

Abstract
Objectives:
Vascular dementia (VaD) accounts for approximately 15%–20% of all dementias, but the relationship of progressive cognitive impairment to neurochemical changes is poorly understood. We have therefore investigated glutamatergic synaptic markers in VaD.
Methods:
We used homogenates prepared from gray matter from 2 neocortical regions (Brodmann area [BA] 9 and BA 20) and Western blotting to determine the concentrations of key components of the glutamatergic neurotransmitter system, vesicular glutamate transporter 1 (VGLUT1) and excitatory amino acid transporter EAAT2 (GLT-1), and the ubiquitous synaptic protein, synaptophysin, in 73 individuals—48 patients with cerebrovascular disease with and without dementia, 10 patients with AD, and 15 controls—in a case-control design.
Results:
VGLUT1 concentrations in BA 20 and BA 9 were correlated with CAMCOG total (Rs 0.525, p = 0.018, n = 20; Rs 0.560, p = 0.002, n = 27) and CAMCOG memory scores (Rs 0.616, p = 0.004, n = 20; Rs 0.675, p = 0.000, n = 27). VGLUT1 concentration in BA 9 differed between the different dementia groups and the stroke no dementia group (1-way analysis of variance F = 6.69, p = 0.001 and Bonferroni p < 0.01 in each case), with subjects with stroke who did not develop dementia exhibiting the highest mean value for VGLUT1.
Conclusions:
These data suggest that loss of glutamatergic synapses is a feature of VaD and Alzheimer disease but the preservation of synapses, in particular glutamatergic synapses, in the frontal cortex against the temporal cortex plays a role in sustaining cognition and protecting against dementia following a stroke.
GLOSSARY
- AD
= Alzheimer disease;
- ANOVA
= analysis of variance;
- BA
= Brodmann area;
- CAA
= cerebral amyloid angiopathy;
- CAMCOG
= Cambridge Assessment of Mental Health for the Elderly, Section B;
- CERAD
= Consortium to Establish a Registry for Alzheimer's Disease;
- DSM-IV
= Diagnostic and Statistical Manual of Mental Disorders, 4th edition;
- GFAP
= glial fibrillary acidic protein;
- H-E
= hematoxylin & eosin;
- LFB
= Luxol fast blue;
- SND
= stroke no dementia;
- VaD
= vascular dementia;
- VGLUT1
= vesicular glutamate transporter 1.
Stroke and dementia are common and often coexistent,1 costly, and devastating to patients and their carers. Vascular dementia (VaD), often accompanied by Alzheimer disease (AD), accounts for 15%–20% of the 25 million people worldwide with dementia.2 Despite the clear need, there are no therapies licensed for VaD.3–5
The neuropathologic and neurochemical basis of cognitive decline in VaD is poorly understood. Evolving attentional and executive impairments are likely associated with the severity of cerebral microvascular disease.6 However, the development of memory impairments, more closely linked to incident dementia,7 is probably associated with the development of concurrent AD pathology.8,9 An improved understanding of the neurochemical changes linked to cognitive impairment and imaging changes10 in patients with cerebrovascular disease is an essential component of developing targeted and effective therapies for patients with VaD.
Glutamate is the principal excitatory amino acid neurotransmitter of the CNS and is involved in most aspects of higher mental function.11 In stroke, much attention has focused on the ability of glutamate antagonists to reduce the size of the infarct and improve functional recovery in animal models. Functionally, the vesicular glutamate transporter (VGLUT1) is associated with synapses involved in long-term potentiation and memory.12 Key unanswered questions include the relationship of glutamatergic alterations to cognition, are whether there are differences between patients with VaD and people who have a stroke but do not progress to develop dementia (SND). We hypothesized that cognitive dysfunction in VaD is associated with loss of presynaptic glutamatergic terminals.
METHODS
Standard protocol approvals, registrations, and patient consents.
All participants or their next of kin gave informed consent for use of postmortem material and for data evaluation for research. The VaD and stroke subjects were originally recruited between 1996 and 2002 from representative hospital-based stroke registers in Tyneside, Wearside, and Cleveland (United Kingdom). The use of postmortem human material for this study was approved by the North West Regional Ethics Committee (ref: 08/H1010/5). The longitudinal study “Memory after Stroke” was approved by the Newcastle & North Tyneside Health Authority Joint Ethics Committee (ref: 99/153).
Subject study groups and brain tissue.
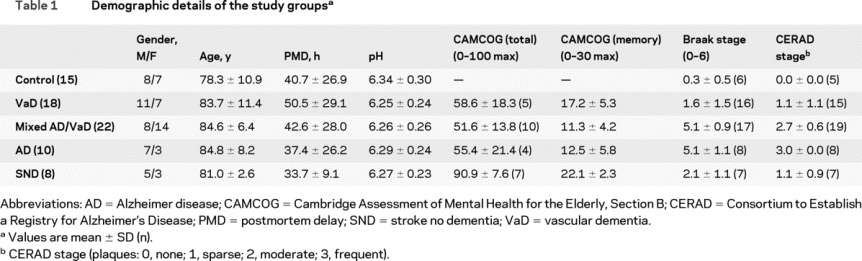
A total of 73 individuals were studied including 48 with cerebrovascular disease (VaD 18, mixed VaD/AD 22, SND 8), 10 with AD, and 15 controls (table 1). Brain specimens from dorsolateral prefrontal cortex (Brodmann area [BA] 9) and inferior/middle temporal cortex (BA 20/21) were obtained from the Newcastle Brain Tissue Resource, Campus for Ageing and Vitality, Newcastle University, UK, and from the MRC London Neurodegenerative Diseases Brain Bank at the Institute of Psychiatry, King's College London, UK, both part of the Brains for Dementia Research network. These brain tissue resources constitute a large collection of frozen and formalin-fixed brains from clinically characterized subjects, a substantial proportion of whom (55%) were from prospective clinical cohort studies with serial standardized evaluations. For individuals who participated in longitudinal clinical studies, standardized cognitive evaluations were completed with the Cambridge Assessment of Mental Health for the Elderly, Section B (CAMCOG), at baseline and at annual intervals until death, undertaken by trained psychology assistants or research nurses. CAMCOG is a 107-point assessment tool, which evaluates a range of cognitive domains and includes a 29-point memory subscale.13 CAMCOG was only completed for those participants who were enrolled in prospective clinical studies. Table 1 shows the age range, gender distribution, postmortem delay, tissue pH, Consortium to Establish a Registry for Alzheimer's Disease (CERAD) description of plaque frequency,14 and Braak stage15 of the participants. In the majority of individuals, bronchopneumonia was recorded as the cause of death. Of the 29 participants in longitudinal studies, 27 completed CAMCOG assessments. None of the individuals was taking a cholinesterase inhibitor, memantine, or antiparkinsonian medication. Only one individual was prescribed an anticholinergic drug. No participants had diagnosis of alcohol abuse or drug dependency. In the poststroke cases, the first cognitive assessment was 3 months after stroke.
Table 1 Demographic details of the study groups
Macroscopic and microscopic pathology was reviewed and assessed by 2 of the authors (R.K. and T.H.) at the Newcastle and London sites, using standardized protocols. Tissue blocks were fixed in 10% formalin and processed into paraffin wax. Blocks were taken as defined by CERAD from the middle frontal gyrus (BA 8/9), superior and middle temporal gyri (BA 21/22), and the anterior hippocampus and parahippocampal gyrus (BA 28, 36), inferior parietal lobule with the intraparietal sulcus (BA 40), midbrain, occipital lobe with the calcarine sulcus (BA 17–19), cingulate gyrus (BA 24), frontal and occipital lobe deep white matter with the watershed areas of the blood supply territories of major arteries, striatum, amygdala, thalamus, cerebellum, pons, and medulla oblongata. Hematoxylin & eosin (H-E) and Luxol fast blue/Nissl (LFB/Nissl) stains, modified Bielschowsky silver impregnation, and immunohistochemistry with antibody raised against phosphorylated tau (AT8, Autogen Bioclear, Wiltshire, UK), β-amyloid (Chemicon, Hampshire, UK), α-synuclein (KM51, Novocastra, Newcastle upon Tyne, UK), ubiquitin (Dako), and glial fibrillary acidic protein (GFAP) (Dako) were applied according to standardized protocols.
H-E was used as standard stain for general neuropathologic assessment of the cytoarchitecture, cellularity, composition, and integrity of neuroanatomic structures represented on the sections; it also visualized cells with acute and chronic ischemic damage and infarcts. LFB/Nissl staining showed the cellular pattern and highlighted myelin loss in areas of white matter rarefaction and infarcts. Bielschowsky silver impregnation and tau immunohistochemistry was applied to assess neuritic plaques and neurofibrillary tangles for CERAD plaque and Braak neurofibrillary tangle staging. The densities of plaques and tangles were determined by semiquantitative scoring as described in the staging protocols. The respective antibodies were applied to label hyperphosphorylated tau, β-amyloid, α-synuclein, ubiquitin, and GFAP to ascertain the neuropathologic diagnosis, and exclude diseases other than AD, VaD, and stroke.
Although the main diagnosis of VaD was made neuropathologically, patients also had to meet DSM-IV clinical criteria for dementia during life. Patients with a stroke according to World Health Organization criteria16 who did not meet National Institute of Neurological Disorders and Stroke–Association Internationale pour la Recherche en l'Enseignement en Neurosciences criteria for VaD or DSM-IV criteria for dementia were diagnosed as SND. Pathologic diagnosis of VaD was defined by the presence of multiple or cystic infarcts involving cortical and subcortical structures, borderzone infarcts, lacunae (<15 mm), microinfarcts (visible by microscopy only), and small vessel disease in subcortical structures in the general absence of neurofibrillary tangles.17 Cerebral amyloid angiopathy (CAA) was assessed with β-amyloid immunohistochemistry6 with an antibody recognizing both Aβ1–40 and Aβ1–42. In the control, VaD, and SND groups, none of the cases had severe CAA. In the AD and VaD and AD groups, although severe CAA was a feature, none of the cases included in this study had CAA-related cerebral hemorrhage and extensive CAA-related cerebral microinfarcts. Clinical evidence of dementia along with the same type or a combination of these lesions at 3 different coronal levels was considered diagnostic for VaD.18 None of the pure VaD cases exhibited tangle burden above Braak stage III15 or more than sparse neuritic plaques as defined by CERAD.14 Concurrent neuropathologic features of AD were characterized by the presence of neuritic plaques in cortical lobes, midbrain, and hippocampal formation according to the CERAD criteria. Tangle burden was graded according to the method of Braak15 and neuritic plaque density following the CERAD criteria. Patients were designated as mixed VaD/AD if they met criteria for VaD and had either a Braak & Braak stage above III or were classified as CERAD-defined probable or definite AD. Control subjects had no evidence of dementia as determined by retrospective assessments with absence of neurologic or psychiatric disorder and confirmed by neuropathologic examination.
Preparation of tissue samples for Western blotting.
Cortical gray matter was dissected free of the meninges and white matter at 0–4°C. Approximately 500 mg of tissue was homogenized in 10 mL ice-cold buffer (50 mm Tris-HCl, 5 mm EGTA, 10 mm EDTA, pH 7.4 containing protease inhibitors: 2 μg/mL leupeptin hemisulfate, 2 μg/mL aprotinin, 1 μg/mL E64, 2 μg/mL pepstatin A, and 20 μg/mL PMSF) using a Teflon/glass power-driven homogenizer. Aliquots were immediately frozen on dry ice and stored at −70°C. Protein concentration was assessed in triplicate using Coomassie Plus protein assay reagent (Pierce Biotechnology Inc.) and measuring absorbency at 640 nm. In preparation for Western blotting, an aliquot was thawed and mixed with concentrated sample buffer (final concentration 0.0625 M Tris-HCl, containing 2% sodium dodecyl sulfate, 5% β-mercaptoethanol, 10% glycerol, and 0.002% bromophenol blue) and then stored at −20°C.
Western blotting.
In contrast to standard techniques, the samples were not boiled, as this causes aggregation of the polytopic membranes.19 Tissue samples of equal protein concentration were then subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis, followed by transfer to nitrocellulose membranes. Membranes were then probed with polyclonal antibodies to VGLUT1 (Synaptic Systems, Germany) at dilutions of 1 in 2,000, and monoclonal antibodies to synaptophysin, β-tubulin III, GLT-1, and β-actin (Sigma Chemical Co., Poole, UK) at dilutions of 1 in 20,000 and 1 in 1,000. Subsequently, membranes were incubated in horseradish peroxidase antibodies before the immunoreactivity of the bands was detected using enhanced chemiluminescence (Enhanced ChemiLuminescence reagent, Amersham Pharmacia Biotech Ltd., UK). Each experiment was performed in triplicate and blots were analyzed using the BioImage Intelligent Quantifier densitometry program (BI Systems Corporation) and integrals of band density obtained.
Statistical analysis.
All values were expressed as a ratio of integral of band density in a sample to that of rat cortex, which was run on each blot for standardization purposes and this value was then corrected to β-actin quantified from the same blot. Statistical analysis was performed with the assistance of SPSS (version 15). The normality of the data was examined using the F ratio and Levene test for equality of variance. Group means were compared using 1-way analysis of variance (ANOVA) followed by Bonferroni multiple range test or Kruskal-Wallis ANOVA and Mann-Whitney U test, as appropriate, with significance level set at p < 0.05. Intercorrelation of neurochemical variables and correlation with clinicopathologic scores were examined using Pearson product moment (r) or Spearman rank correlation (Rs) as appropriate.
RESULTS
There were few correlations between neurochemical variables and demographic factors. VGLUT1 in temporal cortex correlated with tissue pH (Rs = 0.361, p = 0.006, n = 57); however, since tissue pH did not differ between groups, it was not considered further as a cofactor in the statistical analysis. Except for the concentration of EAAT2 (GLT1), no other measure correlated with postmortem delay (Rs = −0.293, p = 0.028, n = 56).
There were no intercorrelations of the neurochemical measures with the exception of VGLUT1 in temporal and frontal cortices (Rs = 0.460, p = 0.001, n = 51). Of particular note, there were no significant correlations between VGLUT1 and synaptophysin concentrations.
In accordance with our hypothesis, consistent correlations were identified between VGLUT1 and cognition. Concentrations of VGLUT1 in temporal cortex correlated with CAMCOG total (Rs = 0.525, p = 0.018, n = 20) and CAMCOG memory (Rs = 0.616, p = 0.004, n = 20) scores (figure 1A), and in frontal cortex with CAMCOG total (Rs = 0.560, p = 0.002, n = 27) and CAMCOG memory (Rs = 0.675, p = 0.000, n = 27) scores (figure 1B).
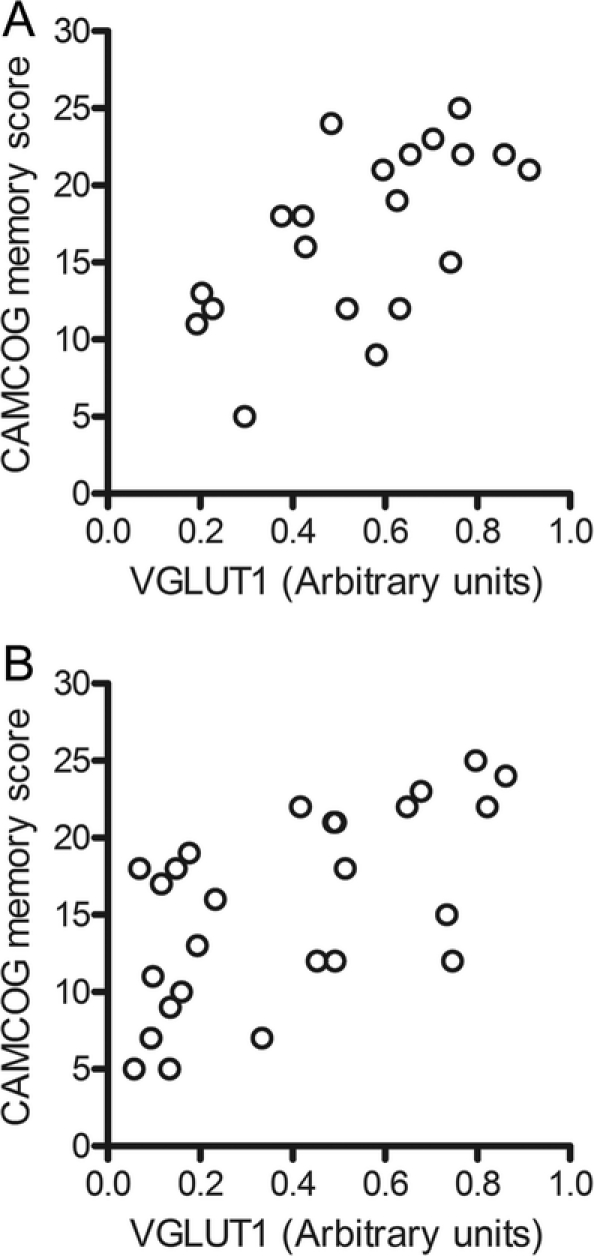
Figure 1 Vesicular glutamate transporter 1 (VGLUT1) and memory score
Scattergrams of VGLUT1 concentrations in Brodmann area (BA) 20 (A) and BA 9 (B) and Cambridge Assessment of Mental Health for the Elderly, Section B (CAMCOG) memory score. Spearman correlation coefficients are Rs 0.616, p = 0.004, n = 20, and Rs 0.675, p = 0.000, n = 27.
Concentrations of synaptophysin and EAAT2 (GLT1) did not correlate with either CAMCOG total score or CAMCOG memory score (p > 0.05).
The concentration of the major glial glutamate transporter EAAT2 was subject to considerable intergroup variation but these differences did not reach significance for either region studied (figures 2 and 3; 1-way ANOVA, p > 0.05). The concentration of synaptophysin in the frontal cortex was lower in all groups compared to controls (figures 2 and 3; 1-way ANOVA, F = 4.762, p = 0.002, and Bonferroni p < 0.05 in each case) but did not reach significance in the AD group (p = 0.051). In the temporal cortex, the concentration of synaptophysin was higher in patients who had stroke but did not develop dementia than in either controls or AD (1-way ANOVA, F = 4.947, p = 0.002, and Bonferroni p = 0.037 and 0.015). Furthermore, the concentration of synaptophysin was lower in the AD group than the VaD group (p = 0.033).

Figure 2 Western blots of synaptic proteins in vascular dementia (VaD)
Representative Western blots of synaptophysin (A, B), EAAT2 (C, D), and vesicular glutamate transporter 1 (E, F) expression in Brodmann area (BA) 9 (A, C, E) and BA 20 (B, D, F) in people with vascular pathology and controls. Protein samples, 10 μg per lane (except EAAT2, 20 μg per lane), were loaded and run on 10% acrylamide sodium dodecyl sulfate gels. Rat cortex (R) was loaded as a positive control and for comparison purposes. Con = control; M = mixed Alzheimer disease and VaD; SND = stroke no dementia.

Figure 3 Changes in synaptic proteins in vascular dementia (VaD)
Concentrations of synaptophysin (A, B), EAAT2 (C, D), and vesicular glutamate transporter 1 (VGLUT1) (E, F) as determined by Western blotting in Brodmann area (BA) 9 (A, C, E) and BA 20 (B, D, F) in people with vascular pathology and controls. AD = Alzheimer disease; Con = control; M = mixed Alzheimer disease and vascular dementia; SND = stroke no dementia; VaD = vascular dementia. Bars represent mean and error bars SEM. Groups that are different from each other are indicated by * (p < 0.05) and ** (p < 0.01); see text for details of statistical analysis.
The specific marker of glutamatergic synapses, VGLUT1, in the temporal cortex was not different among controls, VaD, mixed VaD/AD, AD, and SND groups (figures 2 and 3; 1-way ANOVA, F = 2.334, p = 0.068). However, in the frontal cortex mean VGLUT1 concentration was higher in patients who had stroke but did not develop dementia (SND group) than in all other groups and was elevated in comparison to the VaD, mixed VaD/AD, and AD groups (figures 2 and 3: 1-way ANOVA, F = 7.526, p = 0.000, and Bonferroni p < 0.002 in each case). Given that there was a numerical difference in the mean postmortem delay between the SND group and other groups, we performed a subanalysis of those cases with postmortem delays less than or equal to the maximum in the SND (49 hours) group (table e-1 on the Neurology® Web site at www.neurology.org). This analysis confirmed the robustness of the increase in VGLUT1 in the SND group remaining different from all others (1-way ANOVA, F = 0.78, p = 0.000; Bonferroni post hoc test p < 0.007).
DISCUSSION
The main findings were that there were regionally consistent and significant correlations between VGLUT1 concentration and cognitive scores as determined by CAMCOG and in particular for the memory subscale. Furthermore, VGLUT1 concentration was higher in frontal cortex of subjects who had a stroke but did not develop dementia (SND) as compared to VAD, mixed AD/VaD, and AD. Therefore, the upregulation of VGLUT1 appears to be specifically associated with preserved cognitive function in patients with cerebrovascular disease. These results, taken together with previous findings,20,21 also suggest that loss of glutamatergic synapses is a correlate of cognitive impairment in both VaD and AD. Furthermore, maintenance of the glutamatergic system through protection or plasticity may be an important regulator of cognitive decline in stroke, VaD, and AD.
While there were clear relationships between VGLUT1 and cognition, there were no similar correlations with synaptophysin, and consistent with this observation there was no correlation between VGLUT1 and synaptophysin. This pattern of results suggests that the relationship to cognition is specific to the glutamatergic system and is not a reflection of general synaptic alterations.
Glutamate excitotoxicity, involving excessive activation of receptors and increased intracellular Ca2+ leading to inappropriate activation of Ca2+-dependent processes causing metabolic dysfunction and eventual cell death, has been implicated in the pathophysiology of acute stroke.22 The limited success of translation of successful preclinical studies to stroke patients may be related to the expected adverse effects of NMDA antagonists on normal glutamatergic neurotransmission and perhaps interference with the potential upregulation of trophic factors caused by infarcts that are dependent on glutamate receptors.23 However, little attention has been paid to the role of the glutamatergic system in cognition in the longer term following cerebrovascular insults or the development of dementia in the context of cerebrovascular disease.
CAA is often present in the aging brain with and without dementia. A recent study found no evidence for CAA being an independent risk factor for cognitive decline except in cases with severe CAA lacking neuritic AD pathology6; none of our cases qualified for the latter group. Therefore we do not think CAA had a major impact on the overall results. There is a well-known heterogeneity of vascular lesions in their distribution and etiology and this was reflected in our cases. However, we aimed at studying VaD as a single diagnostic entity, as it is regarded in clinical practice.
Although impairments of attention and executive function are prominent in VaD,7 it has been suggested that progressive memory impairment more closely relates to the development of incident dementia in stroke patients.7–9 The current results are of particular interest given the specific correlation between VGLUT1 and memory function in VaD.
The level of VGLUT expression may affect quantal size24 and hence may contribute to presynaptic plasticity.25 Therefore the increased VGLUT1 expression in SND patients is likely to indicate increased vesicular glutamate storage capacity. In the frontal cortex this increased capacity could reflect more vesicles per synapse or more transporters per vesicle since the more universal marker of synapses remained unchanged. Previous studies have indicated that there is no effect of middle cerebral artery occlusion and reperfusion on levels of VGLUT126 up to 24 hours after onset of ischemia in rats. Our findings suggest that the upregulation of VGLUT1 is part of the delayed, long-term reconstruction of neuronal circuits after ischemic insults in humans.
ACKNOWLEDGMENT
The authors thank Prof. Robert Perry, Dr. Tuomo Polvikoski, and Dr. Evelyn Jaros for help with the collection and classification of some cases. Brain tissue was supplied by the Newcastle Brain Tissue Resource and the MRC London Neurodegenerative Diseases Brain Bank, both part of the Brains for Dementia Research Network.
DISCLOSURE
Dr. Elliott and Dr. Kirvell report no disclosures. Prof. Kalaria has served on a scientific advisory board for the Alzheimer's Research Trust UK; has received funding for travel from the Marabou Foundation, Sweden; serves on editorial advisory boards for Alzheimer's Disease and Associated Disorders, European Neurology, NeuroReport, and Behavioral and Brain Functions; and has received speaker honoraria from Pfizer Inc. Dr. Hortobágyi serves as Associate Editor for Frontiers in Neurodegenerative Diseases; has received a speaker honorarium from the Association of British Neurologists; and receives research support from the Psychiatry Research Trust UK, the Alzheimer's Research Trust UK, and the American Alzheimer's Association USA. Prof. Ballard serves on a scientific advisory board for Myriad Genetics, Inc.; has received funding for travel or speaker honoraria from H. Lundbeck A/S, Novartis, Eisai Inc., and Acadia Pharmaceuticals; serves as a consultant for H. Lundbeck A/S, Novartis, Eisai Inc., Bristol-Myers Squibb, and Acadia Pharmaceuticals; and receives/has received research support from H. Lundbeck A/S, Acadia Pharmaceuticals, Novartis, NIHR (UK), MRC, the Alzheimer's Society, the Alzheimer Research Trust, BUPA Foundation, Research into Ageing, Edmund J Safra Foundation, Wellcome Trust, and Dunhill Medical Trust. Prof. Francis serves on a scientific advisory board for H. Lundbeck A/S; has received speaker honoraria from H. Lundbeck A/S, UBM Medica, and Pfizer Inc/Eisai Inc.; serves as a section editor of the Journal of Neural Transmission; and receives research support from H. Lundbeck A/S, the Alzheimer's Society, the Alzheimer's Research Trust, and the Edmund J. Safra Foundation.
Supplementary Material
Address correspondence and reprint requests to Prof. Paul T. Francis, Wolfson Centre for Age-Related Diseases, Guy's Campus, King's College London, London SE1 1UL, UK paul.francis@kcl.ac.uk
Supplemental data at www.neurology.org
Study funding: Supported by the Dunhill Medical Trust and the Medical Research Council UK.
Disclosure: Author disclosures are provided at the end of the article.
Received April 27, 2010. Accepted in final form July 23, 2010.
REFERENCES
- 1.Hachinski V, Iadecola C, Petersen RC, et al. National Institute of Neurological Disorders and Stroke–Canadian Stroke Network vascular cognitive impairment harmonization standards. Stroke 2006;37:2220–2241. [DOI] [PubMed] [Google Scholar]
- 2.Ferri CP, Prince M, Brayne C, et al. Global prevalence of dementia: a Delphi consensus study. Lancet 2005;366:2112–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Areosa SA, Sherriff F, McShane R. Memantine for dementia. Cochrane Database Syst Rev 2005;CD003154. [DOI] [PubMed]
- 4.Craig D, Birks J. Rivastigmine for vascular cognitive impairment. Cochrane Database Syst Rev 2005;CD004744. [DOI] [PubMed]
- 5.Chapman N, Huxley R, Anderson C, et al. Effects of a perindopril-based blood pressure-lowering regimen on the risk of recurrent stroke according to stroke subtype and medical history: the PROGRESS Trial. Stroke 2004;35:116–121. [DOI] [PubMed] [Google Scholar]
- 6.Attems J, Quass M, Jellinger KA, Lintner F. Topographical distribution of cerebral amyloid angiopathy and its effect on cognitive decline are influenced by Alzheimer disease pathology. J Neurol Sci 2007;257:49–55. [DOI] [PubMed] [Google Scholar]
- 7.Moorhouse P, Rockwood K. Vascular cognitive impairment: current concepts and clinical developments. Lancet Neurol 2008;7:246–255. [DOI] [PubMed] [Google Scholar]
- 8.Lewis H, Beher D, Cookson N, et al. Quantification of Alzheimer pathology in ageing and dementia: age-related accumulation of amyloid-beta(42) peptide in vascular dementia. Neuropathol Appl Neurobiol 2006;32:103–118. [DOI] [PubMed] [Google Scholar]
- 9.Firbank MJ, Burton EJ, Barber R, et al. Medial temporal atrophy rather than white matter hyperintensities predict cognitive decline in stroke survivors. Neurobiol Aging 2007;28:1664–1669. [DOI] [PubMed] [Google Scholar]
- 10.Jagust WJ, Zheng L, Harvey DJ, et al. Neuropathological basis of magnetic resonance images in aging and dementia. Ann Neurol 2008;63:72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Danbolt NC. Glutamate uptake. Prog Neurobiol 2001;65:1–105. [DOI] [PubMed] [Google Scholar]
- 12.Varoqui H, Schafer MK, Zhu H, Weihe E, Erickson JD. Identification of the differentiation-associated Na+/PI transporter as a novel vesicular glutamate transporter expressed in a distinct set of glutamatergic synapses. J Neurosci 2002;22:142–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Williams JG, Huppert FA, Matthews FE, Nickson J. Performance and normative values of a concise neuropsychological test (CAMCOG) in an elderly population sample. Int J Geriatr Psychiatry 2003;18:631–644. [DOI] [PubMed] [Google Scholar]
- 14.Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD) Part II: standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 1991;41:479–486. [DOI] [PubMed] [Google Scholar]
- 15.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991;82:239–259. [DOI] [PubMed] [Google Scholar]
- 16.WHO MONICA Project Principal Investigators. The World Health Organization MONICA Project (monitoring trends and determinants in cardiovascular disease): a major international collaboration. J Clin Epidemiol 1988;41:105–114. [DOI] [PubMed] [Google Scholar]
- 17.Kalaria RN, Kenny RA, Ballard CG, Perry R, Ince P, Polvikoski T. Towards defining the neuropathological substrates of vascular dementia. J Neurol Sci 2004;226: 75–80. [DOI] [PubMed] [Google Scholar]
- 18.Gold G, Bouras C, Canuto A, et al. Clinicopathological validation study of four sets of clinical criteria for vascular dementia. Am J Psychiatry 2002;159:82–87. [DOI] [PubMed] [Google Scholar]
- 19.Bellocchio EE, Hu H, Pohorille A, Chan J, Pickel VM, Edwards RH. The localization of the brain-specific inorganic phosphate transporter suggests a specific presynaptic role in glutamatergic transmission. J Neurosci 1998;18:8648–8659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kirvell SL, Esiri MM, Francis PT. Down regulation of vesicular glutamate transporters precede cell loss and pathology in Alzheimer's disease. J Neurochem 2006;98:939–950. [DOI] [PubMed] [Google Scholar]
- 21.Kashani A, Lepicard E, Poirel O, et al. Loss of VGLUT1 and VGLUT2 in the prefrontal cortex is correlated with cognitive decline in Alzheimer disease. Neurobiol Aging 2008;29:1619–1630. [DOI] [PubMed] [Google Scholar]
- 22.Obrenovitch TP, Urenjak J, Zilkha E, Jay TM. Excitotoxicity in neurological disorders–the glutamate paradox. Int J Dev Neurosci 2000;18:281–287. [DOI] [PubMed] [Google Scholar]
- 23.Mattson MP. Glutamate and neurotrophic factors in neuronal plasticity and disease. Ann NY Acad Sci 2008;1144:97–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilson NR, Kang J, Hueske EV, et al. Presynaptic regulation of quantal size by the vesicular glutamate transporter VGLUT1. J Neurosci 2005;25:6221–6234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takamori S. VGLUTs: ‘exciting’ times for glutamatergic research? Neurosci Res 2006;55:343–351. [DOI] [PubMed] [Google Scholar]
- 26.Vemuganti R. Decreased expression of vesicular GABA transporter, but not vesicular glutamate, acetylcholine and monoamine transporters in rat brain following focal ischemia. Neurochem Int 2005;47:136–142. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.