

Abstract
Methods for efficient and accurate prediction of RNA structure are increasingly valuable, given the current rapid advances in understanding the diverse functions of RNA molecules in the cell. To enhance the accuracy of secondary structure predictions, we developed and refined optimization techniques for the estimation of energy parameters. We build on two previous approaches to RNA free-energy parameter estimation: (1) the Constraint Generation (CG) method, which iteratively generates constraints that enforce known structures to have energies lower than other structures for the same molecule; and (2) the Boltzmann Likelihood (BL) method, which infers a set of RNA free-energy parameters that maximize the conditional likelihood of a set of reference RNA structures. Here, we extend these approaches in two main ways: We propose (1) a max-margin extension of CG, and (2) a novel linear Gaussian Bayesian network that models feature relationships, which effectively makes use of sparse data by sharing statistical strength between parameters. We obtain significant improvements in the accuracy of RNA minimum free-energy pseudoknot-free secondary structure prediction when measured on a comprehensive set of 2518 RNA molecules with reference structures. Our parameters can be used in conjunction with software that predicts RNA secondary structures, RNA hybridization, or ensembles of structures. Our data, software, results, and parameter sets in various formats are freely available at http://www.cs.ubc.ca/labs/beta/Projects/RNA-Params.
Keywords: RNA secondary structure prediction, RNA free-energy parameter estimation, RNA free-energy parameters, RNA thermodynamic models, RNA free-energy models
INTRODUCTION
Thermodynamics-based computational methods for prediction of RNA secondary structure are widely used to advance our understanding of the enzymatic and regulatory roles of cellular RNA (Khvorova et al. 1999; Hutvágner et al. 2001; Lagos-Quintana et al. 2001; Miranda-Rios et al. 2001; Kimb et al. 2004). They also aid in the design of novel RNA molecules that can act as aptamers or enzymes (Tang and Breaker 2000; Penchovsky and Breaker 2005), help regulate gene expression (Bayer and Smolke 2005; Shabalina et al. 2006; Beisel et al. 2008; Lu and Mathews 2008), or respond to molecular inputs according to logic rules (Rinaudo et al. 2007).
Thermodynamics-based methods typically find the structure with minimum free energy (MFE), from the set of all possible structures for a given RNA sequence. Currently, the “Turner” free-energy model of Mathews et al. (1999a, 2004) is the most widely used choice for calculating the free energy of pseudoknot-free structures. However, MFE predictions by the Turner model achieve only 60% F-measure on average, where F-measure is the geometric mean of sensitivity and positive predictive value for base-pair prediction. To achieve more accurate predictions, researchers must provide structural clues that guide the prediction software or use comparative sequence analysis. The former is relatively expensive, requiring experimental analysis of the structure via chemical modification (Wilkinson et al. 2008) or NMR (Hart et al. 2008) to obtain the clues, and the latter is only possible when multiple homologous sequences from several organisms are available (James et al. 1989). For these reasons, methods that improve the accuracy of secondary structure prediction from a single sequence alone are very valuable.
In this work we develop new methods and data sets for deriving optimized energy parameters for the features (i.e., structural fragments, such as stacked pairs and various types of loops) of the Turner free-energy model and variants. We focus on the Turner model because its features are embedded in most available methods for prediction of MFE (Hofacker et al. 1994; Mathews et al. 1999a; Andronescu 2003) and suboptimal (Zuker 1989; Wuchty et al. 1999) secondary structures, partition function calculation (McCaskill 1990), Bayesian statistical prediction approaches (Ding 2006), prediction of oligonucleotide affinity to nucleic acid targets (Mathews et al. 1999b; Rehmsmeier et al. 2004; Lu and Mathews 2008), and simulation of RNA folding kinetics (Flamm et al. 2000; Xayaphoummine et al. 2003; Tang et al. 2008). Thus, improved parameters could immediately be used by all of these methods.
Our work builds on over three decades of parameter estimation for the Turner energy model and its predecessors (Tinoco et al. 1973; Xia et al. 1998; Mathews et al. 1999a, 2004). Two types of training data sets are used in parameter estimation: Structural data sets are comprised of sequence–structure pairs from the literature, and thermodynamic data sets are comprised of sequence–structure–energy triples, where the energies have been determined experimentally.
The current Turner parameters, namely, those of Mathews et al. (1999a, 2004), were estimated using a variety of techniques. For some features, parameters were inferred using linear regression from thermodynamic data (Xia et al. 1998). For features where thermodynamic data is sparse or not available (e.g., for multiloops), genetic (Mathews et al. 1999a) and grid search algorithms (Mathews et al. 2004) were applied primarily to structural data. A disadvantage of this approach is that parameters for some features were already fixed before other parameters were calculated; this limits the range of possibilities considered for the overall parameter set.
Do et al. (2006, 2007) considered a model similar to the Turner model, but in which parameters are not free-energy changes, because they are not normalized. To estimate parameters, they maximize the conditional likelihood of a structural data set and use a gradient-based method to inform the estimation procedure. While the techniques of Do et al. (2006) improve on those used by Mathews et al. (1999a), the Do et al. (2006) parameter set is not informed by thermodynamic data, cannot be used by software that incorporates the Turner model, and does not reliably determine free-energy changes (which are important, for example, in determining siRNA-binding affinities) (Lu and Mathews 2008).
In our earlier work (Andronescu et al. 2007), we described a Constraint Generation (CG) method to estimate Turner parameters. This method uses different types of constraints to ensure that the energy of reference structures are low, relative to alternatives for the same sequence, and also that parameter values respect the thermodynamic data. We applied CG to substantially larger data sets than used in previous work and obtained a 5% improvement in prediction accuracy over the Turner parameters. We also proposed, but did not run on large data sets, a conditional maximum likelihood approach (here called Boltzmann Likelihood) that incorporated thermodynamic data.
In this work we extend earlier approaches in several directions. First, we expand previous data sets to incorporate the latest reference structural and thermodynamic data from available databases and the literature. Second, we describe a Loss-augmented Max-margin Constraint Generation (LAM-CG) method; parameters are constrained so that the more inaccurate a structure is for a sequence in the structural training data set, the greater the margin (difference) between its free energy and that of the reference structure. Third, we provide the first analysis of the Boltzmann Likelihood (BL) method for the Turner model on a large data set. Our BL procedure is informed by thermodynamic as well as structural data. Parameter sets obtained from LAM-CG achieve 68% F-measure, and those obtained from BL achieve 69% F-measure, both significant improvements over our earlier CG parameters (65%) and the original Turner parameters (60%). Fourth, we model relationships between features; these relationships can then be exploited so that reliable estimates of parameters for some features (e.g., those that are covered by thermodynamic data) can improve estimates of closely related parameters. When BL exploits feature relationships, the resulting algorithm, BL-FR, produces parameters with 70% F-measure, slightly better than either BL or LAM-CG. Thus, our best parameters, obtained by BL-FR, improve the accuracy of MFE-based RNA secondary structure prediction by ∼10%, compared with the Turner parameters, and also yield energy predictions that are consistent with available experimental data.
We present several other analyses that provide further insight with respect to: accuracy of our best parameters on subfamilies of RNA structures (such as tRNAs); sensitivity of parameter accuracy to the size and composition of the structural data sets; and accuracy of parameters obtained for variants of the Turner model.
Our data, software, results, and parameter sets in various formats (including Vienna RNA and mfold formats) are freely available at http://www.cs.ubc.ca/labs/beta/Projects/RNA-Params.
MATERIALS AND METHODS
We start by presenting the data sets and accuracy measures we use in this work, followed by a description of the Turner energy model variants that are the focus of our work. Next, we briefly describe the Constraint Generation algorithm that was proposed in our previous work (Andronescu et al. 2007) and present our maximum margin extension. Then, we present the Boltzmann Likelihood (BL) algorithm, which we had briefly discussed previously (Andronescu et al. 2007), but which we had not run on large training data sets. Finally, we propose a novel way to model relationships between the features of the model. The presentation throughout is kept at a high level. Details needed, for example, to reproduce the algorithms, can be found in the Supplemental Material.
Data sets
We collected a structural set denoted by S-Full that contains 3245 RNA sequences with known secondary structures from the RNA STRAND v2.0 database (Andronescu et al. 2008). This set contains a large number of ribosomal RNA molecules (Cannone et al. 2002), transfer RNAs (Sprinzl and Vassilenko 2005), transfer messenger RNAs (Andersen et al. 2006), ribonuclease P RNAs (Brown 1999), signal recognition particle RNAs (Andersen et al. 2006), and secondary structures inferred from tertiary structures (Berman et al. 1992; Westbrook et al. 2003). We applied processing steps to obtain structures that can be predicted by the features of the Turner model and to reduce the uncertainty in the data. These steps include shortening the structures to be at most 700 nucleotides in length and the removal of the minimum number of base pairs that close pseudoknots, of noncanonical base pairs, of structures with unknown nucleotides, and of overly large loops (i.e., 50 for hairpin loops, bulges, and internal loops, and 100 for multiloops; these are removed because we suspect the unpaired bases in such large loops do form structure, but the structure is not yet known). A subset of S-Full was used in our previous work (Andronescu et al. 2007).
Following standard practice for parameter estimation, we split S-Full into two parts: roughly 80% for training (S-Full-Train) and the remaining fraction of ∼20% for testing (S-Full-Test) the accuracy of our parameter sets. Table 1 provides a summary of these and our other data sets. In addition, in order to assess the accuracy of our parameters on large RNA families and on strands that are longer than those used in training, we created the set S-STRAND2, which contains 2518 structures. Unlike the S-Full data set, which contains structures of only up to 700 nucleotides in length, S-STRAND2 also contains long molecules, including 187 small subunit ribosomal RNAs of average length 1276 nucleotides and 52 large subunit ribosomal RNAs of average length 2684 nucleotides (there is a large overlap between S-Full and S-STRAND2). We evaluate the prediction accuracy of our parameter sets and sets from the literature on S-STRAND2. Table 1 shows the number of sequence–secondary structure pairs, average length, and standard deviation of length for these structural sets.
TABLE 1.
Name, description, number of sequences, average length, and standard deviation in length of the structural and thermodynamic data sets used in this work

Secondly, we collected a thermodynamic set denoted by T-Full that contains data from 1291 optical melting experiments, published in 53 research articles (Freier et al. 1986; Sugimoto et al. 1986, 1987; Groebe and Uhlenbeck 1988, 1989; Longfellow et al. 1990; SantaLucia et al. 1990, 1991a,b ; Antao et al. 1991; He et al. 1991; Peritz et al. 1991; Antao and Tinoco 1992; Serra et al. 1993, 1994, 1997, 2004; Walter et al. 1994; Morse and Draper 1995; Wu et al. 1995; Laing and Hall 1996; McDowell and Turner 1996; McDowell et al. 1997; Schroeder et al. 1996, 2003; Xia et al. 1997, 1998; Giese et al. 1998; Kierzek et al. 1999; Meroueh and Chow 1999; Dale et al. 2000; Schroeder and Turner 2000, 2001; Burkard et al. 2001; Diamond et al. 2001; Mathews and Turner 2002; Proctor et al. 2002; Znosko et al. 2002, 2004; Chen et al. 2004, 2005; Mathews et al. 2004; Vecenie and Serra 2004; Bourdélat-Parks and Wartell 2005; O'Toole et al. 2005, 2006; Chen and Turner 2006; Shankar et al. 2006; Vecenie et al. 2006; Badhwar et al. 2007; Davis and Znosko 2007; Tolbert et al. 2007; Christiansen and Znosko 2008) (a subset of this set was used in our previous work) (Andronescu et al. 2007). Because the thermodynamic data is valuable in that it provides free-energy change information, and because it is relatively sparse (i.e., most of the experiments cover different features), we use the entire T-Full for training, and none for testing.
Accuracy measures
We measure the accuracy of a predicted RNA secondary structure relative to a reference secondary structure using the statistical measures sensitivity and positive predictive value (PPV). Sensitivity represents the ratio of correctly predicted base pairs as compared with the base pairs in the reference structures, while PPV represents the fraction of correctly predicted base pairs out of all predicted base pairs:
 |
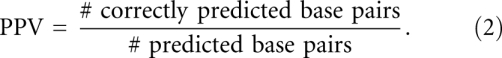 |
A perfect prediction yields sensitivity 1 and PPV 1; conversely, when these measures are 0, there are no correctly predicted base pairs. We use the F-measure to combine the sensitivity and PPV into a single measure. The F-measure is the harmonic mean of the two, which is close to the arithmetic mean when the two numbers are close to each other, but is smaller when one of the numbers is close to 0, thus penalizing predictions for which the sensitivity or PPV are poor:
 |
In addition, we measure the accuracy of the estimated free-energy changes  versus reference free-energy changes e for a set of t thermodynamic data (e.g., T-Full with given sequence, secondary structure, and free-energy change) using the root mean squared error (RMSE):
versus reference free-energy changes e for a set of t thermodynamic data (e.g., T-Full with given sequence, secondary structure, and free-energy change) using the root mean squared error (RMSE):
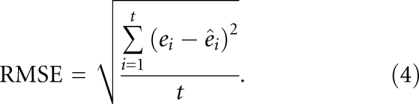 |
RMSE is measured in kcal/mol; the closer to 0 the RMSE value is, the more accurately free-energy changes are estimated.
The Turner model
An RNA free energy model consists of:
A collection of structural features (f1, f2, … , fp) , where p is the number of features of the model. A feature is an RNA secondary structure fragment whose thermodynamics are considered to be important for RNA folding.
A vector of free-energy parameters,
 , where θi is the free-energy change value of feature fi.
, where θi is the free-energy change value of feature fi.A free-energy change function, ΔG(x, y, θ), that defines the free-energy change of a sequence x folded into a specific secondary structure y. Here, we assume that ΔG(x, y, θ) is linear in the parameters θi:
 |
where ci(x, y) is the number of times feature fi occurs in secondary structure y of sequence x, and c(x, y):= (ci(x, y), …, cp(x, y)) denotes the vector of feature counts ci(x, y).
The Turner model (Mathews et al. 1999a, 2004) contains one or more features for each type of loop in an RNA secondary structure. Figure 1A identifies loops in a sample structure, and Figure 1B shows examples of features. Parameters for the features were derived from hundreds of experiments (SantaLucia and Turner 1997) (primarily optical melting experiments of short RNA sequences) as well as from structural data. We consider several variations of the Turner model, as summarized in Table 2. Details are provided in our Supplemental Material.
FIGURE 1.

Secondary structure of a Vimentin 3′ UTR protein-binding RNA region from the Rfam database (Rfam family RF00109, S76850.1/1539-1604). (A) The various types of loops in a pseudoknot-free secondary structure are indicated: stacked pair (two adjacent base pairs stacking onto each other), hairpin loop (HL—a region of unpaired bases closed by a base pair), internal loop (IL—two regions of unpaired bases closed by two base pairs), bulge loop (an internal loop with no unpaired bases on one side), multiloop (three or more stems connected together), and external loop. (B) Marked are examples of features of the Turner model and the corresponding Turner parameter values (Mathews et al. 1999a).
TABLE 2.
Summary of some Turner model variations that are studied in this work

The 1999 version of the Turner model, as described by Mathews et al. (1999a), contains a set of 363 “basic” features, and a set of “extrapolated” features whose parameter values are a function of the parameters for the basic features. We call the model with the basic features only (and not the extrapolated features) the Basic Turner model or M363 model. As one example pertaining to hairpin loops, the basic model has seven features pertaining to hairpin length, i.e., number of unpaired bases: one for each hairpin loop length between 3 and 9. For longer hairpins the length parameter is extrapolated from the length parameter for hairpins of length 9 (via a formula by Jacobson and Stockmayer 1950).
By removing features, we obtain simplified models with fewer features. Since the dangling end features render the prediction and partition function algorithms more complicated (McCaskill 1990; Andronescu 2003; Mathews 2004), we removed the 48 dangling end features from the M363 model to obtain the M315 model. The most parsimonious model that we consider has 79 features and includes significantly fewer terminal mismatch and internal loop features than the M315 model. Conversely, we produced “lavish” models by explicitly considering extrapolated features of the Turner model. For example, all 2 × 2 internal loop sequences have separate parameters, which differs from the 1999 Turner model that extrapolates these numbers from a few simple rules. The Full Turner or M7850 model contains 7850 features (Andronescu 2003); secondary structure prediction software uses tabulated values for exactly these features (Hofacker et al. 1994; Andronescu 2003; Zuker 2003; Mathews 2004). When modeling feature relationships, we use the M7726 model, which is very similar to the M7850 model, but removes some lavish features when available data pertaining to the features are sparse. In the Results section, we explore several further models that lie between the parsimonious and lavish models.
Algorithms
Constraint Generation: The NOM-CG algorithm
The Constraint Generation algorithm, introduced in our previous work (Andronescu et al. 2007), seeks a set of parameters θ such that (1) the reference structure for each sequence x in the training set has a free energy that is lower than the free energy of every other structure, and (2) the predicted free-energy change of structures in the thermodynamic training set is close to the experimentally determined value. Toward this end, the algorithm iteratively generates constraints that help to enforce conditions (1) and (2) above, while keeping values of θ in a reasonable range, and uses a numerical optimization procedure to find θ that satisfies these constraints. Since this variant of the Constraint Generation algorithm does not enforce a large distance between the free energy of the known structure and the free energies of other structures, we call this variant NO-Max-margin CG (NOM-CG).
Loss-Augmented Max-margin CG: The LAM-CG algorithm
Following the “large margin” approach from machine learning (see, e.g., Taskar 2005; Taskar et al. 2005; Do et al. 2008), we do not just require that the free energy of the true structure be less than all the others; instead, we require that the size of the margin or difference be proportional to the similarity between the true structure and the predicted structure. The intuition is that it is desirable for structures that are similar to the reference structure to have similar energies, but the more a structure deviates from the reference structure, the more different its energy should be. Our LAM-CG algorithm, proposed for the first time in this work, formalizes this requirement by changing the constraints of the NOM-CG algorithm.
The Boltzmann Likelihood (BL) algorithm
Our Boltzmann Likelihood (BL) algorithm (Andronescu et al. 2007) maximizes the conditional likelihood of the known structures in the structural set used for training using the Boltzmann function (exponential of the negative MFE normalized by the partition function) as likelihood. This algorithm is similar to the CONTRAfold algorithm of Do et al. (2006) but is informed by thermodynamic as well as structural data.
The BL-FR algorithm: Modeling feature relationships
Many features occur rarely in the training data set, which can cause problems for parameter estimation. This is an example of the “sparse data” problem that is frequently encountered in machine learning. A standard solution is to use hierarchical shrinkage methods (Gelman et al. 2004) that allow parameters to borrow statistical strength from related parameters. We propose a novel way of applying these ideas, and we do so by incorporating them into the Boltzmann Likelihood method of estimating RNA energy parameters from structural and thermodynamic data. We call this extension BL-FR because it is a Boltzmann Likelihood model informed by feature relationships between parameters.
Briefly, we use a graph in which each node corresponds to a feature, see Figure 2 for an example. To derive the graph structure and edge weights, we follow the extrapolation rules used by Mathews et al. (1999a), where the values of the “extrapolated” parameters are linear functions of the values of the “basic” parameters. Here, we use the same linear functions, but in a weaker way, that is, to define a statistical model given by the graph structure and edge weights. A directed edge from feature fi to feature fj indicates that knowledge of the parameter θi can be used by the parameter estimation algorithm in choosing parameter θj. Since the features covered by T-Full are those for which we can most reliably estimate the parameters, features of T-Full are root nodes of our graph. (A feature is covered by a data set if there is at least one experiment in the data set that contains that feature.) If the structural data set has good coverage of a feature that is not in T-Full, then the corresponding parameter value will be determined primarily by the structural data. Otherwise, its value will be determined primarily by the relationship rules. Figure 2 shows an example of a graph for 1 × 1 internal loop features. The root nodes are covered by T-Full, and the child nodes are features that are not covered by T-Full and are connected with other nodes. Details are presented in the Supplemental Material.
FIGURE 2.

Example of relationship graph for one 1 × 1 internal loop. This internal loop is closed by two A-U base pairs, has one U-U mismatch, and the sequence is of type 5′RYY/RYY3′, where R is a purine (A or G) and Y is a pyrimidine (C or U). Therefore, it is connected with the features A-U closure (with unnormalized weight 2, or normalized weight 2/4), U-U mismatch (with unnormalized weight 1) and the corresponding purine-pyrimidine group (with un-normalized weight 1).
RESULTS
We have trained our algorithms, NOM-CG, LAM-CG, BL (for the M363 model), and BL-FR (for the M7726 model), using the structural data set S-Full-Train and the thermodynamic training set T-Full. The result of this training process depends on the values of the hyperparameters of our algorithms (i.e., λ, B, μ, and η for the CG variants, and ρ, μ, and τ0 for the BL variants, see Supplemental Material). To determine hyperparameters resulting in good models, we have performed a series of hold-out validation experiments, in which we trained CG or BL on ∼80% of S-Full-Train and T-Full using various settings of the input arguments and validated on the remaining 20% of S-Full-Train. Details are given in the Supplemental Material and by Andronescu (2008). All results reported in the following have been obtained using the argument values for which the best average F-measure on the validation set was obtained (these argument values are listed in Supplemental Table 3).
Following, we first present a thorough quality evaluation of our best parameter sets and compare the accuracy of secondary structure prediction achieved by the resulting models against those obtained using previous state-of-the-art models, including the Turner99 model (the 1999 version of the Turner model) (Mathews et al. 1999a), CONTRAfold (Do et al. 2006), and our previous parameters (Andronescu et al. 2007). (The 2004 version of the Turner model [Mathews et al. 2004] produces predictions that are <1% more accurate than the 1999 version when measured on our test set S-Full-Test.) This is followed by an analysis of the computation time required to obtain our new parameter sets. Next, we investigate the sensitivity of our algorithms to the training set size. This is followed by an investigation of various extensions of the Turner model, first focusing on additional features and then on relationships between features. Overall, our empirical results (summarized in Fig. 3 and Table 3, and discussed in more detail below) demonstrate clearly that compared with previous energy models, our new models achieve significantly increased accuracy in the prediction of pseudoknot-free RNA secondary structures.
FIGURE 3.

Sensitivity and positive predictive value (PPV) of several parameter sets when measured on S-STRAND2. The points and training sets used for each point are described in Table 3. CONTRAfold uses a parameter γ to set the tradeoff between the sensitivity and PPV (we used values from 1 to 20).
TABLE 3.
Accuracy comparison of various parameter sets

Accuracy analysis
To assess the overall prediction accuracies achieved using our new parameter sets, we evaluated them on our two sets, S-Full-Test and S-STRAND2. While S-Full-Test has no overlap with S-Full-Train, S-STRAND2 does contain structures from S-Full-Train, but also contains long structures and permits analyses on many large classes of RNA molecules. In addition, to assess to which extent the free-energy values predicted using our new parameter sets agree with existing thermodynamic data, we computed the RMSE between the experimentally determined free-energy values in the thermodynamic set T-Full (used for training) and the corresponding predicted values.
As can be seen from the results shown in Table 3, parameter set BL-FR* produces the best results in terms of F-measure on both test sets, S-Full-Test and S-STRAND2. Next best is BL*, followed by the parameter sets obtained from LAM-CG and NOM-CG. All of these parameter sets achieve substantially better prediction accuracies (in terms of F-measure, sensitivity, and PPV) on our test sets than the Turner99 parameters. Two of them, namely, NOM-CG and LAM-CG, also yield a closer fit to the experimentally determined free-energy values in T-Full than the Turner99 parameters, while the fits for the BL variants are only moderately worse. We further note that the difference in F-measure obtained using LAM-CG vs. BL* is quite small.
The remaining results are for previous parameter sets that also yield better prediction accuracy than Turner99. Those have been trained on different sets and are therefore somewhat less comparable to our new sets than those between each other. CG 1.1 is the parameter set we obtained in previous work using an earlier version of CG (which is essentially the same as NOM-CG); this set was published by Andronescu et al. (2007) and was subsequently included as an option in the Vienna RNA Websuite (Gruber et al. 2008). As can be seen when comparing these results with the ones obtained by our new sets, the latter achieve a significant additional increase in prediction accuracy.
The CONTRAfold software by Do et al. (2006) implements an algorithm that is very similar to our BL algorithm; however, it does not use a thermodynamic set. CONTRAfold 1.1 was trained on a small set of 151 RNA secondary structures from the Rfam database, which we denote by S-151Rfam. As can be seen from Table 3, the accuracy obtained by CONTRAfold 1.1 on S-STRAND2 is only slightly higher than that achieved using Turner99 parameters, while on S-Full-Test the accuracy is slightly poorer. A subsequent version, CONTRAfold 2.0, was trained (by Do et al. 2007) on S-Processed, the set we had developed and used for training CG 1.1. CONTRAfold 2.0 achieves an average F-measure of 0.672 on S-STRAND2, which is 0.026 higher than for CG 1.1, possibly because of the differences in the parameter estimation algorithms or energy models (in terms of RNA structural features considered), the maximum-expected accuracy prediction algorithm used by CONTRAfold (Do et al. 2006), or their sophisticated algorithm for multihyperparameter learning (Do et al. 2007). However, CONTRAfold 2.0 does not reach the prediction accuracies achieved by the best new parameter sets presented in this work; in particular, the F-measure on the comprehensive S-STRAND2 set is 0.031 lower than that obtained by the BL-FR* parameters. Furthermore, since CONTRAfold does not directly consider thermodynamic data, it does not predict observed free-energy changes well, as can be seen from the large RMSE values reported in Table 3. Respecting the free energies is important for reasons other than structure prediction, such as small interfering RNA selection using hybridization thermodynamics (Lu and Mathews 2008).
Figure 3 further illustrates these performance results; we plot the average sensitivity versus average positive predictive value (PPV) for some of the parameter sets from Table 3, measured on S-STRAND2. CONTRAfold uses a parameter γ to control the tradeoff between the sensitivity and PPV (we used values from 1 to 20). Again, the improvements in prediction accuracies achieved by the new parameter sets developed in this study are clearly visible.
Further analysis revealed that the differences in prediction accuracy observed between Turner99, BL*, and BL-FR* are quite consistent across large structures (i.e., structures from S-STRAND2 with more than 2000 bases, see Fig. 4; Supplemental Fig. 7). However, when considering short RNAs (with at most 200 bases), a much wider variation of performance differences can be observed (see Fig. 4; Supplemental Fig. 7). We note that while, on average, the prediction accuracy for these small structures is much higher than for longer RNA sequences (about 0.8 for BL* and BL-FR*, and slightly below 0.7 for Turner99), incorrect predictions often have no correctly predicted base pairs (which is partially explained by the fact that there are few potential base pairs, and that the relative contribution of each base pair to the overall free energy is much larger for small structures).
FIGURE 4.

Correlation in prediction accuracy (F-measure) per molecule between our best parameters BL-FR* and the Turner99 parameters, on all the long and short structures in the S-STRAND2 set. (A) Structures of lengths 2000–4000 nucleotides; the correlation coefficient is 0.72. (B) Structures of lengths 0 to 200 nucleotides; the correlation coefficient is 0.59.
We also analyzed the prediction accuracy achieved by various parameter sets on different types of RNAs (see Table 4). For five of the 11 types of RNAs distinguished in S-STRAND2, the predictions using the BL-FR* parameter set are the most accurate, on average. In particular, 5S ribosomal RNAs are predicted substantially more accurately with BL-FR* than with any other parameter set. Furthermore, the predictions obtained with BL-FR* are more accurate than the predictions obtained with our previous parameter set, CG 1.1, for all 11 types of RNAs, and they are more accurate than the Turner parameters for all types except the hammerhead ribozymes, which are predicted only slightly less accurately. CONTRAfold 2.0 gives more accurate predictions for three of the 11 classes (ribonuclease P and large subunit ribosomal RNAs, as well as hammerhead ribozymes). We also note that a parameter set obtained using BL for the Turner model without dangling end features, M315, produces predictions that are about as accurate as those using optimized parameters for the corresponding model with dangling end features, M363. This suggests that the inclusion of dangling ends does not significantly affect accuracy when using BL as the parameter estimation algorithm. Therefore, considering that including dangling end features would require over 1 yr of CPU time for BL training, we decided to omit them from the model M7726 that underlies the BL-FR* parameter set.
TABLE 4.
Prediction accuracy on various classes of RNAs

Next, we analyzed the range and quality of suboptimal structures for all 2518 structures in the S-STRAND2 data set, with the BL* and Turner99 parameters. We predicted all of the suboptimal structures within two specified energy ranges from the MFE: 0 kcal/mol, yielding all secondary structures that have the same free energy as the MFE; and RT = 0.616 kcal/mol, the universal gas constant multiplied by the temperature (310 K). Table 5 shows the results. When using BL*, there are fewer structures whose free energy equals the MFE than when using the Turner99 parameters (3.08 vs. 10.73 structures, on average). This is consistent with the fact that the BL method favors a large margin between the known structures (assumed to be the MFE structures) and suboptimal structures. Looking at these structures in more detail, we notice that most of them are very similar to each other, i.e., have roughly the same abstract shape (that is, 1.04 distinct shapes for BL*, and 1.22 distinct shapes for Turner99, on average, when measuring the third level of abstraction of the RNAshapes software) (Steffen et al. 2006). When considering the larger energy range of 0.616 kcal/mol, there are slightly more suboptimal structures when using BL* (95.85 vs. 83.51, on average), as well as a slightly higher number of abstract shapes (9.84 vs. 7.38). The F-measure of the best suboptimal structure in the set of all suboptimal structures within the RT range is 0.75 for BL* (a 6% increase from the MFE structure), and 0.65 for Turner99.
TABLE 5.
Analysis of predicted suboptimal structures for all molecules in S-STRAND2, using SimFold (Andronescu 2003) with the BL* and Turner99 parameters, for two ranges (0 kcal/mol and RT = 0.616 kcal/mol, where R is the universal gas constant and T is the temperature 310K)

Finally, we analyzed the correlation between the numerical values of the parameters in the Turner99 model, our BL* parameters, and our CG* parameters. We found that the correlation between the CG* and Turner99 parameter values is quite high (with a Pearson correlation coefficient of r = 0.91), while a somewhat weaker correlation (r = 0.78) is observed between the BL* and Turner99 parameter values (see Supplemental Fig. 8). The CG* parameter values are generally within ±2 kcal/mol of the Turner99 values, while some of the BL* values differ by up to slightly >4 kcal/mol from the corresponding Turner99 values. We found that by choosing a higher weight ρ of the thermodynamic set in BL*, we could reduce that difference (Supplemental Fig. 8D) and obtain slightly more accurate predictions of free-energy changes on T-Full; however, this comes at the cost of a slightly reduced average prediction accuracy on our structural test sets. These parameters may be preferred when the focus is more on the estimated free-energy change and less on the predicted secondary structure. Note that the BL-FR* parameter values cannot be easily compared with any of the other sets, since they are based on a different model with a much larger set of features.
Runtime analysis
When using our new energy models, the computational effort expended in obtaining them by running CG and BL, respectively, is irrelevant. However, for anyone interested in running those procedures, for example, on different sets of training data, running times are of interest, and for this reason, we briefly discuss them in the following. All running times we report have been measured in terms of CPU time on a reference machine with a 3GHz Intel Xeon CPU with 1MB cache size and 2GB RAM, running Linux 2.6.16 (OpenSUSE 10.1).
The total running time of our BL and CG procedures on the S-Full-Train structural set is 6–8 CPU months, and 1–3 CPU days, respectively.
CG performs MFE secondary structure prediction for all sequences in the structural training set. For a sequence of length 1000 nucleotides, the MFE prediction (using our SimFold software implemented in C++) takes ∼17 sec, and the CPU time needed to compute the MFE prediction for the entire set S-Full-Train is roughly 57 min. Since this task can be parallelized easily and efficiently, it can be completed in less than a few minutes when run on a cluster of 30 CPUs. In addition, CG solves a quadratic optimization problem with a growing number of constraints at each iteration (we use the commercial package CPLEX 10.1.1 for this task). The CPU time differs between iterations; NOM-CG requires ∼10 min per iteration, while LAM-CG is slower, taking up to 2 h per iteration. Overall, running NOM-CG on S-Full-Train requires slightly over 1 d of CPU time (∼20% of this time is spent for prediction), while running LAM-CG takes ∼3 CPU days (only 2% of this time is spent for prediction, while the remaining 98% are used for optimization).
Unlike CG, BL requires the computation of the partition function and its gradient for each sequence in the training structural set. While the theoretical asymptotic time complexity for this task is the same as that of MFE secondary structure prediction (namely, cubic in the sequence length), in practice (due to different constant terms) computing the partition function and base-pair probabilities takes significantly longer than computing an MFE secondary structure, and computing the gradient of the partition function is even more time-consuming. Using our SimFold package, these three computations take ∼17 CPU minutes for a sequence of length 1000, and ∼1 d for the entire set S-Full-Train. Again, this latter task can be parallelized easily and efficiently, and thus can be completed in around 1 h on a cluster of 30 CPUs. The IPOPT gradient-based solver used in each iteration of BL requires additional partition function and gradient computations; when running BL on S-Full-Train, these computations add up to another 115 CPU days, resulting in a total running time of 200 CPU days (i.e., 6.7 CPU months). It might be possible to achieve significant speedups by using the interior-point stochastic gradient algorithm of Carbonetto et al. (2008) instead of the standard barrier approach that IPOPT uses.
Typically, the number of BL and BL-FR iterations increases with the number of features in the energy model considered. However, the differences are relatively modest, and overall, our most complex model (7802 features) requires only 1.3 times as much running time as our most parsimonious model (79 features).
Sensitivity to the training structural set
Clearly, the results obtained from CG and BL depend on the structural set used for training. To better understand and quantify this dependency, we performed four additional experiments. Considering the substantial computational cost involved in these analyses, we restricted them to BL and LAM-CG.
First, we conducted a fivefold cross-validation analysis by splitting the training set S-Full-Train into five parts of about the same size; each of these parts was then used as a validation set for determining prediction accuracy of models obtained by training BL and LAM-CG on the remaining four parts (Supplemental Table 5). The results from this analysis clearly demonstrate that both parameter estimation methods consistently produce models with substantially higher prediction accuracy than achieved using the Turner parameters. Furthermore, the maximum difference between the F-measures obtained for different validation sets is less than 0.03, which suggests that a difference in F-measure between two algorithms or two models that is greater than 0.03 may be statistically significant.
In our second experiment, we focused on the question as to which extent the quality of the parameters obtained by our estimation methods depends on the amount of structural training data used. We investigated this question by training BL and LAM-CG on increasingly smaller subsets of S-Full-Train (see Supplemental Table 6; Supplemental Fig. 9). Our results indicate that for both methods, the difference in prediction accuracy (average F-measure) obtained by training on all of S-Full-Train versus on only one-eighth of the full set is less than 0.01. This suggests that more data of the same type (i.e., from the same classes of RNAs, or mostly obtained by comparative sequence analysis) would probably not significantly improve the quality of the parameters obtained by our estimation methods.
Third, we eliminated all training data from S-Full-Train that used structural data determined by comparative sequence analysis. This left us with 238 structures from the Protein Data Bank (Westbrook et al. 2003), determined by X-ray crystallography and NMR (secondary structures were generated from the tertiary structures using RNAView) (Yang et al. 2003). The prediction accuracy obtained when training our estimation methods on this data is significantly lower than that achieved by training on a similarly sized subset of S-Full-Train that includes structures from comparative sequence analysis (see Supplemental Table 6). We hypothesize this to be caused primarily by the fact that the average length of the PDB structures is much smaller (50 vs. about 260), and the number of alternative structures for short molecules is not large enough for informative training.
Finally, we removed all small subunit ribosomal RNAs from the training set S-Train-Full (yielding 2070 sequence-structure pairs) and trained BL with the same hyperparameters used to obtain BL* (let BL-no-small-subunit denote this parameter set). The F-measure of S-STRAND2 remained the same as for BL* (0.691). That suggests that, when measured on many RNA families, the parameters are as accurate whether or not a particular class is included in the training (in this case, the small subunit rRNAs). However, when measured only on the subset of 187 small subunit rRNAs from S-STRAND2 (none of them used for training), the F-measure drops from 0.474 for BL* to 0.394 for BL-no-small-subunit, a significant decrease. This suggests that, when tested on molecules of a family that was not used for training, it is likely the performance will be poorer.
Extensions of the Turner model
In what follows we explore several variations of the basic Turner model without dangling ends, M315, by removing and adding some features, as suggested by other models and by experimental research. Although it would be interesting to add the dangling ends in the future, we estimate that this would increase the computation time required for parameter estimation up to >1 yr of CPU time. We take a “parsimonious” approach, in which we keep only 79 features, and a “lavish” approach, which uses an extended set of up to 7802 features. Table 1 gives details of these models, and details about each class of features that we considered can be found in Supplemental Table 7. We used BL (which, as demonstrated earlier, tends to give slightly better results than LAM-CG and NOM-CG) to estimate parameters for these models.
In order to select for the best model, we again performed a hold-out validation experiment, in which we used about four-fifths of S-Full-Train and the thermodynamic set T-Full as training data. The prediction accuracy (average F-measure) was validated on the remaining fifth of S-Full-Train.
We found that the average F-measure of 0.646 for the parsimonious model M79 was worse than the value of 0.684 observed for the optimized M315 model, but still higher than the 0.598 measured for the M363 model with the original Turner parameter values. On the other hand, the accuracy achieved using the most lavish model we considered, M7802, was essentially the same as that using M315 (average F-measure 0.683). Further experiments with models that use “lavish” expansions of only some sets of features (e.g., ones related to loop lengths) indicate that “lavish” features for special hairpin and internal loops lead to the single biggest improvement in average F-measure over the fully parsimonious model. We note that none of the models we considered appear to achieve significant improvements over the optimized basic Turner model, M315 (see Supplemental Table 7).
One possible explanation for this observation could be that the extensions to the energy model we considered are all relatively minor. It is possible that more drastic changes, such as a different energy function or more realistic features for multiloops, would yield further improvements. We note, however, that supporting such features would require significant changes in the algorithms for RNA secondary structure prediction (including partition function and gradient calculations). Another reason might lie in limitations of the structural data that we used. It is possible that this data is biased by artifacts of the comparative sequence analysis methods (recall that most of it was determined in this manner), has too much noise, or that we introduced bias when processing it, for example, by removing pseudoknots.
The use of feature relationships offers a way to deal with the fact that, particularly for the extensions of the Turner model we considered, currently available structural data offers only sparse coverage for some model features. This is reflected in the percentages of features covered by T-Full for M79, M315, and M7802, which are 100%, 75%, and 7%, respectively.
When reducing the size of the structural training set, BL-FR produces substantially better results than BL (see Supplemental Table 8; Supplemental Fig. 10). For example, when trained on one-eighth of S-Full-Train, BL-FR using the lavish model M7802 achieved an average F-measure of 0.677 compared with 0.643 measured for BL using the same model. Furthermore, we observed that for even smaller structural training sets, the accuracy achieved by BL with the lavish model decreased markedly, while a less-pronounced decrease in accuracy was observed for BL-FR. This provides further evidence that, indeed, feature relationships help to offset the detrimental effect of insufficient structural training data.
Based on the insights gained from our analysis of extended models and feature relationships, we created a combined lavish model with 7726 features, described in detail in the Supplemental Material. We trained BL-FR on S-Full-Train and obtained the BL-FR* parameter set that was analyzed in detail in the Accuracy Analysis section. The better average accuracy obtained with the BL-FR* parameter set demonstrates that carefully considering more features and similarities between features can improve the quality of the free-energy change parameters obtained.
DISCUSSION
In this work we presented improved algorithms for RNA free-energy change parameter estimation and a novel way of modeling relationships between the features of the model. We trained our algorithms and evaluated our results on diverse and comprehensive structural and thermodynamic data sets. We obtained free-energy change parameters that facilitate significantly better secondary structure prediction accuracy (F-measure) on average, while the free-energy estimations are close to experimentally determined values.
We have focused on free-energy models that do not include pseudoknots; however, our algorithms can estimate improved parameters for models with pseudoknots as well, as we have recently demonstrated (Andronescu 2008; Andronescu et al. 2010). The parameter sets described in this study can be incorporated into any software that requires energy-based RNA computations without pseudoknots, including:
minimum free energy and suboptimal secondary structure prediction software, such as Mfold (Zuker 2003), RNAstructure (Mathews 2004), and the Vienna RNA package (Hofacker et al. 1994)—our earlier parameters are already part of widely used software such as the RNA Vienna Web Servers (Gruber et al. 2008) and SimFold (Andronescu 2003);
algorithms that focus on probabilities or ensembles of RNA secondary structures and base pairs (such as the aforementioned packages) or perform sampling or clustering of RNA secondary structures, such as RNAshapes (Steffen et al. 2006) and Sfold (Ding and Lawrence 2003);
algorithms that focus on stochastic simulations, RNA cotranscriptional folding, and folding kinetics, such as Kinefold (Xayaphoummine et al. 2003) and Kinwalker (Geis et al. 2008);
algorithms that predict consensus RNA secondary structures common to phylogenetically related molecules, such as Dynalign (Harmanci et al. 2007), PARTS (Harmanci et al. 2008), Foldalign (Havgaard et al. 2007), and RNAalifold (Bernhart et al. 2008);
algorithms that predict secondary structures of interacting RNA molecules, such as the work of Dirks and Pierce (Dirks et al. 2007), PairFold, or MultiFold (Andronescu et al. 2005);
algorithms that measure the hybridization efficiency between probes and targets (Andronescu et al. 2005; Tulpan 2006), or predict the target site accessibility for small interfering RNAs (Lu and Mathews 2008);
algorithms for noncoding RNA discovery in genomes (Washietl et al. 2005; Torarinsson et al. 2006; Uzilov et al. 2006).
In particular, all of the new parameter sets provided on our web page except BL-FR* are fully compatible with the Turner model and our previous set CG 1.1. (Parameter sets that do not include values for dangling end features effectively set those values to zero.) The set of features corresponding to the BL-FR* set include some minor changes, e.g., the addition of features for bulges of size 1 (see details in the Supplemental Material).
We identify three most important directions for future work: combining predictions, improving the quality of data, and improving the model features and energy function, described in the following.
Combining predictions
Although, on average, our best parameters give significantly more accurate results than do previous parameters, we have encountered numerous situations in which one set of parameters produces better predictions for some molecules and another set of parameters produces better predictions for other molecules (see the plots in Supplemental Fig. 7 for examples of different prediction accuracy of individual molecules, and Table 4 for different prediction accuracy averaged on RNA families). To improve the chance of predicting the correct structures, we could borrow the idea used by the software that predicts suboptimal secondary structures (Zuker 1989; Wuchty et al. 1999) and use several parameter sets, several models, or several algorithms to obtain several predictions. For example, if we use the Turner parameters, the BL* and CG* parameters to predict the secondary structures in the set S-STRAND2 and we measure the accuracy of the best structure, we obtain 0.73 average F-measure (whereas the average F-measures of the Turner, LAM-CG and BL parameters are 0.60, 0.68, and 0.69)—this is substantially better, but still far from 1.
Therefore, one direction for future work is to investigate whether there is any correlation between poorly predicted structures and the parameters used, and ideally one would want to come up with an algorithm that chooses the best parameter set or the best prediction algorithm given an input RNA sequence. One could adopt a portfolio-based approach in which multiple predictions are combined or a best algorithm (parameter set or model) is selected on a per-instance basis (Xu et al. 2008).
Improving the quality of data
Other data sets could be used to train our parameter estimation algorithms, in addition to or instead of the structural and thermodynamic data that we have used. Isothermal titration calorimetry can be used for systems where optical melting cannot be performed. Such data could be used in exactly the same way as the optical melting data that we have used in our work. Data from differential scanning calorimetry could be used as well.
Similarly, data from optical tweezers experiments could be considered. Optical tweezers can be used to unfold or refold RNA secondary structures (including pseudoknots), the work required by the unfolding or refolding process can be measured (Hansen et al. 2007), and the free-energy change can be inferred. However, in practice, the inferred free-energy change has a low degree of accuracy. Such data could nevertheless be included in our approaches (with a low weight to account for the large error in these experiments).
We hypothesize that using a moderately large structural data set that is more reliable may yield more accurate free-energy parameters than using a larger structural set of questionable quality. Therefore, we believe that future work should be focused toward a better understanding of which kind of data are more reliable and on collecting and using as much as possible of these data.
Improving the model features and energy function
Our results show that revising the features of the model in addition to considering feature relationships gives an increase in prediction accuracy. However, we could not exceed an accuracy barrier of ∼70.3% when averaged over a large set. We hypothesize that more significant changes in the model might provide a closer approximation to the true model.
First, perhaps a linear energy function for pseudoknot-free structures is too limited, especially for longer structures. Second, there is evidence from the literature that there are non-nearest neighbor effects that are not considered in the Turner model. For example, Kierzek et al. (1999) noted that the stability of AA and UU mismatches is sensitive to the proximity of the mismatch to the end of the helix. Third, coaxial stacking features for multiloops have not been included in our approaches, but are part of the RNAstructure software (Mathews 2004; Lu et al. 2006) and are shown to better represent the true physical model. Furthermore, Mathews and Turner (2002) noted that the asymmetry of the unpaired bases in multiloops should be considered. While it may be challenging to incorporate such contributions in dynamic programming algorithms, it might be possible to implement them more easily by selecting a number of suboptimal structures using an approximated free-energy function and recomputing the exact free-energy change with the more complex model, or by using heuristic approaches.
SUPPLEMENTAL MATERIAL
Supplemental material can be found at http://www.rnajournal.org.
ACKNOWLEDGMENTS
We thank Doug Turner, George Mackie, and Raymond Ng for their insightful comments on an earlier version of this work. We thank Nima Aghaeepour, Aashish Dattani, Monir Hajiaghayi, and Hosna Jabbari for their valuable feedback and contributions to the web page, Alex Brown for help with the code, and the anonymous reviewers for valuable suggestions. This work was supported by NSERC grants to M.A., A.C., and K.P.M; IBM fellowship to M.A.; UBC graduate fellowship to M.A.; MITACS grants to A.C. and H.H.H.; CIFAR grants to K.P.M.; and NIH grant R01GM076485 to D.H.M.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.1950510.
REFERENCES
- Andersen ES, Rosenblad MA, Larsen N, Westergaard JC, Burks J, Wower IK, Wower J, Gorodkin J, Samuelsson T, Zwieb C 2006. The tmRDB and SRPDB resources. Nucleic Acids Res 34: 163–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andronescu MS 2003. “Algorithms for predicting the secondary structure of pairs and combinatorial sets of nucleic acid strands.” Master's thesis, Department of Computer Science, University of British Columbia, Vancouver, BC, Canada [Google Scholar]
- Andronescu MS 2008. “Computational approaches for RNA energy parameter estimation.” PhD thesis, Department of Computer Science, University of British Columbia, Vancouver, BC, Canada [Google Scholar]
- Andronescu M, Zhang Z, Condon A 2005. Secondary structure prediction of interacting RNA molecules. J Mol Biol 345: 987–1001 [DOI] [PubMed] [Google Scholar]
- Andronescu M, Condon A, Hoos HH, Mathews DH, Murphy KP 2007. Efficient parameter estimation for RNA secondary structure prediction. Bioinformatics 23: 19–28 [DOI] [PubMed] [Google Scholar]
- Andronescu M, Bereg V, Hoos HH, Condon A 2008. RNA STRAND: The RNA Secondary Structure and Statistical Analysis Database. BMC Bioinformatics 9: 340 doi: 10.1186/1471-2105-9-340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andronescu M, Pop C, Condon AE 2010. Improved energy parameters for RNA pseudoknotted secondary structure prediction. RNA 16: 26–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antao VP, Tinoco I 1992. Thermodynamic parameters for loop formation in RNA and DNA hairpin tetraloops. Nucleic Acids Res 20: 819–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antao VP, Lai SY, Tinoco I 1991. A thermodynamic study of unusually stable RNA and DNA hairpins. Nucleic Acids Res 19: 5901–5905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badhwar J, Karri S, Cass CK, Wunderlich EL, Znosko BM 2007. Thermodynamic characterization of RNA duplexes containing naturally occurring 1 × 2 nucleotide internal loops. Biochemistry 46: 14715–14724 [DOI] [PubMed] [Google Scholar]
- Bayer TS, Smolke CD 2005. Programmable ligand-controlled riboregulators of eukaryotic gene expression. Nat Biotechnol 23: 337–343 [DOI] [PubMed] [Google Scholar]
- Beisel CL, Bayer TS, Hoff KG, Smolke CD 2008. Model-guided design of ligand-regulated RNAi for programmable control of gene expression. Mol Syst Biol 4: 224 doi: 10.1038/msb.2008.62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Olson WK, Beveridge DL, Westbrook J, Gelbin A, Demeny T, Hsieh SH, Srinivasan AR, Schneider B 1992. The nucleic acid database. A comprehensive relational database of three-dimensional structures of nucleic acids. Biophys J 63: 751–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhart SH, Hofacker IL, Will S, Gruber AR, Stadler PF 2008. RNAalifold: Improved consensus structure prediction for RNA alignments. BMC Bioinformatics 9: 474 doi: 10.1186/1471-2105-9-474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourdélat-Parks BN, Wartell RM 2005. Thermodynamics of RNA duplexes with tandem mismatches containing a uracil-uracil pair flanked by C.G/G.C or G.C/A.U closing base pairs. Biochemistry 44: 16710–16717 [DOI] [PubMed] [Google Scholar]
- Brown J 1999. The Ribonuclease P Database. Nucleic Acids Res 27: 314 doi: 10.1093/nar/27.1.314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkard ME, Xia T, Turner DH 2001. Thermodynamics of RNA internal loops with a guanosine-guanosine pair adjacent to another noncanonical pair. Biochemistry 40: 2478–2483 [DOI] [PubMed] [Google Scholar]
- Cannone JJ, Subramanian S, Schnare MN, Collett JR, D'Souza LM, Du Y, Feng B, Lin N, Madabusi LV, Müller KM, et al. 2002. The comparative RNA web (CRW) site: An online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics 3: 2 doi: 10.1186/1471-2105-3-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbonetto P, Schmidt M, de Freitas N 2008. An interior-point stochastic approximation method and an L1-regularized delta rule. In Advances in neural information processing systems 21, pp. 233–240. MIT Press, Cambridge, MA [Google Scholar]
- Chen G, Turner DH 2006. Consecutive GA pairs stabilize medium-size RNA internal loops. Biochemistry 45: 4025–4043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Znosko BM, Jiao X, Turner DH 2004. Factors affecting thermodynamic stabilities of RNA 3 × 3 internal loops. Biochemistry 43: 12865–12876 [DOI] [PubMed] [Google Scholar]
- Chen G, Znosko BM, Kennedy SD, Krugh TR, Turner DH 2005. Solution structure of an RNA internal loop with three consecutive sheared GA pairs. Biochemistry 44: 2845–2856 [DOI] [PubMed] [Google Scholar]
- Christiansen ME, Znosko BM 2008. Thermodynamic characterization of the complete set of sequence symmetric tandem mismatches in RNA and an improved model for predicting the free energy contribution of sequence asymmetric tandem mismatches. Biochemistry 47: 4329–4336 [DOI] [PubMed] [Google Scholar]
- Dale T, Smith R, Serra MJ 2000. A test of the model to predict unusually stable RNA hairpin loop stability. RNA 6: 608–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis AR, Znosko BM 2007. Thermodynamic characterization of single mismatches found in naturally occurring RNA. Biochemistry 46: 13425–13436 [DOI] [PubMed] [Google Scholar]
- Diamond JM, Turner DH, Mathews DH 2001. Thermodynamics of three-way multibranch loops in RNA. Biochemistry 40: 6971–6981 [DOI] [PubMed] [Google Scholar]
- Ding Y 2006. Statistical and Bayesian approaches to RNA secondary structure prediction. RNA 12: 323–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Lawrence CE 2003. A statistical sampling algorithm for RNA secondary structure prediction. Nucleic Acids Res 31: 7280–7301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirks RM, Bois JS, Schaeffer JM, Winfree E, Pierce NA 2007. Thermodynamic analysis of interacting nucleic acid strands. SIAM Rev 49: 65–88 [Google Scholar]
- Do CB, Woods DA, Batzoglou S 2006. CONTRAfold: RNA secondary structure prediction without physics-based models. Bioinformatics 22: e90–e98 [DOI] [PubMed] [Google Scholar]
- Do CB, Foo C-S, Ng AY 2007. Efficient multiple hyperparameter learning for log-linear models. In Advances in neural information processing systems 20, pp. 377–384 MIT Press, Cambridge, MA [Google Scholar]
- Do CB, Foo CS, Batzoglou S 2008. A max-margin model for efficient simultaneous alignment and folding of RNA sequences. Bioinformatics 24: 68–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flamm C, Fontana W, Hofacker IL, Schuster P 2000. RNA folding at elementary step resolution. RNA 6: 325–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freier SM, Kierzek R, Caruthers MH, Neilson T, Turner DH 1986. Free energy contributions of G.U and other terminal mismatches to helix stability. Biochemistry 25: 3209–3213 [DOI] [PubMed] [Google Scholar]
- Geis M, Flamm C, Wolfinger MT, Tanzer A, Hofacker IL, Middendorf M, Mandl C, Stadler PF, Thurner C 2008. Folding kinetics of large RNAs. J Mol Biol 379: 160–173 [DOI] [PubMed] [Google Scholar]
- Gelman A, Carlin J, Stern H, Rubin D 2004. Bayesian data analysis. 2nd ed Chapman and Hall, New York [Google Scholar]
- Giese MR, Betschart K, Dale T, Riley CK, Rowan C, Sprouse KJ, Serra MJ 1998. Stability of RNA hairpins closed by wobble base pairs. Biochemistry 37: 1094–1100 [DOI] [PubMed] [Google Scholar]
- Groebe DR, Uhlenbeck OC 1988. Characterization of RNA hairpin loop stability. Nucleic Acids Res 16: 11725–11735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groebe DR, Uhlenbeck OC 1989. Thermal stability of RNA hairpins containing a four-membered loop and a bulge nucleotide. Biochemistry 28: 742–747 [DOI] [PubMed] [Google Scholar]
- Gruber ARR, Lorenz R, Bernhart SHH, Neuböc R, Hofacker IL 2008. The Vienna RNA Websuite. Nucleic Acids Res; 36: W70–W74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen TM, Reihani SN, Oddershede LB, Sorensen MA 2007. Correlation between mechanical strength of messenger RNA pseudoknots and ribosomal frameshifting. Proc Natl Acad Sci 104: 5830–5835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmanci AO, Sharma G, Mathews DH 2007. Efficient pairwise RNA structure prediction using probabilistic alignment constraints in Dynalign. BMC Bioinformatics 8: 130 doi: 10.1186/1471-2105-8-130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmanci AO, Sharma G, Mathews DH 2008. PARTS: Probabilistic alignment for RNA joinT secondary structure prediction. Nucleic Acids Res 36: 2406–2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart JM, Kennedy SD, Mathews DH, Turner DH 2008. NMR-assisted prediction of RNA secondary structure: Identification of a probable pseudoknot in the coding region of an R2 retrotransposon. J Am Chem Soc 130: 10233–10239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havgaard JH, Torarinsson E, Gorodkin J 2007. Fast pairwise structural RNA alignments by pruning of the dynamical programming matrix. PLoS Comput Biol 3: 1896–1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Kierzek R, SantaLucia J, Walter AE, Turner DH 1991. Nearest-neighbor parameters for G·U mismatches: 5′GU3′/3′UG5′ is destabilizing in the contexts CGUG/GUGC, UGUA/AUGU, and AGUU/UUGA but stabilizing in GGUC/CUGG. Biochemistry 30: 11124–11132 [DOI] [PubMed] [Google Scholar]
- Hofacker IL, Fontana W, Stadler PF, Bonhoeffer LS, Tacker M, Schuster P 1994. Fast folding and comparison of RNA secondary structures. Monatsh Chem 125: 167–188 [Google Scholar]
- Hutvágner G, McLachlan J, Pasquinelli AE, Bálint E, Tuschl T, Zamore PD 2001. A cellular function for the RNA-interference enzyme dicer in the maturation of the let-7 small temporal RNA. Science 293: 834–838 [DOI] [PubMed] [Google Scholar]
- Jacobson H, Stockmayer W 1950. Intramolecular reaction in polycondensations. I. The theory of linear systems. J Chem Phys 18: 1600–1606 [Google Scholar]
- James BD, Olsen GJ, Pace NR 1989. Phylogenetic comparative analysis of RNA secondary structure. Methods Enzymol 180: 227–239 [DOI] [PubMed] [Google Scholar]
- Khvorova A, Kwak Y-G, Tamkun M, Majerfeld I, Yarus M 1999. RNAs that bind and change the permeability of phospholipid membranes. Proc Natl Acad Sci 96: 10649–10654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kierzek R, Burkard ME, Turner DH 1999. Thermodynamics of single mismatches in RNA duplexes. Biochemistry 38: 14214–14223 [DOI] [PubMed] [Google Scholar]
- Kimb J, Krichevskyb A, Grad Y, Hayesb GD 2004. Identification of many microRNAs that copurify with polyribosomes in mammalian neurons. Proc Natl Acad Sci 101: 360–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T 2001. Identification of novel genes coding for small expressed RNAs. Science 294: 853–858 [DOI] [PubMed] [Google Scholar]
- Laing LG, Hall KB 1996. A model of the iron responsive element RNA hairpin loop structure determined from NMR and thermodynamic data. Biochemistry 35: 13586–13596 [DOI] [PubMed] [Google Scholar]
- Longfellow CE, Kierzek R, Turner DH 1990. Thermodynamic and spectroscopic study of bulge loops in oligoribonucleotides. Biochemistry 29: 278–285 [DOI] [PubMed] [Google Scholar]
- Lu ZJ, Mathews DH 2008. Efficient siRNA selection using hybridization thermodynamics. Nucleic Acids Res 36: 640–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu JZ, Turner DH, Mathews DH 2006. A set of nearest neighbor parameters for predicting the enthalpy change of RNA secondary structure formation. Nucleic Acids Res 34: 4912–4924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews D 2004. Using an RNA secondary structure partition function to determine confidence in base pairs predicted by free energy minimization. RNA 10: 1178–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews DH, Turner DH 2002. Experimentally derived nearest-neighbor parameters for the stability of RNA three- and four-way multibranch loops. Biochemistry 41: 869–880 [DOI] [PubMed] [Google Scholar]
- Mathews DH, Sabina J, Zuker M, Turner DH 1999a. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J Mol Biol 288: 911–940 [DOI] [PubMed] [Google Scholar]
- Mathews DH, Burkard ME, Freier SM, Wyatt JR, Turner DH 1999b. Predicting oligonucleotide affinity to nucleic acid targets. RNA 5: 1458–1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews DH, Disney MD, Childs JL, Schroeder SJ, Zuker M, Turner DH 2004. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc Natl Acad Sci 101: 7287–7292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaskill JS 1990. The equilibrium partition function and base pair binding probabilities for RNA secondary structure. Biopolymers 29: 1105–1119 [DOI] [PubMed] [Google Scholar]
- McDowell JA, Turner DH 1996. Investigation of the structural basis for thermodynamic stabilities of tandem GU mismatches: Solution structure of (rGAGGUCUC)2 by two-dimensional NMR and simulated annealing. Biochemistry 35: 14077–14089 [DOI] [PubMed] [Google Scholar]
- McDowell JA, He L, Chen X, Turner DH 1997. Investigation of the structural basis for thermodynamic stabilities of tandem GU wobble pairs: NMR structures of (rGGAGUUCC)2 and (rGGAUGUCC)2. Biochemistry 36: 8030–8038 [DOI] [PubMed] [Google Scholar]
- Meroueh M, Chow CS 1999. Thermodynamics of RNA hairpins containing single internal mismatches. Nucleic Acids Res 27: 1118–1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda-Rios J, Navarro M, Soberon M 2001. A conserved RNA structure (thi box) is involved in regulation of thiamin biosynthetic gene expression in bacteria. Proc Natl Acad Sci 98: 9736–9741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morse SE, Draper DE 1995. Purine-purine mismatches in RNA helices: Evidence for protonated G.A pairs and next-nearest neighbor effects. Nucleic Acids Res 23: 302–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Toole AS, Miller S, Serra MJ 2005. Stability of 3′ double nucleotide overhangs that model the 3′ ends of siRNA. RNA 11: 512–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Toole AS, Miller S, Haines N, Zink MC, Serra MJ 2006. Comprehensive thermodynamic analysis of 3′ doublenucleotide overhangs neighboring Watson-Crick terminal base pairs. Nucleic Acids Res 34: 3338–3344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penchovsky R, Breaker RR 2005. Computational design and experimental validation of oligonucleotide-sensing allosteric ribozymes. Nat Biotechnol 23: 1424–1433 [DOI] [PubMed] [Google Scholar]
- Peritz AE, Kierzek R, Sugimoto N, Turner DH 1991. Thermodynamic study of internal loops in oligoribonucleotides: Symmetric loops are more stable than asymmetric loops. Biochemistry 30: 6428–6436 [DOI] [PubMed] [Google Scholar]
- Proctor DJ, Schaak JE, Bevilacqua JM, Falzone CJ, Bevilacqua PC 2002. Isolation and characterization of a family of stable RNA tetraloops with the motif YNMG that participate in tertiary interactions. Biochemistry 41: 12062–12075 [DOI] [PubMed] [Google Scholar]
- Rehmsmeier M, Steffen P, Hochsmann M, Giegerich R 2004. Fast and effective prediction of microRNA/target duplexes. RNA 10: 1507–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaudo K, Bleris L, Maddamsetti R, Subramanian S, Weiss R, Benenson Y 2007. A universal RNAi-based logic evaluator that operates in mammalian cells. Nat Biotechnol 25: 795–801 [DOI] [PubMed] [Google Scholar]
- SantaLucia J, Turner DH 1997. Measuring the thermodynamics of RNA secondary structure formation. Biopolymers 44: 309–319 [DOI] [PubMed] [Google Scholar]
- SantaLucia J, Kierzek R, Turner DH 1990. Effects of GA mismatches on the structure and thermodynamics of RNA internal loops. Biochemistry 29: 8813–8819 [DOI] [PubMed] [Google Scholar]
- SantaLucia J, Kierzek R, Turner DH 1991a. Functional group substitutions as probes of hydrogen bonding between GA mismatches in RNA internal loops. J Am Chem Soc 113: 4313–4322 [Google Scholar]
- SantaLucia J, Kierzek R, Turner DH 1991b. Stabilities of consecutive A.C, C.C, G.G, U.C, and U.U mismatches in RNA internal loops: Evidence for stable hydrogen-bonded U.U and C.C.+ pairs. Biochemistry 30: 8242–8251 [DOI] [PubMed] [Google Scholar]
- Schroeder SJ, Turner DH 2000. Factors affecting the thermodynamic stability of small asymmetric internal loops in RNA. Biochemistry 39: 9257–9274 [DOI] [PubMed] [Google Scholar]
- Schroeder SJ, Turner DH 2001. Thermodynamic stabilities of internal loops with GU closing pairs in RNA. Biochemistry 40: 11509–11517 [DOI] [PubMed] [Google Scholar]
- Schroeder S, Kim J, Turner DH 1996. G.A and U.U mismatches can stabilize RNA internal loops of three nucleotides. Biochemistry 35: 16105–16109 [DOI] [PubMed] [Google Scholar]
- Schroeder SJ, Fountain MA, Kennedy SD, Lukavsky PJ, Puglisi JD, Krugh TR, Turner DH 2003. Thermodynamic stability and structural features of the J4/5 Loop in a Pneumocystis carinii Group I Intron. Biochemistry 42: 14184–14196 [DOI] [PubMed] [Google Scholar]
- Serra MJ, Lyttle MH, Axenson TJ, Schadt CA, Turner DH 1993. RNA hairpin loop stability depends on closing base pair. Nucleic Acids Res 21: 3845–3849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra MJ, Axenson TJ, Turner DH 1994. A model for the stabilities of RNA hairpins based on a study of the sequence dependence of stability for hairpins of six nucleotides. Biochemistry 33: 14289–14296 [DOI] [PubMed] [Google Scholar]
- Serra MJ, Barnes TW, Betschart K, Gutierrez MJ, Sprouse KJ, Riley CK, Stewart L, Temel RE 1997. Improved parameters for the prediction of RNA hairpin stability. Biochemistry 36: 4844–4851 [DOI] [PubMed] [Google Scholar]
- Serra MJ, Smolter PE, Westhof E 2004. Pronounced instability of tandem IU base pairs in RNA. Nucleic Acids Res 32: 1824–1828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabalina S, Spiridonov AN, Ogurtsov AY 2006. Computational models with thermodynamic and composition features improve siRNA design. BMC Bioinformatics 7: 65 doi: 10.1186/1471-2105-7-65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar N, Kennedy SD, Chen G, Krugh TR, Turner DH 2006. The NMR structure of an internal loop from 23S ribosomal RNA differs from its structure in crystals of 50s ribosomal subunits. Biochemistry 45: 11776–11789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprinzl M, Vassilenko KS 2005. Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res 33: 139–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffen P, Voss B, Rehmsmeier M, Reeder J, Giegerich R 2006. RNAshapes: An integrated RNA analysis package based on abstract shapes. Bioinformatics 22: 500–503 [DOI] [PubMed] [Google Scholar]
- Sugimoto N, Kierzek R, Freier SM, Turner DH 1986. Energetics of internal GU mismatches in ribooligonucleotide helixes. Biochemistry 25: 5755–5759 [DOI] [PubMed] [Google Scholar]
- Sugimoto N, Kierzek R, Turner DH 1987. Sequence dependence for the energetics of dangling ends and terminal base pairs in ribonucleic acid. Biochemistry 26: 4554–4558 [DOI] [PubMed] [Google Scholar]
- Tang J, Breaker RR 2000. Structural diversity of self-cleaving ribozymes. Proc Natl Acad Sci 97: 5784–5789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Thomas S, Tapia L, Giedroc DP, Amato NM 2008. Simulating RNA folding kinetics on approximated energy landscapes. J Mol Biol 381: 1055–1067 [DOI] [PubMed] [Google Scholar]
- Taskar B 2005. “Learning structured prediction models: A large margin approach.” PhD thesis, Stanford University, CA [Google Scholar]
- Taskar B, Chatalbashev V, Koller D, Guestrin C 2005. Learning structured prediction models: A large margin approach. In Proceedings of the 22nd International Conference on Machine Learning. pp. 896–903, Bonn, Germany [Google Scholar]
- Tinoco I Jr, Borer PN, Dengler B, Levin MD, Uhlenbeck OC, Crothers DM, Bralla J 1973. Improved estimation of secondary structure in ribonucleic acids. Nat New Biol 246: 40–41 [DOI] [PubMed] [Google Scholar]
- Tolbert BS, Kennedy SD, Schroeder SJ, Krugh TR, Turner DH 2007. NMR structures of (rGCUGAGGCU)2 and (rGCGGAUGCU)2: Probing the structural features that shape the thermodynamic stability of GA pairs. Biochemistry 46: 1511–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torarinsson E, Sawera M, Havgaard JH, Fredholm M, Gorodkin J 2006. Thousands of corresponding human and mouse genomic regions unalignable in primary sequence contain common RNA structure. Genome Res 16: 885–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulpan DC 2006. “Effective heuristic methods for DNA strand design.” PhD thesis,Department of Computer Science, University of British Columbia, Vancouver, BC, Canada [Google Scholar]
- Uzilov AV, Keegan JM, Mathews DH 2006. Detection of non-coding RNAs on the basis of predicted secondary structure formation free energy change. BMC Bioinformatics 7: 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vecenie CJ, Serra MJ 2004. Stability of RNA hairpin loops closed by AU base pairs. Biochemistry 43: 11813–11817 [DOI] [PubMed] [Google Scholar]
- Vecenie CJ, Morrow CV, Zyra A, Serra MJ 2006. Sequence dependence of the stability of RNA hairpin molecules with six nucleotide loops. Biochemistry 45: 1400–1407 [DOI] [PubMed] [Google Scholar]
- Walter AE, Wu M, Turner DH 1994. The stability and structure of tandem GA mismatches in RNA depend on closing base pairs. Biochemistry 33: 11349–11354 [DOI] [PubMed] [Google Scholar]
- Washietl S, Hofacker IL, Stadler PF 2005. Fast and reliable prediction of noncoding RNAs. Proc Natl Acad Sci 102: 2454–2459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westbrook J, Feng Z, Chen L, Yang H, Berman HM 2003. The Protein Data Bank and structural genomics. Nucleic Acids Res 31: 489–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson KA, Gorelick RJ, Vasa SM, Guex N, Rein A, Mathews DH, Giddings MC, Weeks KM 2008. High-throughput SHAPE analysis reveals structures in HIV-1 genomic RNA strongly conserved across distinct biological states. PLoS Biol 6: e96 doi: 10.1371/journal.pbio.0060096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, McDowell JA, Turner DH 1995. A periodic table of symmetric tandem mismatches in RNA. Biochemistry 34: 3204–3211 [DOI] [PubMed] [Google Scholar]
- Wuchty S, Fontana W, Hofacker IL, Schuster P 1999. Complete suboptimal folding of RNA and the stability of secondary structures. Biopolymers 49: 145–165 [DOI] [PubMed] [Google Scholar]
- Xayaphoummine A, Bucher T, Thalmann F, Isambert H 2003. Prediction and statistics of pseudoknots in RNA structures using exactly clustered stochastic simulations. Proc Natl Acad Sci 100: 15310–15315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia T, McDowell JA, Turner DH 1997. Thermodynamics of nonsymmetric tandem mismatches adjacent to G.C base pairs in RNA. Biochemistry 36: 12486–12497 [DOI] [PubMed] [Google Scholar]
- Xia T, SantaLucia J, Burkard ME, Kierzek R, Schroeder SJ, Jiao X, Cox C, Turner DH 1998. Thermodynamic parameters for an expanded nearest-neighbor model for formation of RNA duplexes with Watson-Crick base pairs. Biochemistry 37: 14719–14735 [DOI] [PubMed] [Google Scholar]
- Xu L, Hutter F, Hoos HH, Leyton-Brown K 2008. SATzilla: Portfolio-based algorithm selection for SAT. J Artif Intell Res 32: 565–606 [Google Scholar]
- Yang H, Jossinet F, Leontis N, Chen L, Westbrook J, Berman H, Westhof E 2003. Tools for the automatic identification and classification of RNA base pairs. Nucleic Acids Res 31: 3450–3460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Znosko BM, Silvestri SB, Volkman H, Boswell B, Serra MJ 2002. Thermodynamic parameters for an expanded nearest-neighbor model for the formation of RNA duplexes with single nucleotide bulges. Biochemistry 41: 10406–10417 [DOI] [PubMed] [Google Scholar]
- Znosko BM, Kennedy SD, Wille PC, Krugh TR, Turner DH 2004. Structural features and thermodynamics of the J4/5 loop from the Candida albicans and Candida dubliniensis group I introns. Biochemistry 43: 15822–15837 [DOI] [PubMed] [Google Scholar]
- Zuker M 1989. On finding all suboptimal foldings of an RNA molecule. Science 244: 48–52 [DOI] [PubMed] [Google Scholar]
- Zuker M 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31: 3406–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]