: 10.1111/j.1471-4159.2009.06345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halaris A. Comorbidity between depression and cardiovascular disease. Int Angiol. 2009;28:92–99. [PubMed] [Google Scholar]
- Harrison NA, Brydon L, Walker C, Gray MA, Steptoe A, Critchley HD. Inflammation causes mood changes through alterations in subgenual cingulate activity and mesolimbic connectivity. Biol Psychiatry. 2009;66(5):407–414. doi: 10.1016/j.biopsych.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashioka S, Klegeris A, Monji A, Kato T, Sawada M, McGeer PL, Kanba S. Antidepressants inhibit interferon-gamma-induced microglial production of IL-6 and nitric oxide. Exp Neurol. 2007;206:33–42. doi: 10.1016/j.expneurol.2007.03.022. [DOI] [PubMed] [Google Scholar]
- Howren MB, Lamkin DM, Suls J. Associations of depression with C-reactive protein, IL-1, and IL-6: a meta-analysis. Psychosom Med. 2009;71:171–186. doi: 10.1097/PSY.0b013e3181907c1b. [DOI] [PubMed] [Google Scholar]
- Huckans M, Seelye A, Parcel T, Mull L, Woodhouse J, Bjornson D, Fuller BE, Loftis JM, Morasco BJ, Sasaki AW, Storzbach D, Hauser P. The cognitive effects of hepatitis C in the presence and absence of a history of substance use disorder. J Int Neuropsychol Soc. 2009;15:69–82. doi: 10.1017/S1355617708090085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ide S, Sora I, Ikeda K, Minami M, Uhl GR, Ishihara K. Reduced emotional and corticosterone responses to stress in mu-opioid receptor knockout mice. Neuropharmacology. 2010;58(1):241–247. doi: 10.1016/j.neuropharm.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto K, Kakiuchi C, Bundo M, Ikeda K, Kato T. Molecular characterization of bipolar disorder by comparing gene expression profiles of postmortem brains of major mental disorders. Mol Psychiatry. 2004;9:406–416. doi: 10.1038/sj.mp.4001437. [DOI] [PubMed] [Google Scholar]
- Jackson JC, Gordon SM, Hart RP, Hopkins RO, Ely EW. The association between delirium and cognitive decline: a review of the empirical literature. Neuropsychol Rev. 2004;14:87–98. doi: 10.1023/b:nerv.0000028080.39602.17. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Deacon R, Anthony DC, Campbell SJ. Inhibition of peripheral TNF can block the malaise associated with CNS inflammatory diseases. Neurobiol Dis. 2008;32:125–132. doi: 10.1016/j.nbd.2008.06.017. [DOI] [PubMed] [Google Scholar]
- Kaneko N, Kudo K, Mabuchi T, Takemoto K, Fujimaki K, Wati H, Iguchi H, Tezuka H, Kanba S. Suppression of cell proliferation by interferon-alpha through interleukin-1 production in adult rat dentate gyrus. Neuropsychopharmacology. 2006;31:2619–2626. doi: 10.1038/sj.npp.1301137. [DOI] [PubMed] [Google Scholar]
- Karakelides H, Irving BA, Short KR, O'Brien P, Nair KS. Age, obesity, and sex effects on insulin sensitivity and skeletal muscle mitochondrial function. Diabetes. 2009 Oct 15; doi: 10.2337/db09-0591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly A, Vereker E, Nolan Y, Brady M, Barry C, Loscher CE, Mills KH, Lynch MA. Activation of p38 plays a pivotal role in the inhibitory effect of lipopolysaccharide and interleukin-1 beta on long term potentiation in rat dentate gyrus. J Biol Chem. 2003;278:19453–19462. doi: 10.1074/jbc.M301938200. [DOI] [PubMed] [Google Scholar]
- Kentner AC, James JS, Miguelez M, Bielajew C. Investigating the hedonic effects of interferon-alpha on female rats using brain-stimulation reward. Behav Brain Res. 2007;177:90–99. doi: 10.1016/j.bbr.2006.10.033. [DOI] [PubMed] [Google Scholar]
- Khairova RA, Machado-Vieira R, Du J, Manji HK. A potential role for pro-inflammatory cytokines in regulating synaptic plasticity in major depressive disorder. Int J Neuropsychopharmacol. 2009;12:561–578. doi: 10.1017/S1461145709009924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiank C, Holtfreter B, Starke A, Mundt A, Wilke C, Schutt C. Stress susceptibility predicts the severity of immune depression and the failure to combat bacterial infections in chronically stressed mice. Brain Behav Immun. 2006;20:359–368. doi: 10.1016/j.bbi.2005.10.151. [DOI] [PubMed] [Google Scholar]
- Kim C, Sano Y, Todorova K, Carlson BA, Arpa L, Celada A, Lawrence T, Otsu K, Brissette JL, Arthur JS, Park JM. The kinase p38 alpha serves cell type-specific inflammatory functions in skin injury and coordinates pro- and anti-inflammatory gene expression. Nat Immunol. 2008;9:1019–1027. doi: 10.1038/ni.1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konsman JP, Veeneman J, Combe C, Poole S, Luheshi GN, Dantzer R. Central nervous action of interleukin-1 mediates activation of limbic structures and behavioural depression in response to peripheral administration of bacterial lipopolysaccharide. Eur J Neurosci. 2008;28:2499–2510. doi: 10.1111/j.1460-9568.2008.06549.x. [DOI] [PubMed] [Google Scholar]
- Koo JW, Duman RS. Interleukin-1 receptor null mutant mice show decreased anxiety-like behavior and enhanced fear memory. Neurosci Lett. 2009;456:39–43. doi: 10.1016/j.neulet.2009.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus MR, Al Taie O, Schafer A, Pfersdorff M, Lesch KP, Scheurlen M. Serotonin-1A receptor gene HTR1A variation predicts interferon-induced depression in chronic hepatitis C. Gastroenterology. 2007;132:1279–1286. doi: 10.1053/j.gastro.2007.02.053. [DOI] [PubMed] [Google Scholar]
- Kraus MR, Schäfer A, Schöttker K, Keicher C, Weissbrich B, Hofbauer I, Scheurlen M. Therapy of interferon-induced depression in chronic hepatitis C with citalopram: a randomised, double-blind, placebo-controlled study. Gut. 2008;57(4):531–536. doi: 10.1136/gut.2007.131607. [DOI] [PubMed] [Google Scholar]
- Kroes RA, Panksepp J, Burgdorf J, Otto NJ, Moskal JR. Modeling depression: social dominance-submission gene expression patterns in rat neocortex. Neuroscience. 2006;137:37–49. doi: 10.1016/j.neuroscience.2005.08.076. [DOI] [PubMed] [Google Scholar]
- Krueger JM, Majde JA. Humoral links between sleep and the immune system: research issues. Ann N Y Acad Sci. 2003;992:9–20. doi: 10.1111/j.1749-6632.2003.tb03133.x. [DOI] [PubMed] [Google Scholar]
- Kubera M, Simbirtsev A, Mathison R, Maes M. Effects of repeated fluoxetine and citalopram administration on cytokine release in C57BL/6 mice. Psychiatry Res. 2000;96:255–266. doi: 10.1016/s0165-1781(00)00184-0. [DOI] [PubMed] [Google Scholar]
- Kumsta R, Entringer S, Koper JW, van Rossum EF, Hellhammer DH, Wust S. Glucocorticoid receptor gene polymorphisms and glucocorticoid sensitivity of subdermal blood vessels and leukocytes. Biol Psychol. 2008;79:179–184. doi: 10.1016/j.biopsycho.2008.04.007. [DOI] [PubMed] [Google Scholar]
- Lambert MJ, Hatch DR, Kingston MD, Edwards BC. Zung, Beck, and Hamilton Rating Scales as measures of treatment outcome: a meta-analytic comparison. J Consult Clin Psychol. 1986;54(1):54–59. doi: 10.1037//0022-006x.54.1.54. [DOI] [PubMed] [Google Scholar]
- Langer JA, Pestka S. Interferon receptors. Immunology Today. 1988;9:393–400. doi: 10.1016/0167-5699(88)91241-8. [DOI] [PubMed] [Google Scholar]
- Lau A, von Dossow V, Sander M, MacGuill M, Lanzke N, Spies C. Alcohol use disorder and perioperative immune dysfunction. Anesth Analg. 2009;108:916–920. doi: 10.1213/ane.0b013e318193fd89. [DOI] [PubMed] [Google Scholar]
- Lee KJ, Kim YB, Kim JH, Kwon HC, Kim DK, Cho SW. The alteration of enterochromaffin cell, mast cell, and lamina propria T lymphocyte numbers in irritable bowel syndrome and its relationship with psychological factors. J Gastroenterol Hepatol. 2008;23:1689–1694. doi: 10.1111/j.1440-1746.2008.05574.x. [DOI] [PubMed] [Google Scholar]
- Leonard BE. Inflammation, depression and dementia: are they connected? Neurochem Res. 2007;32:1749–1756. doi: 10.1007/s11064-007-9385-y. [DOI] [PubMed] [Google Scholar]
- Leslie DL, Kozma L, Martin A, Landeros A, Katsovich L, King RA, Leckman JF. Neuropsychiatric disorders associated with streptococcal infection: a case–control study among privately insured children. J Am Acad Child Adolesc Psychiatry. 2008;47(10):1166–1172. doi: 10.1097/CHI.0b013e3181825a3d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewitus GM, Wilf-Yarkoni A, Ziv Y, Shabat-Simon M, Gersner R, Zangen A, Schwartz M. Vaccination as a novel approach for treating depressive behavior. Biol Psychiatry. 2009;65:283–288. doi: 10.1016/j.biopsych.2008.07.014. [DOI] [PubMed] [Google Scholar]
- Lim CM, Kim SW, Park JY, Kim C, Yoon SH, Lee JK. Fluoxetine affords robust neuroprotection in the postischemic brain via its anti-inflammatory effect. J Neurosci Res. 2009;87:1037–1045. doi: 10.1002/jnr.21899. [DOI] [PubMed] [Google Scholar]
- Lindqvist D, Janelidze S, Hagell P, Erhardt S, Samuelsson M, Minthon L, Hansson O, Björkqvist M, Träskman-Bendz L, Brundin L. Interleukin-6 is elevated in the cerebrospinal fluid of suicide attempters and related to symptom severity. Biol Psychiatry. 2009;66(3):287–292. doi: 10.1016/j.biopsych.2009.01.030. [DOI] [PubMed] [Google Scholar]
- Loftis JM, Hauser P. The phenomenology and treatment of interferon-induced depression. J Affect Disord. 2004;82:175–190. doi: 10.1016/j.jad.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Loftis JM, Wall JM, Pagel RL, Hauser P. Administration of pegylated interferon-alpha-2a or -2b does not induce sickness behavior in Lewis rats. Psychoneuroendocrinology. 2006a;31:1289–1294. doi: 10.1016/j.psyneuen.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Loftis JM, Hauser P, Macey TA, Lowe JD. Can rodents be used to model interferon-alpha-induced depressive symptoms? Prog Neuropsychopharmacol Biol Psychiatry. 2006b;30:1364–1365. doi: 10.1016/j.pnpbp.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Loftis JM, Huckans M, Ruimy S, Hinrichs DJ, Hauser P. Depressive symptoms in patients with chronic hepatitis C are correlated with elevated plasma levels of interleukin-1beta and tumor necrosis factor-alpha. Neurosci Lett. 2008;430:264–268. doi: 10.1016/j.neulet.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loftis JM, Murphy-Crews A, Menasco DJ, Huckans MS, Strater M. Cytokine-induced depression: effects of interleukin-1β and corticotrophin-releasing factor antagonism on biochemical and behavioral indicators of “depression” in the rat. Brain, Behavior, and Immunity. 2009a;23:S46. 2009. [Google Scholar]
- Lotrich FE, Ferrell RE, Rabinovitz M, Pollock BG. Risk for depression during interferon-alpha treatment is affected by the serotonin transporter polymorphism. Biol Psychiatry. 2009b;65:344–348. doi: 10.1016/j.biopsych.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccioni RB, Rojo LE, Fernandez JA, Kuljis RO. The role of neuroimmunomodulation in Alzheimer's disease. Ann N Y Acad Sci. 2009;1153:240–246. doi: 10.1111/j.1749-6632.2008.03972.x. [DOI] [PubMed] [Google Scholar]
- Maes M, Scharpe S, Meltzer HY, Bosmans E, Suy E, Calabrese J, Cosyns P. Relationships between interleukin-6 activity, acute phase proteins, and function of the hypothalamic–pituitary–adrenal axis in severe depression. Psychiatry Res. 1993;49:11–27. doi: 10.1016/0165-1781(93)90027-e. [DOI] [PubMed] [Google Scholar]
- Maes M, Bosmans E, Meltzer HY. Immunoendocrine aspects of major depression. Relationships between plasma interleukin-6 and soluble interleukin-2 receptor, prolactin and cortisol. Eur Arch Psychiatry Clin Neurosci. 1995;245:172–178. doi: 10.1007/BF02193091. [DOI] [PubMed] [Google Scholar]
- Maes M, Bosmans E, De Jongh R, Kenis G, Vandoolaeghe E, Neels H. Increased serum IL-6 and IL-1 receptor antagonist concentrations in major depression and treatment resistant depression. Cytokine. 1997;9:853–858. doi: 10.1006/cyto.1997.0238. [DOI] [PubMed] [Google Scholar]
- Maes M, Yirmyia R, Noraberg J, Brene S, Hibbeln J, Perini G, Kubera M, Bob P, Lerer B, Maj M. The inflammatory & neurodegenerative (I&ND) hypothesis of depression: leads for future research and new drug developments in depression. Metab Brain Dis. 2009;24:27–53. doi: 10.1007/s11011-008-9118-1. [DOI] [PubMed] [Google Scholar]
- Makino M, Kitano Y, Komiyama C, Hirohashi M, Kohno M, Moriyama M, Takasuna K. Human interferon-alpha induces immobility in the mouse forced swimming test: involvement of the opioid system. Brain Res. 2000;852:482–484. doi: 10.1016/s0006-8993(99)02235-0. [DOI] [PubMed] [Google Scholar]
- Malberg JE, Eisch AJ, Nestler EJ, Duman RS. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci. 2000;20:9104–9110. doi: 10.1523/JNEUROSCI.20-24-09104.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado MD, Perez-San-Gregorio MA, Reiter RJ. The role of melatonin in the immuno-neuro-psychology of mental disorders. Recent Pat CNS Drug Discov. 2009;4:61–69. doi: 10.2174/157488909787002564. [DOI] [PubMed] [Google Scholar]
- Malemud CJ, Miller AH. Pro-inflammatory cytokine-induced SAPK/MAPK and JAK/STAT in rheumatoid arthritis and the new anti-depression drugs. Expert Opin Ther Targets. 2008;12:171–183. doi: 10.1517/14728222.12.2.171. [DOI] [PubMed] [Google Scholar]
- Marchand F, Perretti M, McMahon SB. Role of the immune system in chronic pain. Nat Rev Neurosci. 2005;6:521–532. doi: 10.1038/nrn1700. [DOI] [PubMed] [Google Scholar]
- Martin AL, Brown RE. The lonely mouse: Verification of a separation-induced model of depression in female mice. Behav Brain Res. 2010;207(1):196–207. doi: 10.1016/j.bbr.2009.10.006. [DOI] [PubMed] [Google Scholar]
- McAfoose J, Baune BT. Evidence for a cytokine model of cognitive function. Neurosci Biobehav Rev. 2009;33:355–366. doi: 10.1016/j.neubiorev.2008.10.005. [DOI] [PubMed] [Google Scholar]
- McNally L, Bhagwagar Z, Hannestad J. Inflammation, glutamate, and glia in depression: a literature review. CNS Spectr. 2008;13:501–510. doi: 10.1017/s1092852900016734. [DOI] [PubMed] [Google Scholar]
- Merali Z, Brennan K, Brau P, Anisman H. Dissociating anorexia and anhedonia elicited by interleukin-1beta: antidepressant and gender effects on responding for “free chow” and “earned” sucrose intake. Psychopharmacology (Berl) 2003;165:413–418. doi: 10.1007/s00213-002-1273-1. [DOI] [PubMed] [Google Scholar]
- Miller GE, Rohleder N, Cole SW. Chronic interpersonal stress predicts activation of pro- and anti-inflammatory signaling pathways 6 months later. Psychosom Med. 2009a;71:57–62. doi: 10.1097/PSY.0b013e318190d7de. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry. 2009b;65(9):732–741. doi: 10.1016/j.biopsych.2008.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor TR, Huang Q, Witt AE. Cytokine–Purine interactions in traumatic stress, behavioral depression, and sickness. CNS Neurol Disord Drug Targets. 2006;5:547–560. doi: 10.2174/187152706778559282. [DOI] [PubMed] [Google Scholar]
- Morasco BJ, Rifai MA, Loftis JM, Indest DW, Moles JK, Hauser P. A randomized trial of paroxetine to prevent interferon-alpha-induced depression in patients with hepatitis C. J Affect Disord. 2007;103(1-3):83–90. doi: 10.1016/j.jad.2007.01.007. [DOI] [PubMed] [Google Scholar]
- Moreau M, Andre C, O'Connor JC, Dumich SA, Woods JA, Kelley KW, Dantzer R, Lestage J, Castanon N. Inoculation of Bacillus Calmette-Guerin to mice induces an acute episode of sickness behavior followed by chronic depressive-like behavior. Brain Behav Immun. 2008;22:1087–1095. doi: 10.1016/j.bbi.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mount MP, Lira A, Grimes D, Smith PD, Faucher S, Slack R, Anisman H, Hayley S, Park DS. Involvement of interferon-gamma in microglial-mediated loss of dopaminergic neurons. J Neurosci. 2007;27:3328–3337. doi: 10.1523/JNEUROSCI.5321-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller N, Schwarz MJ. The immune-mediated alteration of serotonin and glutamate: towards an integrated view of depression. Mol Psychiatry. 2007;12:988–1000. doi: 10.1038/sj.mp.4002006. [DOI] [PubMed] [Google Scholar]
- Muller N, Schwarz MJ. COX-2 inhibition in schizophrenia and major depression. Curr Pharm Des. 2008;14:1452–1465. doi: 10.2174/138161208784480243. [DOI] [PubMed] [Google Scholar]
- Mulsant BH, Ganguli M. Epidemiology and diagnosis of depression in late life. J Clin Psychiatry. 1999;60(Suppl 20):9–15. [PubMed] [Google Scholar]
- Munoz L, Ranaivo HR, Roy SM, Hu W, Craft JM, McNamara LK, Chico LW, Van Eldik LJ, Watterson DM. A novel p38 alpha MAPK inhibitor suppresses brain proinflammatory cytokine up-regulation and attenuates synaptic dysfunction and behavioral deficits in an Alzheimer's disease mouse model. J Neuroinflammation. 2007;4:21. doi: 10.1186/1742-2094-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myint AM, Steinbusch HW, Goeghegan L, Luchtman D, Kim YK, Leonard BE. Effect of the COX-2 inhibitor celecoxib on behavioural and immune changes in an olfactory bulbectomised rat model of depression. Neuroimmunomodulation. 2007;14:65–71. doi: 10.1159/000107420. [DOI] [PubMed] [Google Scholar]
- Navarre BM, Laggart JD, Craft RM. Anhedonia in postpartum rats. Physiol Behav. 2009 doi: 10.1016/j.physbeh.2009.10.011. 2009 Oct 20. (Electronic publication ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nery FG, Monkul ES, Hatch JP, Fonseca M, Zunta-Soares GB, Frey BN, Bowden CL, Soares JC. Celecoxib as an adjunct in the treatment of depressive or mixed episodes of bipolar disorder: a double-blind, randomized, placebocontrolled study. Hum Psychopharmacol. 2008;20023(2):87–94. doi: 10.1002/hup.912. [DOI] [PubMed] [Google Scholar]
- Neurauter G, Schrocksnadel K, Scholl-Burgi S, Sperner-Unterweger B, Schubert C, Ledochowski M, Fuchs D. Chronic immune stimulation correlates with reduced phenylalanine turnover. Curr Drug Metab. 2008;9:622–627. doi: 10.2174/138920008785821738. [DOI] [PubMed] [Google Scholar]
- Neveu PJ, Bluthé RM, Liège S, Moya S, Michaud B, Dantzer R. Interleukin-1-induced sickness behavior depends on behavioral lateralization in mice. Physiol Behav. 1998;63(4):587–590. doi: 10.1016/s0031-9384(97)00495-2. [DOI] [PubMed] [Google Scholar]
- O'Brien SM, Scully P, Fitzgerald P, Scott LV, Dinan TG. Plasma cytokine profiles in depressed patients who fail to respond to selective serotonin reuptake inhibitor therapy. J Psychiatr Res. 2007;41:326–331. doi: 10.1016/j.jpsychires.2006.05.013. [DOI] [PubMed] [Google Scholar]
- O'Mahony SM, Myint AM, van den HD, Desbonnet L, Steinbusch H, Leonard BE. Gestational stress leads to depressive-like behavioural and immunological changes in the rat. Neuroimmunomodulation. 2006;13:82–88. doi: 10.1159/000096090. [DOI] [PubMed] [Google Scholar]
- O'Mahony SM, Marchesi JR, Scully P, Codling C, Ceolho AM, Quigley EM, Cryan JF, Dinan TG. Early life stress alters behavior, immunity, and microbiota in rats: implications for irritable bowel syndrome and psychiatric illnesses. Biol Psychiatry. 2009;65:263–267. doi: 10.1016/j.biopsych.2008.06.026. [DOI] [PubMed] [Google Scholar]
- Orre IJ, Murison R, Dahl AA, Ueland T, Aukrust P, Fosså SD. Levels of circulating interleukin-1 receptor antagonist and C-reactive protein in long-term survivors of testicular cancer with chronic cancer-related fatigue. Brain Behav Immun. 2009;23(6):868–874. doi: 10.1016/j.bbi.2009.04.003. [DOI] [PubMed] [Google Scholar]
- Orsal AS, Blois SM, Bermpohl D, Schaefer M, Coquery N. Administration of interferon-alpha in mice provokes peripheral and central modulation of immune cells, accompanied by behavioral effects. Neuropsychobiology. 2008;58:211–222. doi: 10.1159/000201718. [DOI] [PubMed] [Google Scholar]
- Ovaskainen Y, Koponen H, Jokelainen J, Keinanen-Kiukaanniemi S, Kumpusalo E, Vanhala M. Depressive symptomatology is associated with decreased interleukin-1 beta and increased interleukin-1 receptor antagonist levels in males. Psychiatry Res. 2009;167:73–79. doi: 10.1016/j.psychres.2007.12.004. [DOI] [PubMed] [Google Scholar]
- Owen BM, Eccleston D, Ferrier IN, Young AH. Raised levels of plasma interleukin-1beta in major and postviral depression. Acta Psychiatr Scand. 2001;103:226–228. doi: 10.1034/j.1600-0447.2001.00162.x. [DOI] [PubMed] [Google Scholar]
- Padmos RC, Hillegers MH, Knijff EM, Vonk R, Bouvy A, Staal FJ, de Ridder D, Kupka RW, Nolen WA, Drexhage HA. A discriminating messenger RNA signature for bipolar disorder formed by an aberrant expression of inflammatory genes in monocytes. Arch Gen Psychiatry. 2008;65:395–407. doi: 10.1001/archpsyc.65.4.395. [DOI] [PubMed] [Google Scholar]
- Pandey DK, Mahesh R, Kumar AA, Rao VS, Arjun M, Rajkumar R. A novel 5-HT(2A) receptor antagonist exhibits antidepressant-like effects in a battery of rodent behavioural assays: Approaching early-onset antidepressants. Pharmacol Biochem Behav. 2009 doi: 10.1016/j.pbb.2009.09.018. Oct 2. (Electronic publication ahead of print) [DOI] [PubMed] [Google Scholar]
- Parachikova A, Agadjanyan MG, Cribbs DH, Blurton-Jones M, Perreau V, Rogers J, Beach TG, Cotman CW. Inflammatory changes parallel the early stages of Alzheimer disease. Neurobiol Aging. 2007;28:1821–1833. doi: 10.1016/j.neurobiolaging.2006.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pariante CM. Glucocorticoid receptor function in vitro in patients with major depression. Stress. 2004;7(4):209–219. doi: 10.1080/10253890500069650. [DOI] [PubMed] [Google Scholar]
- Parnet P, Amindari S, Wu C, Brunke-Reese D, Goujon E, Weyhenmeyer JA, Dantzer R, Kelley KW. Expression of type I and type II interleukin-1 receptors in mouse brain. Brain Res Mol Brain Res. 1994;27:63–70. doi: 10.1016/0169-328x(94)90185-6. [DOI] [PubMed] [Google Scholar]
- Penninx BW, Geerlings SW, Deeg DJ, van Eijk JT, van Tilburg W, Beekman AT. Minor and major depression and the risk of death in older persons. Arch Gen Psychiatry. 1999;56:889–895. doi: 10.1001/archpsyc.56.10.889. [DOI] [PubMed] [Google Scholar]
- Pitychoutis PM, Nakamura K, Tsonis PA, Papadopoulou-Daifoti Z. Neurochemical and behavioral alterations in an inflammatory model of depression: sex differences exposed. Neuroscience. 2009;159:1216–1232. doi: 10.1016/j.neuroscience.2009.01.072. [DOI] [PubMed] [Google Scholar]
- Plata-Salaman CR, Oomura Y, Kai Y. Tumor necrosis factor and interleukin-1 beta: suppression of food intake by direct action in the central nervous system. Brain Res. 1988;448:106–114. doi: 10.1016/0006-8993(88)91106-7. [DOI] [PubMed] [Google Scholar]
- Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, Knapp DJ, Crews FT. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55:453–462. doi: 10.1002/glia.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raison CL, Borisov AS, Majer M, Drake DF, Pagnoni G, Woolwine BJ, Vogt GJ, Massung B, Miller AH. Activation of central nervous system inflammatory pathways by interferon-alpha: relationship to monoamines and depression. Biol Psychiatry. 2009;65:296–303. doi: 10.1016/j.biopsych.2008.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resler G, Lavie R, Campos J, Mata S, Urbina M, Garcia A, Apitz R, Lima L. Effect of folic acid combined with fluoxetine in patients with major depression on plasma homocysteine and vitamin B12, and serotonin levels in lymphocytes. Neuroimmunomodulation. 2008;15:145–152. doi: 10.1159/000151527. [DOI] [PubMed] [Google Scholar]
- Rothermundt M, Arolt V, Peters M, Gutbrodt H, Fenker J, Kersting A, Kirchner H. Inflammatory markers in major depression and melancholia. J Affect Disord. 2001;63:93–102. doi: 10.1016/s0165-0327(00)00157-9. [DOI] [PubMed] [Google Scholar]
- Sammut S, Goodall G, Muscat R. Acute interferon-alpha administration modulates sucrose consumption in the rat. Psychoneuroendocrinology. 2001;26:261–272. doi: 10.1016/s0306-4530(00)00051-2. [DOI] [PubMed] [Google Scholar]
- Sammut S, Bethus I, Goodall G, Muscat R. Antidepressant reversal of interferon-alpha-induced anhedonia. Physiol Behave. 2002;75:765–772. doi: 10.1016/s0031-9384(02)00677-7. [DOI] [PubMed] [Google Scholar]
- Schobitz B, de Kloet ER, Sutanto W, Holsboer F. Cellular localization of interleukin 6 mRNA and interleukin 6 receptor mRNA in rat brain. Eur J Neurosci. 1993;5:1426–1435. doi: 10.1111/j.1460-9568.1993.tb00210.x. [DOI] [PubMed] [Google Scholar]
- Schwartz M, Ziv Y. Immunity to self and self-maintenance: a unified theory of brain pathologies. Trends Immunol. 2008;29:211–219. doi: 10.1016/j.it.2008.01.003. [DOI] [PubMed] [Google Scholar]
- Simen BB, Duman CH, Simen AA, Duman RS. TNFalpha signaling in depression and anxiety: behavioral consequences of individual receptor targeting. Biol Psychiatry. 2006;59:775–785. doi: 10.1016/j.biopsych.2005.10.013. [DOI] [PubMed] [Google Scholar]
- Soczynska JK, Kennedy SH, Goldstein BI, Lachowski A, Woldeyohannes HO, McIntyre RS. The effect of tumor necrosis factor antagonists on mood and mental health-associated quality of life: novel hypothesis-driven treatments for bipolar depression? Neurotoxicology. 2009;30(4):497–521. doi: 10.1016/j.neuro.2009.03.004. [DOI] [PubMed] [Google Scholar]
- Song C, Horrobin DF, Leonard BE. The comparison of changes in behavior, neurochemistry, endocrine, and immune functions after different routes, doses and durations of administrations of IL-1beta in rats. Pharmacopsychiatry. 2006;39(3):88–99. doi: 10.1055/s-2006-941557. [DOI] [PubMed] [Google Scholar]
- Song C, Halbreich U, Han C, Leonard BE, Luo H. Imbalance between pro- and anti-inflammatory cytokines, and between Th1 and Th2 cytokines in depressed patients: the effect of electroacupuncture or fluoxetine treatment. Pharmacopsychiatry. 2009a;42(5):182–188. doi: 10.1055/s-0029-1202263. [DOI] [PubMed] [Google Scholar]
- Song C, Zhang XY, Manku M. Increased phospholipase A2 activity and inflammatory response but decreased nerve growth factor expression in the olfactory bulbectomized rat model of depression: effects of chronic ethyleicosapentaenoate treatment. J Neurosci. 2009b;29:14–22. doi: 10.1523/JNEUROSCI.3569-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart JC, Rand KL, Muldoon MF, Kamarck TW. A prospective evaluation of the directionality of the depression–inflammation relationship. Brain Behav Immun. 2009;23:936–944. doi: 10.1016/j.bbi.2009.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su S, Miller AH, Snieder H, Bremner JD, Ritchie J, Maisano C, Jones L, Murrah NV, Goldberg J, Vaccarino V. Common genetic contributions to depressive symptoms and inflammatory markers in middle-aged men: the Twins Heart Study. Psychosom Med. 2009;71:152–158. doi: 10.1097/PSY.0b013e31819082ef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiergiel AH, Dunn AJ. Effects of interleukin-1beta and lipopolysaccharide on behavior of mice in the elevated plus-maze and open field tests. Pharmacol Biochem Behav. 2007;86:651–659. doi: 10.1016/j.pbb.2007.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiergiel AH, Smagin GN, Johnson LJ, Dunn AJ. The role of cytokines in the behavioral responses to endotoxin and influenza virus infection in mice: effects of acute and chronic administration of the interleukin-1-receptor antagonist (IL-1ra) Brain Res. 1997;776:96–104. doi: 10.1016/s0006-8993(97)01009-3. [DOI] [PubMed] [Google Scholar]
- Tache Y, Brunnhuber S. From Hans Selye's discovery of biological stress to the identification of corticotropin-releasing factor signaling pathways: implication in stress-related functional bowel diseases. Ann N Y Acad Sci. 2008;1148:29–41. doi: 10.1196/annals.1410.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe Y, Nishibori T, Su L, Arduini RM, Baker DP, David M. Cutting edge: role of STAT1, STAT3, and STAT5 in IFN-alpha beta responses in T lymphocytes. J Immunol. 2005;174:609–613. doi: 10.4049/jimmunol.174.2.609. [DOI] [PubMed] [Google Scholar]
- Taylor MW, Feng GS. Relationship between interferon-gamma, indoleamine 2,3-dioxygenase, and tryptophan catabolism. FASEB J. 1991;5:2516–2522. [PubMed] [Google Scholar]
- Traks T, Koido K, Eller T, Maron E, Kingo K, Vasar V, Vasar E, Koks S. Polymorphisms in the interleukin-10 gene cluster are possibly involved in the increased risk for major depressive disorder. BMC Med Genet. 2008;9:111. doi: 10.1186/1471-2350-9-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao CW, Lin YS, Chen CC, Bai CH, Wu SR. Cytokines and serotonin transporter in patients with major depression. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30:899–905. doi: 10.1016/j.pnpbp.2006.01.029. [DOI] [PubMed] [Google Scholar]
- Turner EH, Loftis JM, Blackwell AD. Serotonin a la carte: supplementation with the serotonin precursor 5-hydroxytryptophan. Pharmacol Ther. 2006;109:325–338. doi: 10.1016/j.pharmthera.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Uguz F, Akman C, Kucuksarac S, Tufekci O. Anti-tumor necrosis factor-alpha therapy is associated with less frequent mood and anxiety disorders in patients with rheumatoid arthritis. Psychiatry Clin Neurosci. 2009;63:50–55. doi: 10.1111/j.1440-1819.2008.01905.x. [DOI] [PubMed] [Google Scholar]
- Vaidya VA, Siuciak JA, Du F, Duman RS. Hippocampal mossy fiber sprouting induced by chronic electroconvulsive seizures. Neuroscience. 1999;89:157–166. doi: 10.1016/s0306-4522(98)00289-9. [DOI] [PubMed] [Google Scholar]
- van den Biggelaar AH, Gussekloo J, de Craen AJ, Frolich M, Stek ML, van der Mast RC, Westendorp RG. Inflammation and interleukin-1 signaling network contribute to depressive symptoms but not cognitive decline in old age. Exp Gerontol. 2007;42:693–701. doi: 10.1016/j.exger.2007.01.011. [DOI] [PubMed] [Google Scholar]
- Van Lieshout RJ, Bienenstock J, MacQueen GM. A review of candidate pathways underlying the association between asthma and major depressive disorder. Psychosom Med. 2009;71:187–195. doi: 10.1097/PSY.0b013e3181907012. [DOI] [PubMed] [Google Scholar]
- Varghese AK, Verdu EF, Bercik P, Khan WI, Blennerhassett PA, Szechtman H, Collins SM. Antidepressants attenuate increased susceptibility to colitis in a murine model of depression. Gastroenterology. 2006;130:1743–1753. doi: 10.1053/j.gastro.2006.02.007. [DOI] [PubMed] [Google Scholar]
- Wang JY, Zeng XY, Fan GX, Yuan YK, Tang JS. mu- but not delta- and kappa-opioid receptor mediates the nucleus submedius interferon-alpha-evoked antinociception in the rat. Neurosci Lett. 2006;397:254–258. doi: 10.1016/j.neulet.2005.12.046. [DOI] [PubMed] [Google Scholar]
- Wang J, Campbell IL, Zhang H. Systemic interferon-alpha regulates interferon-stimulated genes in the central nervous system. Mol Psychiatry. 2008;13:293–301. doi: 10.1038/sj.mp.4002013. [DOI] [PubMed] [Google Scholar]
- Wang H, Buchner M, Moser MT, Daniel V, Schiltenwolf M. The role of IL-8 in patients with fibromyalgia: a prospective longitudinal study of 6 months. Clin J Pain. 2009;25:1–4. doi: 10.1097/AJP.0b013e31817e13a3. [DOI] [PubMed] [Google Scholar]
- Williams LJ, Pasco JA, Jacka FN, Henry MJ, Dodd S, Berk M. Depression and bone metabolism. A review. Psychother Psychosom. 2009;78:16–25. doi: 10.1159/000162297. [DOI] [PubMed] [Google Scholar]
- Wirtz PH, Redwine LS, Linke S, Hong S, Rutledge T, Greenberg BH, Mills PJ. Circulating levels of soluble intercellular adhesion molecule-1 (sICAM-1) independently predict depressive symptom severity after 12 months in heart failure patients. Brain Behav Immun. 2009 Feb 13; doi: 10.1016/j.bbi.2009.02.004. [DOI] [PubMed] [Google Scholar]
- Wood LJ, Nail LM, Gilster A, Winters KA, Elsea CR. Cancer chemotherapy-related symptoms: evidence to suggest a role for proinflammatory cytokines. Oncol Nurs Forum. 2006;33:535–542. doi: 10.1188/06.ONF.535-542. [DOI] [PubMed] [Google Scholar]
- Xu Y, Ku B, Cui L, Li X, Barish PA, Foster TC, Ogle WO. Curcumin reverses impaired hippocampal neurogenesis and increases serotonin receptor 1A mRNA and brain-derived neurotrophic factor expression in chronically stressed rats. Brain Res. 2007;1162:9–18. doi: 10.1016/j.brainres.2007.05.071. [DOI] [PubMed] [Google Scholar]
- Yaka R, Salomon S, Matzner H, Weinstock M. Effect of varied gestational stress on acquisition of spatial memory, hippocampal LTP and synaptic proteins in juvenile male rats. Behav Brain Res. 2007;179:126–132. doi: 10.1016/j.bbr.2007.01.018. [DOI] [PubMed] [Google Scholar]
- Zautra AJ, Davis MC, Reich JW, Nicassario P, Tennen H, Finan P, Kratz A, Parrish B, Irwin MR. Comparison of cognitive behavioral and mindfulness meditation interventions on adaptation to rheumatoid arthritis for patients with and without history of recurrent depression. J Consult Clan Psychol. 2008;76:408–421. doi: 10.1037/0022-006X.76.3.408. [DOI] [PubMed] [Google Scholar]
- Zhu J, Bengtsson BO, Mix E, Ekerling L, Thorell LH, Olsson T, Link H. Clomipramine and imipramine suppress clinical signs and T and B cell response to myelin proteins in experimental autoimmune neuritis in Lewis rats. J Autoimmun. 1998;11:319–327. doi: 10.1006/jaut.1998.0209. [DOI] [PubMed] [Google Scholar]