

Abstract
Gene duplication is probably the most important mechanism for generating new gene functions. However, gene duplication has been overlooked as a potentially effective way to resolve genetic conflicts. Here, we analyze the entire set of Drosophila melanogaster nuclearly encoded mitochondrial duplicate genes and show that both RNA- and DNA-mediated mitochondrial gene duplications exhibit an unexpectedly high rate of relocation (change in location between parental and duplicated gene) as well as an extreme tendency to avoid the X chromosome. These trends are likely related to our observation that relocated genes tend to have testis-specific expression. We also infer that these trends hold across the entire Drosophila genus. Importantly, analyses of gene ontology and functional interaction networks show that there is an overrepresentation of energy production-related functions in these mitochondrial duplicates. We discuss different hypotheses to explain our results and conclude that our findings substantiate the hypothesis that gene duplication for male germline function is likely a mechanism to resolve intralocus sexually antagonistic conflicts that we propose are common in testis. In the case of nuclearly encoded mitochondrial duplicates, our hypothesis is that past sexually antagonistic conflict related to mitochondrial energy function in Drosophila was resolved by gene duplication.
Keywords: nuclearly encoded mitochondrial functions, gene duplication, male-specific expression, intralocus sexual antagonism
Introduction
Forty years ago, gene duplication was put forth as the most important molecular mechanism for the origin of new genes (Ohno 1970). Now, we have a plethora of examples of how gene duplication contributes to the origin of new gene functions (True and Carroll 2002; Taylor and Raes 2004; Hurley et al. 2005). Current genome data regarding copy number polymorphism have revealed that, in fact, segmental duplication is a highly dynamic process in all the taxa in which it has been studied: for example, humans (Conrad et al. 2009), mice (Graubert et al. 2007), and flies (Emerson et al. 2008). However, only recent studies have begun to investigate which gene functions experience the highest rates of duplicate retention. In Drosophila melanogaster, the largest expansions of gene families seem to correspond to genes with functions related to external stimuli, behavior, sex, and reproduction among others (Hahn et al. 2007; Heger and Ponting 2007). The reasons for these biases remain unknown.
Intriguingly, recent work has revealed that 15 nuclearly encoded genes with mitochondrial function originated through retroposition (i.e., RNA-mediated duplication) and show testis-biased expression in D. melanogaster (Bai et al. 2007). Another study revealed additional testis-biased duplicated genes with mitochondrial functions involved in oxidative phosphorylation (a.k.a. OXPHOS) in the same species (Tripoli et al. 2005). These data prompted us to explore whether gene duplication is a major mechanism underlying genetic innovation for mitochondrial functions in the male germline or in fertilization in Drosophila and to try to understand the processes driving these duplications.
In this study, we analyzed the entire set of nuclearly encoded mitochondrial genes in D. melanogaster, including RNA- and DNA-mediated duplication mechanisms. Our results point to a large contribution of gene duplication to mitochondrial functions during spermatogenesis and to strong selective forces (revealed by particular tendencies) underlying the origin and evolution of these genes. We found that nuclearly encoded mitochondrial gene duplicates that are retained in the D. melanogaster genome preferentially relocate from the parental copy (i.e., they are usually located in different Muller elements [Powell 1997] compared with their parental genes), strongly avoid the X chromosome and have testis-specific expression that differs from the parental copy. In addition, we found that nuclearly encoded mitochondrial gene duplicates are characterized by an overrepresentation of oxidative energy-producing functions. We discuss different hypotheses to explain our results and conclude that these patterns of duplicate retention and sex-biased expression might be explained as the outcome to the conflict that emerges if males, in particular their testis/sperm, have different functional needs than females. In the case of nuclearly encoded mitochondrial gene duplicates, we hypothesize the past existence of sexually antagonistic conflict related to mitochondrial function in Drosophila that was resolved through gene duplication and sex-specific expression of duplicated genes. In support of this postulated sexually antagonistic conflict related to mitochondrial functions in Drosophila, there have been studies (Rand et al. 2001) that found that mitochondrial–nuclear genotypes exhibit antagonistic sex-specific effects in this species.
Materials and Methods
Gene Family Annotation
Using the FlyMine webpage (Lyne et al. 2007), we found 535 genes with the gene ontology (GO) designation mitochondria in the D. melanogaster genome. Of these genes, 498 are nuclearly encoded, and the rest (37) are encoded by the mitochondrial genome. The protein sequences for the 498 nuclearly encoded mitochondrial genes were downloaded from FlyBase (http://flybase.org/) to perform BlastP searches (Altschul et al. 1997) against the annotated protein sequence databases of the 12 sequenced Drosophila species genomes (D. melanogaster, D. simulans, D. sechellia, D. yakuba, D. erecta, D. ananassae, D. pseudoobscura, D. persimilis, D. willistoni, D. mojavensis, D. virilis, and D. grimshawi). Duplicate gene annotation was conducted using computational approaches that were similar to the ones used in a previous study (Bai et al. 2007). Here, we annotate a gene family in all species where it is present if the alignment between two different sequences is as long as 50% of the length of each sequence and the amino acid identity level in the aligned region is at least 50% in 1 of the 12 species. The final orthology and paralogy relationships among similar sequences were assigned by means of phylogenetic analysis and synteny block comparison. This procedure allowed us to find 123 duplicate genes belonging to 53 families. Fourteen of these 123 genes were not annotated as mitochondrial genes at the time we downloaded the data (i.e., they were not included in the 498 gene set). Analyses of their protein sequences using the programs Mitoprot (Claros and Vincens 1996) and Predotar (Small et al. 2004) showed that: 8 of the 14 genes (CG17928, CG4393, CG6255, CG6888, Jafrac1, Jafrac2, l(2)03709, and Scs-alpha) bear mitochondrial targeting sequences (MTSs); and two other genes (CG33177 and CG33178) have no MTSs, probably because they are located in the mitochondrial outer membrane, similar to their parental gene (Mgstl). These analyses also indicated that Hsc70-3 is preprocessed in the endoplasmic reticulum (Predotar analysis predicts this with a probability of 99%) and then sent to the mitochondria (as are its four paralogous genes) and that CG17597 has 98.8% similarity with its paralogous gene CG17320, which has GO: mitochondria. In addition, the results of these analyses show that Rpt3 and Rpt3R have no MTSs but there are data suggesting the relationship of Rpt3 with testis mitochondria (Belote and Zhong 2009). Thus, we decided to consider these 14 genes as mitochondrial genes, resulting in a final data set of 512 nuclearly encoded mitochondrial genes (123 of which are duplicates that belong to 53 families). Finally, analyses predicted only two putative cases (out of the 123 duplicate genes) of subcellular relocation (Hsc70-3 and Jafrac2), in which the encoded proteins may be preprocessed in the endoplasmic reticulum before their relocation to the mitochondria. We still considered these two genes as nuclearly encoded mitochondrial genes.
Gene Duplication Events and Losses
The presence/absence of the 123 duplicate genes in the 12 Drosophila species (see below; supplementary table 1, Supplementary Material online), allowed us to assign the parental gene (see details below), infer when the duplication occurred and calculate the duplication rate for the D. melanogaster nuclearly encoded mitochondrial genes during the last 63 My (i.e., minimum evolutionary time for the divergence between the Drosophila and Sophophora subgenera; Tamura et al. 2004). Additionally, Heger and Ponting (2008) accurately assigned orthology and paralogy relationships for every D. melanogaster gene and for its predicted homologs in the rest of the Drosophila species, so we downloaded and analyzed their data (Heger and Ponting 2008) and found a total of 1,808 duplicate genes in D. melanogaster, that were clustered into 657 different gene families. Every family was individually analyzed manually to identify and date every duplication event and further estimate a gene duplication rate for the D. melanogaster lineage during the evolution of the Sophophora subgenus. We also analyzed Dfam data (Hahn et al. 2007) downloaded from http://www.indiana.edu/∼hahnlab/fly/DfamDB/ and found a total of 2,288 duplicate genes out of 14,449 nuclear genes for D. melanogaster.
When a putative loss was detected (i.e., an orthologous gene was not found in a particular species), two further analyses were performed to confirm this: 1) TBlastN searches against the whole nucleotide genome sequence and 2) check for gaps in the syntenic block where the orthologous gene in this species should have been found. When the TBlastN search did not produce additional hits and no gaps were found in the orthologous syntenic region, we then assumed that a loss had occurred in that species lineage.
RNA- and DNA-Mediated Duplicates
Following previous approaches (Bai et al. 2007; Meisel et al. 2009; Vibranovski, Zhang, and Long 2009), the closest related multiexonic genes within the gene families were inferred to be the parental genes of the single-exon genes that were assumed to have originated by retroduplication. When both copies proved to be multiexonic, detection of the parental gene was not trivial. Additional TBlastN searches in Insecta (“nr” and “wgs” databases) and phylogenetic and synteny block analyses were performed to infer the parental gene. In most of these cases, these analyses allowed us to determine which sequence of the pair was the parental gene. However, we could not establish the parental gene in seven families (CG2014/CG9172, CG18193/CG9603, CG31075/Aldh, Cyp12a5/Cyp12a4, Pepck/CG10924, Cyt-b5-r/CG17928, and l(2)37Cc/l(2)03709). Data related to these seven families were not included in analyses that required ascertaining the parental gene copy.
Relocation Pattern of the Duplicates
To analyze the relocation pattern between paralogous genes in a single species, it is necessary to know which gene is the parental gene and which is the duplicated gene as well as what Muller element they are linked to. The relocations (changes in location between the parental and duplicated gene) were grouped into three classes: 1) [X → A], meaning that the parental and duplicated gene are located in the X chromosome and in an autosomal arm, respectively; 2) [A → X], in which the relocation occurred from an autosomal arm to the X chromosome; and 3) [Ai → Aj], in which the relocation involves different autosomal arms. Once we assigned the relocation pattern for the orthologous genes in every species, we further established the original relocation pattern for a gene family (supplementary table 2, Supplementary Material online), which was inferred by applying the maximum parsimony principle to the relocation patterns of the single species. To test whether departures from the expected frequencies [X → A], [A → X], and [Ai → Aj] exist for the original relocation pattern, we first analyzed the chromosome distribution of the single copy and parental genes altogether. Because no significant deviation from the expected number of genes linked to each Muller element was found (X2 = 0.91; P = 0.97; degrees of freedom [df] = 5), the expectations of “all branches analysis” from Vibranovski, Zhang, and Long (2009) were used to compare our data.
Expression Data
For this analysis, we used Drosophila gene expression data compiled in FlyAtlas (Chintapalli et al. 2007). We considered a gene to have testis-biased expression when it is upregulated in that tissue (i.e., its expression level in testis is higher than its average expression level in the whole fly) and downregulated in the rest of the adult tissues (following the nomenclature from FlyAtlas). We did not find genes having an expression bias for any adult tissues other that the testis. We also looked for changes in gene expression and potential associations with the relocation pattern in other Drosophila species. Using data from Zhang et al. (2007), we analyzed whether our set of genes and their orthologs in D. simulans, D. yakuba, D. ananassae, D. pseudoobscura, D. mojavensis, and D. virilis present or do not present male-biased expression. From the data obtained using these six species, we inferred the genus expression pattern for each gene family (supplementary table 2, Supplementary Material online), which we defined as the pattern observed for the majority of the species that we examined, including at least D. mojavensis or D. virilis for old families. We then assigned the corresponding genus expression pattern for every original relocation pattern to ascertain whether a correlation between the two patterns exists when the entire Drosophila genus is considered.
Analyses of Tandem Duplicate Gene Expression
We considered that two paralogous genes are arranged in tandem if they do not have any type of overlap and no gene is located between them. We analyzed the Dfam database (Hahn et al. 2007; http://www.indiana.edu/∼hahnlab/fly/DfamDB) and found a total of 190 families of size 2 (genes) conforming to this definition. We inferred that a duplicate gene in a family exhibits testis-biased expression that did not exist in the parental gene when only one of the two genes has testis-biased expression (as defined in the previous section).
Evolutionary Rate Analyses
We used the branch models implemented in the Codeml program of the PAML package (Yang 2007) to calculate a single (dN/dS), or w ratio, for each gene family. A single evolutionary ratio (w0) was calculated for every tree type (supplementary fig. 1, Supplementary Material online). This model (model 0) assumes that every gene in the tree is evolving at the same evolutionary rate. Model 0 was used as the null hypothesis (i.e., parental and duplicated lineages evolve at equal evolutionary rates) to test a priori-defined alternative evolutionary models. The way that the alternative models were tested depended on the gene family and gene relationships (i.e., tree type). Tree type I (the most common type of tree, with 43 families belonging to this type; supplementary fig. 1, Supplementary Material online) describes a gene family with only two genes, the parental and the duplicated gene. For these families, only one alternative model (model 1) was tested, for which two different evolutionary rates were estimated, one for the parental (w0) and another for the duplicated gene (w1). Trees of type II represent gene families in which three different genes are present (supplementary fig. 1, Supplementary Material online). In this case, the first alternative model (model 1) that was tested estimates one evolutionary rate (w0) for the parental gene and another single evolutionary rate (w1) for the other two duplicated genes. If model 1 was significantly more likely than model 0, then model 2, in which one independent evolutionary rate was calculated for each gene (i.e., w0, w1, and w2; supplementary fig. 1, Supplementary Material online), was tested against model 1. The evolutionary models for tree types III, IV, and V were similarly tested as explained for tree type I and II (supplementary fig. 1, Supplementary Material online), allowing us to infer whether parental and duplicated genes evolve at different (dN/dS) ratios.
To avoid the overestimation of the (dN/dS) ratio caused by saturation of synonymous substitutions, the evolutionary rates were estimated using only the melanogaster subgroups species' sequences (Singh et al. 2008). The phylogenetic tree provided to PAML was ((D. melanogaster, (D. simulans, D. sechellia)), (D. yakuba, D. erecta)).
GO and Interactome Analysis
Our nuclearly encoded mitochondrial gene set was split in two different subsets: duplicate and nonduplicate gene subsets. We ran FatiGO (Al-Shahrour et al. 2004) implemented in Babelomics (Al-Shahrour et al. 2006) to test whether particular GOs were overrepresented or underrepresented in the duplicate gene subset compared with the nonduplicate gene subset. Briefly, for each GO, the program applies Fisher’s exact test for 2 × 2 contingency tables and returns adjusted P values based on three different correction methods for multiple testing (for further details, see Al-Shahrour et al. 2004). We considered significant GO terms to be those with adjusted P values ≤ 0.01.
We extracted the functional network information for 144 mitochondrial genes from Costello et al.’s (2009) data and generated a graph with 952 interactions. We used UVCLUSTER (Arnau et al. 2005) to calculate the secondary distances among the 144 proteins and to generate a hierarchical representation (i.e., a tree) of the interaction graph. We then used TreeTracker (Marco and Marín 2007) to test whether any cluster from the tree had a significant enrichment of duplicate genes.
Results
Genomic Features of the Nuclearly Encoded Mitochondrial Genes
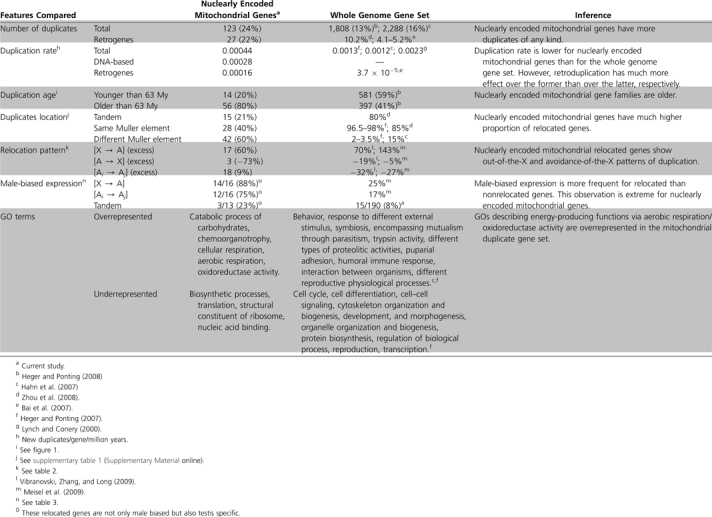
The complete set of 498 nuclearly encoded genes with mitochondria GO for D. melanogaster was downloaded from FlyMine (Lyne et al. 2007). After BlastP searches using these genes against the D. melanogaster genome, an additional set of 14 computationally verified (see Materials and Methods) nuclearly encoded mitochondrial genes was also included in our initial set. All of these 512 genes have been predicted or suggested to be mitochondrial in different studies, and we predicted a mitochondrial-targeting signal for most of them (Hartl et al. 1989) using two different programs, Mitoprot (Claros and Vincens 1996) and Predotar (Small et al. 2004; data not shown). Out of these 512 genes, 123 had enough protein similarity to at least one other gene to be clustered into 53 gene families, as described in the Materials and Methods. This leads to an estimate of 24% (123/512) of genes belonging to a gene family (table 1).
Table 1.
Distinctive Features of Nuclearly Encoded Mitochondrial Duplicates Compared with Whole Genome Duplicate Gene Set
 |
We also analyzed the congruence of our FlyMine GO retrieval with the MitoDrome database (Sardiello et al. 2003). We observed that 94% of MitoDrome database (i.e., 271 genes) was included in our gene set. This result shows that our mitochondrial data set is congruent with the MitoDrome database but also more comprehensive.
Phylogenetic analyses and synteny block comparisons allowed us to detect the orthologous genes of the 123 duplicate genes in 11 sequenced Drosophila genomes (see Materials and Methods). We inferred that a total of 70 duplication events gave rise to the 123 duplicates. Surprisingly, the vast majority of duplications (60%) involve two different Muller elements (i.e., indicating relocation of the duplicated gene), and a high percentage (39%) could be concluded to have originated by retroposition with certainty (table 1). These results were totally unexpected because most lineage-specific duplication occurs within the same Muller element in Drosophila (85–98%; Hahn et al. 2007; Heger and Ponting 2007) or by tandem duplication (about 80% on average for D. melanogaster and D. yakuba lineages; Zhou et al. 2008), and retrogenes are only a small fraction (4–10%; table 1) of duplicate genes in Drosophila (Bai et al. 2007; Zhou et al. 2008).
Next, we dated all the 70 mitochondrial gene duplication events (fig. 1) by analysis of the presence/absence of genes in other sequenced genomes (see Materials and Methods; supplementary table 1, Supplementary Material online). We found that a total of 14 duplication events (i.e., 20%) took place during the last 63 My, in the lineages leading to D. melanogaster (Tamura et al. 2004). Fifty-six duplication events were observed to have taken place before the diversification of the Drosophila genus (fig. 1), which is in agreement with previous observations made for OXPHOS genes in the Insecta (Porcelli et al. 2007). However, when we analyzed the 657 gene families contained in Heger and Ponting’s (2007) data using the same procedure, we calculated 581 duplication events (i.e., 59%) within the last 63 My of the 978 duplication events inferred (fig. 1). These ratios are significantly different (Fisher’s exact test; P = 5 × 10−13), revealing that many duplicated genes are young, as was previously described in Heger and Ponting (2007), though this is not true for mitochondrial-duplicated genes (for individual comparisons, see table 1).
FIG. 1.—

Proportion of duplication events inferred to happen in a particular branch (nuclearly encoded mitochondrial genes/nuclear genes). Scale approximately reflects the evolutionary time measured in million years, based on Tamura et al. (2004).
Tripoli et al. (2005) analyzed 78 OXPHOS genes in D. melanogaster and D. pseudoobscura that were orthologous to humans and estimated an OXPHOS gene origination rate of ∼0.0978 (4.5/46) per million years (i.e., approximately one new OXPHOS gene every 10 My). It is worth mentioning that their estimation for the duplication rate, like ours, does not take into account the rate of gene loss, which is higher than the duplication rate in Drosophila (Hahn et al. 2007). We detected 14 duplication events in the lineages leading to D. melanogaster after the split of the subgenera Drosophila and Sophophora (i.e., at least 63 Ma; Tamura et al. 2004; see supplementary table 1, Supplementary Material online), five of which are OXPHOS genes (i.e., CG17856, CG11423, CG31477, CG12027, and CG33503). This result leads to an estimate of 0.0794 OXPHOS gene duplicates per million years (5/63). It is likely that this discrepancy is due to the fact that these other authors dated the split between the melanogaster and obscura groups to be only 46 My old (no reference was given), whereas it is probably almost 10 My older (i.e., 54.9 My old; Tamura et al. 2004). Correcting this date, their results lead to a new estimate of 0.0820 new OXPHOS genes per million years, which is very close to our estimate of 0.0794 new OXPHOS genes per million years.
Altogether, our analyses show that there is a significantly higher relocation bias observed for nuclearly encoded mitochondrial-duplicated genes, a much lower duplication rate of mitochondrial genes compared with the rest of the nuclear genes, with the exception of retrogenes, and a higher long-term retention of mitochondrial-duplicated genes compared with the nonmitochondrial-duplicated genes.
Avoidance of the X Chromosome by Nuclearly Encoded Mitochondrial Genes in Drosophila
Previous studies reported that the relocation pattern (i.e., change in location between parental and duplicated gene) of duplicate genes exhibits a nonrandom distribution in Drosophila (Betrán et al. 2002; Bai et al. 2007; Meisel et al. 2009; Vibranovski, Zhang, and Long 2009). These studies reported that relocation from the X chromosome to any autosomal arm, [X → A], is significantly more frequent than expected by chance, whereas any other relocation pattern ([Ai → Aj] and [A → X]), is significantly underrepresented. All of these studies concluded that selective forces might be operating across the whole Drosophila genome, which would favor the [X → A] relocation pattern (see table 1).
We analyzed the relocation pattern in each Drosophila species genome to infer the ancestral gene locations and original relocation pattern (see Materials and Methods and supplementary table 1, Supplementary Material online). Because single copy and parental mitochondrial genes are randomly distributed in the D. melanogaster genome (see Materials and Methods), we used the same expectations for the location of RNA- and DNA-mediated duplicated genes as was used in Vibranovski, Zhang, and Long (2009). These expectations were calculated considering the number of genes in a donor chromosome and the amount of euchromatin in the acceptor chromosome, following Betrán et al. (2002), and assuming that there is dosage compensation in the germline in the case of RNA-mediated duplication (Betrán et al. 2002). We used the averaged expectation for the total duplicated genes (i.e., weighted average of the expectations for RNA- and DNA-mediated duplicated genes considering the frequency of the two types of duplicates). We tested whether the observed relocation frequencies are equal to the expected ones (table 2; note that four relocations were not included because we could not infer the direction of the relocation). We found that [X → A] and [Ai → Aj] relocations are 60.4 and 9.1% more frequent than expected (P = 0.0108 after applying Bonferroni’s correction), respectively, whereas [A → X] relocations are highly underrepresented (72.5% less than expected; table 2). We observed similar results for the independently analyzed sets of retrogenes and DNA-based duplicates, although due to the smaller sample size, the trend was not significant (not shown). Our result for [X → A] relocations is in agreement with, but slightly lower than, the result described in previous whole-genome analyses (table 1). Nevertheless, unlike these previous studies (Betrán et al. 2002; Bai et al. 2007; Meisel et al. 2009; Vibranovski, Zhang, and Long 2009), we found a moderate excess of [Ai → Aj] relocations and a much larger paucity of [A → X] relocations (table 1). We conclude that in addition to an out-of-the-X pattern, our results for mitochondrial duplicate genes suggest an avoidance-of-the-X pattern.
Table 2.
Analysis of Relocation Patterns
| Relocation Pattern | |||
| X → A | A → X | Ai → Aj | |
| Expected (No.) | 10.6 | 10.9 | 16.5 |
| Observed (No.) | 17 | 3 | 18 |
| Excess (%) | 60.4 | −72.5 | 9.1 |
| Gadj = 11.25; df = 2, P = 0.0036; *P = 0.0108 | |||
NOTE.—*After applying Bonferroni’s correction. X: X chromosome; A, autosome; excess = ([O – E]/E) × 100.
Complementary Expression between Parental and Testis-Specific Duplicated Genes
Previous studies have reported that functional retrogenes often show testis-specific or testis-biased expression not only in Drosophila (Betrán et al. 2002; Bai et al. 2007) but also in mammals (Emerson et al. 2004), including humans (Emerson et al. 2004; Vinckenbosch et al. 2006). At least in Drosophila (Meisel et al. 2009; Vibranovski, Zhang, and Long 2009), this is also true for DNA-based duplicates, and it has been inferred that selective forces might be driving these patterns (Betrán et al. 2002; Emerson et al. 2004; Bai et al. 2007; Ellegren and Parsch 2007; Vibranovski, Lopes, et al. 2009; Vibranovski, Zhang, and Long 2009).
We wanted to determine whether this characteristic also holds true for nuclearly encoded mitochondrial genes. To do so, we used Chintapalli et al.’s (2007) data and analyzed the expression pattern of the 123 mitochondrial duplicate genes in D. melanogaster (see Materials and Methods). Our first observation was that all nuclearly encoded duplicates having biased expression are testis-biased (i.e., we did not find any gene having biased expression for any other tissue; supplementary table 3, Supplementary Material online). Hence, we decided to split the data into families with and without genes showing testis-biased expression. Duplicated genes in both groups were observed to have a much lower expression level in the whole fly than the parental genes, which are more highly expressed (whole-body comparisons in fig. 2). Detailed analysis of nuclearly encoded mitochondrial-duplicated genes with testis-biased expression revealed that in fact, they are not expressed in the rest of the tissues of the fly (the average expression of this set of genes in the whole fly excluding testes was found to be very low, 6.43 ± 2.33). This effect could be clearly seen when we compare the level of expression between mitochondrial paralogs in the ovary. In the ovary (as well as in any other nontestis adult tissues; data not shown), nuclearly encoded mitochondrial-duplicated genes with testis-biased expression are not expressed at all (0.36 ± 0.26), whereas the respective parental genes are highly expressed (827.09 ± 105.23; P = 2 × 10−14; Wilcoxon rank sum test). However, we observed that nuclearly encoded mitochondrial-duplicated genes with testis-biased expression have an extremely high level of expression in testis (1,046 ± 227; i.e., within the 5% of maximum for expressed genes in testis), whereas their respective parental genes have a much lower level of expression in the same tissue (289.44 ± 54.95; t = 5.6261, df = 71, P = 3 × 10-9; Wilcoxon rank sum test). We refer to this pattern of expression between mitochondrial paralogous genes (i.e., high expression of duplicated genes in testis but very low expression in other tissues where the parental genes are highly expressed) as complementary. As shown in figure 2, there is no complementary expression in mitochondrial families that do not contain genes with testis-biased expression.
FIG. 2.—

Averaged level of expression (±standard error of the mean) of parental (black bars) and duplicated (gray bars) genes in testis (T), ovary (O), and whole D. melanogaster body (W). The data were also divided in families with (right) and without (left) genes with testis-specific expression. Dotted gray bars indicate whole genome average level of expression in different tissues. Asterisk: P < 0.0001.
We wanted to go further and elucidate whether the complementary expression observed between nuclearly encoded mitochondrial paralogs is also observed in different areas of the testis. A testis in Drosophila can be divided into three regions with respect to the relative abundance of cells in mitotic (apical part of the testis), meiotic (middle part of the testis), and postmeiotic (posterior region of the testis, in which mature sperm are located) phases. For this, we used the recently published data from Vibranovski, Lopes, et al. (2009). These authors analyzed the expression of most of D. melanogaster genes in detail in the aforementioned regions of the testis. As before, we separately analyzed two groups of mitochondrial gene families: those in which at least one gene has testis-specific expression and families in which none of the members have testis-specific expression (fig. 3). We observed that genes with testis-specific expression and their parental genes are expressed at different levels in different testis region (P = 6.4570 × 10−6 and P = 0.0132, respectively; analysis of variance test; fig. 3). However, they show inverted trends of expression, whereas duplicated genes (testis-specific; black squares, fig. 3) and parental genes (nontestis specific; gray squares, fig. 3) have the same level of expression in mitotic cells (t = 0.8602, df = 62, P = 0.3930), their respective expression levels become complementary as the germ cells approach the final state of mature sperm (t = 5.8451, df = 62, P < 0.0001 for meiotic cells; t = 6.4718, df = 62, P < 0.0001 for postmeiotic cells). In addition, the expression profile described for testis-specific genes is the same for [X → A] and [Ai → Aj] duplicated genes (supplementary fig. 3, Supplementary Material online), indicating that the same evolutionary forces are likely to be working on both types of relocated genes. On the contrary, for families in which none of the members have testis-specific expression, no differences in expression were detected either among the different regions in the testis or between paralogous genes (black and gray diamonds, fig. 3).
FIG. 3.—

Average expression level (±standard error of the mean) of duplicates in the mitotic (Mit), meiotic (Mei), and postmeiotic (Post Mei) cells of the D. melanogaster testis. The data set was divided in families with (squares) and without (diamonds) testis-specific genes. Black, duplicated genes; Gray, parental genes.
We conclude that the nuclearly encoded mitochondrial duplicates that have complementary expression in adult tissues also exhibit complementary expression in different spermatogenesis stages, showing important differences between meiosis and postmeiosis, in which the new genes are highly expressed, but parental genes are expressed at low levels. This trend holds true and is of equal intensity regardless of the relocation patterns (i.e., [X → A] and [Ai → Aj]).
Most Relocated Nuclearly Encoded Mitochondrial Genes Exhibit Testis-Specific Expression in D. melanogaster
We further explored whether the expression and relocation patterns described in the previous sections are correlated. Our analyses revealed a significant difference between relocated and nonrelocated genes in their frequency of testis-specific expression: whereas 83% of the relocated duplicated genes have testis-specific expression, only 38% of nonrelocated (i.e., located in the same Muller element) duplicated genes are testis-specific (table 3 and supplementary table 3 [Supplementary Material online]; Fisher’s exact test; P = 0.0004 after applying Bonferroni’s correction). We also analyzed tandem duplicates (see Materials and Methods) and found that only 23% of these families contain a duplicate gene that shows testis-specific expression, whereas the other gene is expressed in the whole adult body (table 1). When the whole-genome duplicate gene set was analyzed for comparison, we found that only 8% of tandem duplicates show this new testis-biased expression (table 1), whereas 21% of relocated genes show testis-biased expression (Meisel et al. 2009; table 1). These results indicate that relocation correlates with testis-specific expression and that this genomic feature is extreme for nuclearly encoded mitochondrial genes.
Table 3.
Correlation between Relocation and Testis-Specific Expression
| Expression Pattern | ||
| Test | Testisa | Nontestisb |
| Relocation vs. no relocation test | ||
| Relocation | 29 | 6 |
| No relocation | 6 | 10 |
| P = 0.0002; *P = 0.0004 | ||
| [X → A] vs. [Ai → Aj] test | ||
| X → A | 14 | 2 |
| Ai → Aj | 12 | 4 |
| P = 0.0860; *P = 0.1719 | ||
NOTE.—*After applying Bonferroni’s correction. X: X chromosome; A, autosome.
The parental gene (which is nontestis specific) generates a copy that express testis specifically.
Both, parental and duplicated genes, are nontestis specific.
We next analyzed independently relocated genes to test whether duplicates with an [X → A] pattern have a higher probability than duplicates with an [Ai → Aj] pattern of being expressed testis specifically. We found that this probability is not significantly different between the two types of relocations (table 3 and supplementary table 3 [Supplementary Material online]; Fisher’s exact test; P = 0.1719 after applying Bonferroni’s correction). Furthermore, relocated genes achieve similar and extremely high (see previous section) levels of expression regardless of the relocation pattern (supplementary fig. 2, Supplementary Material online). Interestingly, Meisel et al. (2009) also did not find differences between the proportions of testis-expressed genes between these relocation patterns when the whole nuclear gene set was analyzed (table 1).
Testis-Specific Mitochondrial Duplicates Have Preserved Their Pattern of Expression
How the pattern of expression of new genes evolves remains largely a mystery, especially for retrogenes because they do not inherit the promoter regions from their parental genes. Previous studies have reported that functional retrogenes often show testis-specific expression not only in Drosophila (Betrán et al. 2002; Bai et al. 2007) but also in mammals (Emerson et al. 2004), including humans (Emerson et al. 2004; Vinckenbosch et al. 2006). The “out-of-the-testis” hypothesis suggests that expression in testis is the first step for functional retrogenes (Vinckenbosch et al. 2006). This initial testis-specific function of new retrogenes would allow them to be preserved in the organism when they were initially inserted to later evolve new or wider expression patterns.
We wanted to test whether duplicated genes that originally show testis-specific expression have conserved that expression pattern in other Drosophila species or whether that expression pattern has changed over time. As described above for D. melanogaster, we looked for changes in the expression pattern between parental and duplicated genes associated with their relocation pattern in other Drosophila species. We analyzed data from Zhang et al. (2007) to examine whether the D. melanogaster orthologous genes in D. simulans, D. yakuba, D. ananassae, D. pseudoobscura, D. mojavensis, and D. virilis, have sex-biased expression (supplementary table 2, Supplementary Material online) to infer their genus expression pattern (see Materials and Methods). Although Zhang et al. (2007) analyzed gene expression for whole fly, we assumed that a mitochondrial gene with male-biased expression has testis-biased expression and potentially testis-specific expression for three reasons: first, it is well known that male-biased expression is mostly due to testis expression in Drosophila (Parisi et al. 2004; Ellegren and Parsch 2007; Zhang et al. 2007); second, several studies have suggested that, at least in mice (Yang et al. 2006, Mank et al. 2008) and chicken (Mank et al. 2008), expression differences between sexes are mostly generated by particular tissues instead of by differences in broader expression patterns (Ellegren and Parsch 2007); and third, because all mitochondrial genes in D. melanogaster that are testis biased are, in fact (as we describe above), testis specific.
Assuming this, we could infer acquisition (i.e., not inherited from the parental gene) of testis-specific/testis-biased expression for 29 and 22 relocated duplicates in D. melanogaster and in the Drosophila genus, respectively (supplementary table 2, Supplementary Material online). For six and eight genes, relocation did not involve testis-specific/testis-biased expression in D. melanogaster and in the Drosophila genus, respectively. We tested whether the bias for testis-specific expression observed for relocated genes in D. melanogaster holds for the rest of the Drosophila genus. As we found a lack of significant differences (P = 0.1087; Fisher’s exact test), we concluded that the bias in the expression pattern observed for relocated duplicates in D. melanogaster is also present with the same intensity in the rest of the species of the Drosophila genus. This means that, once relocated duplicates acquire testis-specific/testis-biased expression, they maintain this specific pattern of expression. Our results do not support the out-of-the-testis hypothesis, and they indicate that it is likely that these new genes emerge and are maintained in the genome because of their testis-specific function. Testis-specific data need to be provided in species other than D. melanogaster to further support this conclusion.
Higher Evolutionary Rate of Duplicated Genes with Testis-Specific Expression
Gene duplication increases gene expression diversification and that diversification seems to correlate positively with different measures of the rate of coding region evolution (Li et al. 2005 and references therein). For each gene family, we used the Codeml program implemented in the PAML package (Yang 2007) to test equal rates of protein evolution ([dN/dS] ratio) between parental and duplicated genes (see Materials and Methods). To avoid estimation errors induced by saturation at synonymous sites (see Materials and Methods), we only used sequences from species belonging to the melanogaster subgroup (Singh et al. 2008). We found significant differences between parental and derived genes in 27 families (supplementary table 4, Supplementary Material online). Figure 4A shows the average value of (dN/dS) for parental and duplicated genes. Although both type of genes are evolving at low rates, duplicated genes evolve almost twice as fast as parental genes ([dN/dS] of 0.073 and 0.044, respectively; P = 0.0032; Wilcoxon rank sum test, after applying Bonferroni’s correction). We further independently analyzed gene families with and without gene expression differentiation (i.e., gene families in which the parental gene is broadly expressed, but the duplicated gene has testis-specific expression in Drosophila, and gene families in which all members have broad expression). We observed (fig. 4B) that duplicates with testis-specific expression are evolving 2.2 times faster than their parental genes ([dN/dS] of 0.082 and 0.037, respectively; P = 5 × 10−5; Wilcoxon rank sum test, after applying Bonferroni’s correction). However, these differences are not observed in families in which both parental and duplicated genes have broad expression patterns ([dN/dS] of 0.066 and 0.056 for parental and duplicated genes, respectively; P = 1; Wilcoxon rank sum test, after applying Bonferroni’s correction).
FIG. 4.—

Average evolutionary rates (±standard error of the mean) for duplicate (D) and parental (P) lineages. In (A), we show the results compiled for all families for which we could establish the parental lineage. In (B), the same data set is divided in families with and without testis-specific genes to show that only duplicated lineages that evolved testis-specific expression have higher evolutionary rates than their respective parental lineages.
We conclude that only those duplicated genes that show testis-specific expression patterns are evolving faster than their respective parental genes but still under purifying selection.
Nuclearly Encoded Mitochondrial Duplicates Are Enriched for Energy Functions
We used FatiGO (Al-Shahrour et al. 2004) implemented in Babelomics (Al-Shahrour et al. 2006) to test whether there is a functional enrichment in the nuclearly encoded mitochondrial duplicate gene set compared with the rest (nonduplicate) of the nuclearly encoded mitochondrial genes. Briefly, the program assigns the corresponding GO terms to each gene included in both gene sets and tests if there is an overrepresentation and/or underrepresentation of particular GOs in one of the sets with respect to the other one. Table 4 shows the results obtained by FatiGO for GO:biological process and GO:molecular function. For high GO levels (levels three to five in GO:biological process and level three for GO:molecular function), representing general processes or functions, the set of duplicate genes is significantly enriched in energy-producing functions. For instance, 26% of the nuclearly encoded mitochondrial genes were classified into the catabolic process ontology category (biological process: level 3, GO: 0009056), 77% of which (i.e., 20/26) are duplicates, whereas 23% are single copy genes. This difference remained significant after correcting for multiple testing (adjusted P value = 3.33 × 10−3; see Materials and Methods). In addition, the results obtained for lower levels (level six and seven for GO:biological process; table 4), show carbohydrate catabolism and aerobic respiration as the overrepresented GOs related to energy production. Interestingly, GOs related to macromolecular biosynthesis, ribosome-related function, and nucleic acid binding are significantly underrepresented in the duplicate gene set (table 4).
Table 4.
Overrepresented and Underrepresented Gos in the Set of Nuclearly Encoded Mitochondrial Duplicate Genes Compared With the Nuclearly Encoded Mitochondrial Nonduplicate Gene Set
| Ontology Level | Ontology | Adjusted P Valuea | Duplicate Versus Nonduplicate Genesb |
| Overrepresented ontologies in duplicate genes | |||
| Biological process | |||
| Level 3 | Catabolic process (GO:0009056) | 3.35 × 10−3 | 20.19% vs. 5.99% |
| Level 4 | Carbohydrate metabolic process (GO:0005975) | 1.54 × 10−5 | 21.57% vs. 3.53% |
| Generation of precursor metabolites and energy (GO:0006091) | 7.30 × 10−4 | 51.96% vs. 27.92% | |
| Cellular catabolic process (GO:0044248) | 1.46 × 10−3 | 18.63% vs. 4.59% | |
| Level 5 | Chemoorganotrophy (GO:0015980) | 6.00 × 10−6 | 25.88% vs. 3.95% |
| Cofactor catabolic process (GO:0051187) | 6.69 × 10−4 | 16.47% vs. 2.37% | |
| Level 6 | Cellular carbohydrate metabolic process (GO:0044262) | 8.28 × 10−7 | 34.38% vs. 4.85% |
| Acetyl-CoA metabolic process (GO:0006084) | 1.36 × 10−4 | 21.88% vs. 2.91% | |
| Coenzyme catabolic process (GO:0009109) | 1.36 × 10−4 | 21.88% vs. 2.91% | |
| Cellular respiration (GO:0045333) | 1.36 × 10−4 | 21.88% vs. 2.91% | |
| Dicarboxylic acid metabolic process (GO:0043648) | 9.38 × 10−4 | 12.50% vs. 0.49% | |
| Level 7 | Aerobic respiration (GO:0009060) | 1.46 × 10−3 | 26.92% vs. 4.23% |
| Acetyl-CoA catabolic process (GO:0046356) | 1.46 × 10−3 | 26.92% vs. 4.23% | |
| Tricarboxylic acid cycle intermediate metabolic process (GO:0006100) | 5.06 × 10−3 | 15.38% vs. 0.70% | |
| Molecular function | |||
| Level 3 | Oxidoreductase activity (GO:0016491) | 6.61 × 10−3 | 44.68% vs. 25.33% |
| Underrepresented ontologies in duplicate genes | |||
| Biological process | |||
| Level 3 | Biosynthetic process (GO:0009058) | 5.43 × 10−4 | 12.5% vs. 34.15% |
| Level 4 | Cellular biosynthetic process (GO:0044249) | 9.11 × 10−4 | 12.75% vs. 33.57% |
| Cellular macromolecule metabolic process (GO:0044260) | 2.28 × 10−3 | 10.78% vs. 28.98% | |
| Protein metabolic process (GO:0019538) | 2.34 × 10−3 | 11.76% vs. 30.39% | |
| Level 5 | Macromolecule biosynthetic process (GO:0009059) | 7.10 × 10−5 | 2.35% vs. 23.72% |
| Level 6 | Translation (GO:0006412) | 8.50 × 10−7 | 0% vs. 28.64% |
| Molecular function | |||
| Level 3 | Structural constituent of ribosome (GO:0003735) | 4.99 × 10−4 | 0% vs. 13.49% |
| Nucleic acid binding (GO:0003676) | 2.13 × 10−3 | 0% vs. 10.53% | |
Only differences significant at one per cent are selected.
Percentage of genes in duplicate and nonduplicate gene sets having this particular GO.
Because functional networks reflect the functional structure of the cell (Cusick et al. 2005), we also analyzed the functional interaction network for D. melanogaster mitochondrial genes. From Costello et al.’s (2009) data, we extracted a network of 144 mitochondrial proteins (22 of which are duplicates) with 952 interactions. We calculated the secondary distance tree of the network using the program UVCLUSTER (Arnau et al. 2005) and then we used the program TreeTracker (Marco and Marín 2007) to compare the hierarchical representation of the interactome with the dichotomic partition (duplicate or nonduplicate) of the mitochondrial gene set. This procedure allowed us to detect the OXPHOS protein cluster and a small cluster represented by protein-folding proteins as the only groups with a significant overrepresentation of duplicate genes (supplementary fig. 4, Supplementary Material online).
Both types of analyses (GO and functional interaction network) detect a significant overrepresentation of oxidative energy production functions in the duplicate gene set compared with the rest of the nuclearly encoded mitochondrial genes. We conclude that mitochondrial-duplicated genes holding these functions (at least 52% of them because some genes in some ontology levels may not be represented in lower levels; table 4) are more successful in being maintained in the Drosophila genome (i.e., favored by selection). Interestingly, these overrepresented GOs in mitochondrial-duplicated genes are not detected (see table 1) when analyzing the whole genome gene set (Hahn et al. 2007; Heger and Ponting 2007), revealing that, when separated per GO, mitochondrial-duplicated genes are only a small fraction of all duplicates, but they represent an interesting set of genes, as shown in this study.
Discussion
Mitochondria Are Different in Testis and Sperm in Drosophila
We have analyzed the entire set of nuclearly encoded mitochondrial genes in D. melanogaster and found that mitochondrial genes, although having much lower duplication rates compared with the rest of the nuclear genes (with the exception of retrogenes), exhibit a higher proportion of duplicate genes and of older duplicate genes, which is likely due to these genes persisting longer than other duplicated genes (table 1). Reduced evolvability caused by functional stoichiometric constraints has been suggested to explain the low duplication rate of a set of Drosophila OXPHOS genes (Tripoli et al. 2005). We also found that mitochondrial duplicates have several distinctive and revealing duplication patterns: 1) a high rate of relocation except to the X chromosome; 2) a high rate of duplication out of the X though we found less of an excess of this than observed for the whole genome relocation analyses (see table 1); and 3) no underrepresentation of [Ai → Aj] duplicates, which differs from what was found in previous studies (see table 1). Detailed analyses of the gene expression levels in adult tissues showed that relocated genes are expressed testis specifically.
Previous studies have reported that relocated duplicates in the Drosophila genome (both, RNA- and DNA-based) show a significant excess of an [X → A] relocation pattern and that they show testis-biased expression (Betrán et al. 2002; Bai et al. 2007; Meisel et al. 2009; Vibranovski, Zhang, and Long 2009; see table 1). Although it has been substantiated that selection is involved in the association between relocation and testis-biased expression (Vinckenbosch et al. 2006; Fontanillas et al. 2007; Vibranovski, Zhang, and Long 2009), this association might often be facilitated by the relocation mechanism (e.g., retrocopies might insert close to germline genes or often be transcribed in testis; Loppin et al. 2005; Vinckenbosch et al. 2006; Bai et al. 2007). Our analyses show that nuclearly encoded mitochondrial duplicate genes have a much higher rate of relocation (at least 60% of the duplication events involved relocation) compared with that observed for the total set of nuclear gene duplicates. In addition, most relocated duplicates (83%) have testis-biased expression in D. melanogaster, that is, much more often than the average for the whole set of nuclear gene duplicates or tandem duplicates (table 1). When we analyzed the expression pattern of tandem duplicate genes and compared it with that for relocated duplicates across the whole genome, we found that only 8% of tandem duplicates acquire testis-biased expression, whereas 21% of relocated genes show testis-biased expression (Meisel et al. 2009; table 1). We infer that there has been selection to preferentially retain relocated duplicates because they often exhibit testis-specific expression. Mitochondrial genes might be taking special advantage of this not yet completely understood genomic feature.
Additionally, there could also be some selection for relocated duplicates because they might be sheltered from homogenizing mechanisms, such as nonallelic gene conversion (Casola et al. 2010). Under this hypothesis, if a functionally different protein is required in the testis, there will be selection for duplicates that relocate away from their parental gene, facilitating differentiation. Thus, differentiation could be an additional target of selection for the nuclearly encoded mitochondrial-duplicated genes.
The testis-specific expression is also characterized by the complementary expression pattern observed between relocated and parental genes (fig. 2). Thus, although testis-specific nuclearly encoded mitochondrial genes are not expressed in any other tissue apart from testis, the respective parental genes are expressed at low levels in testis but at high levels in the rest of the somatic tissues and the female germline. Interestingly, this is true for both [X → A] and [Ai → Aj] relocated genes (supplementary fig. 2, Supplementary Material online). In addition, further analyses of gene expression data in six other Drosophila species allowed us to determine that these genes retained a male-biased expression pattern through time. It is well known that male-biased expression is mostly due to testis expression in Drosophila (Parisi et al. 2004; Ellegren and Parsch 2007; Zhang et al. 2007), and other studies have suggested that expression differences between sexes, at least in mice (Yang et al. 2006, Mank et al. 2008) and chicken (Mank et al. 2008), are mostly generated by particular tissues instead of by differences in broader expression patterns (Ellegren and Parsch 2007). These results and the fact that most nuclearly encoded mitochondrial relocated genes are testis-specific in D. melanogaster (current study) indicate to us that our analyses are probably revealing testis-specific functions for the mitochondrial relocated genes that have been retained through time.
These functions are probably relevant in mature sperm and may not be supplied by their parental genes because we found a positive correlation between sperm developmental phase and the level of expression for relocated genes but a negative correlation for parental genes (fig. 3). Interestingly, both types of relocation patterns (i.e., [X → A] and [Ai → Aj]), contribute with the same intensity to the differences in expression between parental and duplicated genes during sperm development (supplementary fig. 3, Supplementary Material online). In agreement with this prediction, we found that proteins encoded by 19 mitochondrial-duplicated genes are present in the sperm proteome (Dorus et al. 2006), whereas only six proteins encoded by parental genes were found.
Finally, our analyses of evolutionary rates reveal that mitochondrial duplicates with testis-specific expression evolve faster than parental genes but are still under strong purifying selection ([dN/dS] = 0.082; 95% confidence interval [CI]: 0.0640 – 0.0992). However, they seem to evolve at similar rates ([dN/dS] = 0.104; 95% CI: 0.093 – 0.114) to those observed for testis-specific genes in general (Haerty et al. 2007). These data are in agreement with those genes having important male functions. These differences in the evolutionary rates may be due to a relaxation of functional constraints or positive selection (Haerty et al. 2007).
What Selective Forces Might Explain This Excess of Testis Mitochondrial Duplicates?
Different hypotheses have been suggested to explain the duplication pattern of male-biased genes (Betrán et al. 2002; Bai et al. 2007; Meisel et al. 2009; Vibranovski, Zhang, and Long 2009) and/or demasculinization of the X chromosome (Parisi et al. 2003; Sturgill et al. 2007) in Drosophila. The X chromosome inactivation hypothesis postulates that genes required during the meiotic phase of spermatogenesis will be selected to be located in an autosome (i.e., an out-of-the-X pattern) because some X-borne genes in D. melanogaster seems to have a reduced level of expression in that phase (meiotic sexual chromosome inactivation or MSCI; Hense et al. 2007; Vibranovski, Lopes, et al. 2009). Another hypothesis suggests that because dosage compensation in Drosophila occurs through hypertranscription of the X chromosome in males and there could be a limit to an additional increase of expression of X-linked genes in males (Vicoso and Charlesworth 2009), highly expressed genes will evolve male-biased expression more often when located in autosomes than when located in the X chromosome. Consequently, relocation from the X chromosome to the autosomes might also be beneficial to those genes that need to be highly expressed in males.
Some of our observations are incompatible with these hypotheses, and this helped us to rule them out as being completely explanatory. In the case of mitochondrial duplicates, we think that our results cannot be completely explained by the MSCI hypothesis because of the presence of many autosome-to-autosome duplicates with testis- and sperm-specific expression (supplementary figs. 2 and 3, Supplementary Material online). This observation, as well as the finding that parental genes (even autosomal ones) are expressed at a lower level in testis (fig. 2) and that duplicated genes are only needed during spermatogenesis (fig. 3), can also not be explained by the dosage compensation hypothesis. In addition, neither of these hypotheses explains the overrepresentation of energetic functions in the duplicate set. Finally, the previously mentioned fact that parental genes (even autosomal ones) are expressed at a lower level in testis does not support a hypothesis that involves only selection for an increase in the level of expression.
Thus, our hypothesis is that because of the way spermatogenesis proceeds, the way sperm are formed and the way fertilization occurs, testes might require a special set of mitochondrial genes that might not be the most beneficial for the soma and/or the female germline. Spermatogenesis in Drosophila is a complex process that requires coordination of cell division cycles and morphological changes to produce mature sperm (Fuller 1993), in which mitochondria undergo some of the most dramatic changes in morphology of any Drosophila cell type. In primary spermatocytes, multiple mitochondria are found near the nuclear membrane, but after the meiotic divisions occur, these mitochondria fuse, forming the characteristic spherical Nebenkern (onion-stage) structure. The large fused mitochondrial structure splits in half as the spermatids mature, and the two derivatives of the mitochondria elongate with the developing axoneme. Adenosine triphosphate production from the remnants of this mitochondrial structure provides energy for the movement of mature sperm during fertilization (Fuller 1993). In particular, our data show that oxidative energy-producing functions are overrepresented among the mitochondrial duplicate genes compared with the rest of the (single copy) nuclearly encoded mitochondrial genes (at least 52% of the mitochondrial duplicate genes are related to energy-producing functions). It has been reported that human mitochondria accumulate a high number of mtDNA mutations after sperm differentiation (Reynier et al. 1998), probably due to increased activity of the mitochondrial energy-producing complexes (Ruiz-Pesini et al. 1998). Interestingly, a 10% reduction of the maximum membrane potential reduces the total reactive oxygen species (ROS) production by 90% (Andreyev et al. 2005), that is, working over the maximum threshold of energy production not only generates more energy but also higher levels of ROS. Although we do not have data demonstrating these same trends in Drosophila, it is reasonable to imagine that a similar situation exists. This leads us to postulate that protein modifications related to high energy-producing sperm cells might be selected for in Drosophila because increasing energy production would make the sperm more competitive. These modifications might produce high levels of ROS, but their potentially damaged mtDNA would not be passed to the next generation (Allen 1996; Burt and Trivers 2006). Furthermore, testis specialization for high-energy production would be possible even if it damages the nuclear genome because sperm competition might select for individuals that rapidly produce large amounts of sperm despite the associated high mutation rate (Blumenstiel 2007). However, high energy-producing alleles or duplicates could be detrimental when expressed in the soma or ovary because the high ROS production rate would cause faster aging in the former case (Rand 2005) and inheritance of damaged mitochondria by the offspring in the latter (Allen 1996; Burt and Trivers 2006). This situation would generate intralocus sexually antagonistic conflict that might be resolved through the fixation and maintenance of testis-specific duplicated genes. In agreement with the postulated existence of sexually antagonistic conflict for mitochondrial function, there are studies (Rand et al. 2001; Dowling et al. 2007) that reveal that mitochondrial-nuclear genotypes have antagonistic sex-specific effects in Drosophila and beetles.
In this intralocus sexually antagonistic conflict model, different alleles of nuclearly encoded mitochondrial genes that are beneficial for males but detrimental to females would often end up being present at intermediate frequencies in the population (Rice 1984; Fry 2009; Patten and Haig 2009). Such antagonism could promote the emergence of the duplicated genes under the model proposed by Proulx and Phillips (2006). Given that relocation facilitates attaining testis-specific expression and specialization of these genes, relocated duplicates would allow for the specialization of testis/sperm mitochondria, and parental genes will not be needed in those tissues; which will help to alleviate the antagonism. Interestingly, a very recent study (Innocenti and Morrow 2010) on the contribution of sex-biased expression to sexual antagonism showed that only 10% of the sex-biased transcripts in D. melanogaster have sexually antagonistic effects, and they rarely reside in the ovary or testis. This result seems to confirm that sex-biased expression (or in our extreme case, testis-specific expression of duplicated genes) would be the outcome of intralocus sexual antagonism (Ellegren and Parsch 2007; Bonduriansky and Chenoweth 2009). At the same time, factors such as male meiotic X inactivation (Hense et al. 2007; Vibranovski, Lopes, et al. 2009), increasing the level of expression of X-linked genes in testes (Vicoso and Charlesworth 2009) and/or particular sexually antagonistic situations (Rice 1984; Patten and Haig 2009) may determine the observed excess of relocation of the duplicated genes from the X chromosome to autosomes (i.e., out-of-the-X) and the avoidance of the X chromosome.
As in the case of mitochondrial-duplicated genes, we also hypothesize that other types of genes might exhibit these extreme patterns because testis specialization might be under selection for other functions. We propose that retention of testis-specific duplicated genes is common because intralocus sexually antagonistic conflicts are common in testis (i.e., testis is a very different tissue and under very strong selective pressures to specialize and evolve quickly due to male–male competition, sexual antagonism, and sexual selection). Testis-specific proteasome genes might exemplify this (Belote and Zhong 2009). Proteasomes represent protein-degrading machineries. In D. melanogaster, 12 of the 33 genes that make up the 26S proteasome subunit have testis-specific duplicates. Interestingly, most of these are not only relocated duplicates but are also been generated recurrently from the same parental gene. In addition, there are also multiple testis-specific duplicated genes for the 19S cap proteasome subunit (Belote and Zhong 2009), and the authors argued that a specialized proteasome may possibly have been selected for sperm individualization. As in the case of mitochondria, we propose that when specialization of the proteasome was needed in testis, alleles that benefited males because they were good for the testis began to segregate in the population and later duplicated.
Further experimental evidence supporting our hypotheses needs to be obtained. Regarding mitochondrial functions, for instance, higher levels of free radicals in the male germline and mutations in sperm mitochondria should be observed, as has been observed in mature human sperm (Reynier et al. 1998). If the new genes have different functions, as predicted, their replacement with somatic paralogs might affect fitness (i.e., fertility should be lower), and if we replace somatic copies with testis-specific ones, the life span should be shorter. Many duplicates might also have a function related to the many morphological changes that occur in the germline, and swapping the paralogs should also have deleterious effects. Because the expression of the testis-specific forms is virtually nonexistent in other tissues, we postulate that even their ectopic expression might decrease life span. In addition, we also predict that this conflict might exist in other species and envision that many genomes will be found where testis-biased mitochondrial duplicates are overrepresented. Finally, we predict that a great deal of sexually antagonistic variation will map to genes that are housekeeping genes for which testis/sperm would benefit from a specialized duplicate. These predictions remain to be tested.
Supplementary Material
Supplementary figures S1–S4 and table S1–S4 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
We want to thank Jeff Demuth, David Rand, Marta Wayne, and an anonymous reviewer for comments on this work. This work was supported by the National Institutes of Health (GM071813) and University of Texas at Arlington startup funds to E.B. and by Texas Wesleyan University for C.C.
References
- Allen JF. Separate sexes and the mitochondrial theory of ageing. J Theor Biol. 1996;180:135–140. doi: 10.1006/jtbi.1996.0089. [DOI] [PubMed] [Google Scholar]
- Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Shahrour F, Díaz-Uriarte R, Dopazo J. FatiGO: a web tool for finding significant associations of Gene Ontology terms with groups of genes. Bioinformatics. 2004;20:578–580. doi: 10.1093/bioinformatics/btg455. [DOI] [PubMed] [Google Scholar]
- Al-Shahrour F, et al. BABELOMICS: a systems biology perspective in the functional annotation of genome-scale experiments. Nucleic Acids Res. 2006;34:W472–W476. doi: 10.1093/nar/gkl172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreyev AY, Kushnareva YE, Starkov AA. Mitochondrial metabolism of reactive oxygen species. Biochemistry Mosc. 2005;70:200–214. doi: 10.1007/s10541-005-0102-7. [DOI] [PubMed] [Google Scholar]
- Arnau V, Mars S, Marín I. Iterative cluster analysis of protein interaction data. Bioinformatics. 2005;21:364–378. doi: 10.1093/bioinformatics/bti021. [DOI] [PubMed] [Google Scholar]
- Bai Y, Casola C, Feschotte C, Betrán E. Comparative genomics reveals a constant rate of origination and convergent acquisition of functional retrogenes in Drosophila. Genome Biol. 2007;8:R11. doi: 10.1186/gb-2007-8-1-r11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belote JM, Zhong L. Duplicated proteasome subunit genes in Drosophila and their roles in spermatogenesis. Heredity. 2009;103:23–31. doi: 10.1038/hdy.2009.23. [DOI] [PubMed] [Google Scholar]
- Betrán E, Thornton K, Long M. Retroposed new genes out of the X in Drosophila. Genome Res. 2002;12:1854–1859. doi: 10.1101/gr.604902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenstiel JP. Sperm competition can drive a male-biased mutation rate. J Theor Biol. 2007;249:624–632. doi: 10.1016/j.jtbi.2007.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonduriansky R, Chenoweth SF. Intralocus sexual conflict. Trends Ecol Evol. 2009;24:280–288. doi: 10.1016/j.tree.2008.12.005. [DOI] [PubMed] [Google Scholar]
- Burt A, Trivers R. Genes in conflict: the biology of selfish genetic elements. Cambridge (MA): Harvard University Press; 2006. pp. 161–162. [Google Scholar]
- Casola C, Ganote CL, Hahn MW. Nonallelic gene conversion in the genus Drosophila. Genetics. 2010;185:95–103. doi: 10.1534/genetics.110.115444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chintapalli VR, Wang J, Dow JAT. Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat Genet. 2007;39:715–720. doi: 10.1038/ng2049. [DOI] [PubMed] [Google Scholar]
- Claros MG, Vincens P. Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur J Biochem. 1996;241:779–786. doi: 10.1111/j.1432-1033.1996.00779.x. [DOI] [PubMed] [Google Scholar]
- Conrad DF, et al. Origins and functional impact of copy number variation in the human genome. Nature. 2009;464:704–712. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello JC, et al. Gene networks in Drosophila melanogaster: integrating experimental data to predict gene function. Genome Biol. 2009;10:R97. doi: 10.1186/gb-2009-10-9-r97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusick ME, Klitgord N, Vidal M, Hill DE. Interactome: gateway into systems biology. Hum Mol Genet. 2005;2:R171–181. doi: 10.1093/hmg/ddi335. [DOI] [PubMed] [Google Scholar]
- Dorus S, et al. Genomic and functional evolution of the Drosophila melanogaster sperm proteome. Nat Genet. 2006;38:1440–1445. doi: 10.1038/ng1915. [DOI] [PubMed] [Google Scholar]
- Dowling DK, Friberg U, Arnqvist G. A comparison of nuclear and cytoplasmic genetic effects on sperm competitiveness and female remating in a seed beetle. J Evol Biol. 2007;20:2113–2125. doi: 10.1111/j.1420-9101.2007.01433.x. [DOI] [PubMed] [Google Scholar]
- Ellegren H, Parsch J. The evolution of sex-biased genes and sex-biased gene expression. Nat Rev Genet. 2007;8:689–698. doi: 10.1038/nrg2167. [DOI] [PubMed] [Google Scholar]
- Emerson JJ, Cardoso-Moreira M, Borevitz JO, Long M. Natural selection shapes genome-wide patterns of copy-number polymorphism in Drosophila melanogaster. Science. 2008;320:1629–1631. doi: 10.1126/science.1158078. [DOI] [PubMed] [Google Scholar]
- Emerson JJ, Kaessmann H, Betrán E, Long M. Extensive gene traffic on the mammalian X chromosome. Science. 2004;303:537–540. doi: 10.1126/science.1090042. [DOI] [PubMed] [Google Scholar]
- Fontanillas P, Hartl DL, Reuter M. Genome organization and gene expression shape the transposable element distribution in the Drosophila melanogaster euchromatin. PLoS Genet. 2007;3:2256–2267. doi: 10.1371/journal.pgen.0030210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry JD Forthcoming. The genomic location of sexually antagonistic variation: some cautionary comments. Evolution. 2009;64(5):1510–1516. doi: 10.1111/j.1558-5646.2009.00898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller M. Bate M, Martinez Arias A, editors. The development of Drosophila melanogaster. Vol. 1. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 1993. Spermatogenesis; pp. 71–147. [Google Scholar]
- Graubert TA, et al. A high-resolution map of segmental DNA copy number variation in the mouse genome. PLoS Genet. 2007;3:e3. doi: 10.1371/journal.pgen.0030003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haerty W, et al. Evolution in the fast lane: rapidly evolving sex-related genes in Drosophila. Genetics. 2007;177:1321–1335. doi: 10.1534/genetics.107.078865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn MW, Han MV, Han S. Gene family evolution across 12 Drosophila genomes. PLoS Genet. 2007;3:e197. doi: 10.1371/journal.pgen.0030197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl FU, Pfanner N, Nicholson DW, Neupert W. Mitochondrial protein import. Biochim Biophys Acta. 1989;988:1–45. doi: 10.1016/0304-4157(89)90002-6. [DOI] [PubMed] [Google Scholar]
- Heger A, Ponting CP. Evolutionary rate analyses of orthologs and paralogs from 12 Drosophila genomes. Genome Res. 2007;17:1837–1849. doi: 10.1101/gr.6249707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heger A, Ponting CP. OPTIC: orthologous and paralogous transcripts in clades. Nucleic Acids Res. 2008;36:D267–D270. doi: 10.1093/nar/gkm852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hense W, Baines JF, Parsch J. X chromosome inactivation during Drosophila spermatogenesis. PLoS Biol. 2007;5:e273. doi: 10.1371/journal.pbio.0050273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley I, Hale ME, Prince VE. Duplication events and the evolution of segmental identity. Evol Dev. 2005;7:556–567. doi: 10.1111/j.1525-142X.2005.05059.x. [DOI] [PubMed] [Google Scholar]
- Innocenti P, Morrow EH. The sexually antagonistic genes of Drosophila melanogaster. PLoS Biol. 2010;8:e1000335. doi: 10.1371/journal.pbio.1000335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Yang J, Gu X. Expression divergence between duplicate genes. Trends Genet. 2005;21:602–607. doi: 10.1016/j.tig.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Loppin B, Lepetit D, Dorus S, Couble P, Karr TL. Origin and neofunctionalization of a Drosophila paternal effect gene essential for zygote viability. Curr Biol. 2005;15:87–93. doi: 10.1016/j.cub.2004.12.071. [DOI] [PubMed] [Google Scholar]
- Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000;290:1151–1155. doi: 10.1126/science.290.5494.1151. [DOI] [PubMed] [Google Scholar]
- Lyne R, et al. FlyMine: an integrated database for Drosophila and Anopheles genomics. Genome Biol. 2007;8:R129. doi: 10.1186/gb-2007-8-7-r129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mank JE, Hultin-Rosenberg L, Zwahlen M, Ellegren H. Pleiotropic constraint hampers the resolution of sexual antagonism in vertebrate gene expression. Am Nat. 2008;171:35–43. doi: 10.1086/523954. [DOI] [PubMed] [Google Scholar]
- Marco A, Marín I. A general strategy to determine the congruence between a hierarchical and a non-hierarchical classification. BMC Bioinformatics. 2007;8:442. doi: 10.1186/1471-2105-8-442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisel RP, Han MV, Hahn MW. A complex suite of forces drives gene traffic from Drosophila X Chromosomes. Genome Biol Evol. 2009;1:176–188. doi: 10.1093/gbe/evp018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno S. Evolution by gene duplication. New York: Springer; 1970. [Google Scholar]
- Parisi M, et al. Paucity of genes on the Drosophila X chromosome showing male-biased expression. Science. 2003;299:697–700. doi: 10.1126/science.1079190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi M, et al. A survey of ovary-, testis-, and soma-biased gene expression in Drosophila melanogaster adults. Genome Biol. 2004;5:R40. doi: 10.1186/gb-2004-5-6-r40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patten MM, Haig D. Maintenance or loss of genetic variation under sexual and parental antagonism at a sex-linked locus. Evolution. 2009;63:2888–2895. doi: 10.1111/j.1558-5646.2009.00764.x. [DOI] [PubMed] [Google Scholar]
- Porcelli D, Barsanti P, Pesole G, Caggese C. The nuclear OXPHOS genes in insecta: a common evolutionary origin, a common cis-regulatory motif, a common destiny for gene duplicates. BMC Evol Biol. 2007;7:215. doi: 10.1186/1471-2148-7-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell JR. Progress and prospects in Evolutionary Biology: the Drosophila model. New York: Oxford University Press; 1997. [Google Scholar]
- Proulx SR, Phillips PC. Allelic divergence precedes and promotes gene duplication. Evolution. 2006;60:881–892. [PubMed] [Google Scholar]
- Rand DM. Mitochondrial genetics of aging: intergenomic conflict resolution. Sci Aging Knowledge Environ. 2005;2005:re5. doi: 10.1126/sageke.2005.45.re5. [DOI] [PubMed] [Google Scholar]
- Rand DM, Clark AG, Kann LM. Sexually antagonistic cytonuclear fitness interactions in Drosophila melanogaster. Genetics. 2001;159:173–187. doi: 10.1093/genetics/159.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynier P, et al. Long PCR analysis of human gamete mtDNA suggests defective mitochondrial maintenance in spermatozoa and supports the bottleneck theory for oocytes. Biochem Biophys Res Commun. 1998;252:373–377. doi: 10.1006/bbrc.1998.9651. [DOI] [PubMed] [Google Scholar]
- Rice WR. Sex chromosomes and the evolution of sexual dimorphism. Evolution. 1984;38:735–742. doi: 10.1111/j.1558-5646.1984.tb00346.x. [DOI] [PubMed] [Google Scholar]
- Ruiz-Pesini E, et al. Correlation of sperm motility with mitochondrial enzymatic activities. Clin Chem. 1998;44:1616–1620. [PubMed] [Google Scholar]
- Sardiello M, Licciulli F, Catalano D, Attimonelli M, Caggese C. MitoDrome: a database of Drosophila melanogaster nuclear genes encoding proteins targeted to the mitochondrion. Nucleic Acids Res. 2003;31:322–324. doi: 10.1093/nar/gkg123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh ND, Larracuente AM, Clark AG. Contrasting the efficacy of selection on the X and autosomes in Drosophila. Mol Biol Evol. 2008;25:454–467. doi: 10.1093/molbev/msm275. [DOI] [PubMed] [Google Scholar]
- Small I, Peeters N, Legeai F, Lurin C. Predotar: a tool for rapidly screening proteomes for N-terminal targeting sequences. Proteomics. 2004;4:1581–1590. doi: 10.1002/pmic.200300776. [DOI] [PubMed] [Google Scholar]
- Sturgill D, Zhang Y, Parisi M, Oliver B. Demasculinization of X chromosomes in the Drosophila genus. Nature. 2007;450:238–241. doi: 10.1038/nature06330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Subramanian S, Kumar S. Temporal patterns of fruit fly (Drosophila) evolution revealed by mutation clocks. Mol Biol Evol. 2004;21:36–44. doi: 10.1093/molbev/msg236. [DOI] [PubMed] [Google Scholar]
- Taylor JS, Raes J. Duplication and divergence: the evolution of new genes and old ideas. Annu Rev Genet. 2004;38:615–643. doi: 10.1146/annurev.genet.38.072902.092831. [DOI] [PubMed] [Google Scholar]
- Tripoli G, D'Elia D, Barsanti P, Caggese C. Comparison of the oxidative phosphorylation (OXPHOS) nuclear genes in the genomes of Drosophila melanogaster, Drosophila pseudoobscura and Anopheles gambiae. Genome Biol. 2005;6:R11. doi: 10.1186/gb-2005-6-2-r11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- True JR, Carroll SB. Gene co-option in physiological and morphological evolution. Annu Rev Cell Dev Biol. 2002;18:53–80. doi: 10.1146/annurev.cellbio.18.020402.140619. [DOI] [PubMed] [Google Scholar]
- Vibranovski MD, Lopes HF, Karr TL, Long M. Stage-specific expression profiling of Drosophila spermatogenesis suggests that meiotic sex chromosome inactivation drives genomic relocation of testis-expressed genes. PLoS Genet. 2009;5:e1000731. doi: 10.1371/journal.pgen.1000731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vibranovski MD, Zhang Y, Long M. General gene movement off the X chromosome in the Drosophila genus. Genome Res. 2009;19:897–903. doi: 10.1101/gr.088609.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicoso B, Charlesworth B. The deficit of male-biased genes on the D. melanogaster X chromosome is expression-dependent: a consequence of dosage compensation? J Mol Evol. 2009;68:576–583. doi: 10.1007/s00239-009-9235-4. [DOI] [PubMed] [Google Scholar]
- Vinckenbosch N, Dupanloup I, Kaessmann H. Evolutionary fate of retroposed gene copies in the human genome. Proc Natl Acad Sci U S A. 2006;103:3220–3225. doi: 10.1073/pnas.0511307103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, et al. Tissue-specific expression and regulation of sexually dimorphic genes in mice. Genome Res. 2006;16:995–1004. doi: 10.1101/gr.5217506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Sturgill D, Parisi M, Kumar S, Oliver B. Constraint and turnover in sex-biased gene expression in the genus Drosophila. Nature. 2007;450:233–237. doi: 10.1038/nature06323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, et al. On the origin of new genes in Drosophila. Genome Res. 2008;18:1446–1455. doi: 10.1101/gr.076588.108. [DOI] [PMC free article] [PubMed] [Google Scholar]