

Abstract
The GlmS ribozyme is believed to exploit a general acid-base catalytic mechanism in the presence of glucosamine-6-phosphate (GlcN6P) to accelerate self-cleavage by approximately six orders of magnitude. The general acid and general base are not known, and the role of the GlcN6P cofactor is even less well understood. The amine group of GlcN6P has the ability to either accept or donate a proton and could therefore potentially act as an acid or a base. In order to decipher the role of GlcN6P in the self-cleavage of glmS, we have determined the preferred protonation state of the amine group in the wild-type and an inactive G40A mutant using molecular dynamics simulations and free energy calculations. Here we show that, upon binding of GlcN6P to wild-type glmS, the pKa of the amine moiety is altered by the active site environment, decreasing by about 2.2 from a solution pKa of about 8.2. On the other hand, we show that the pKa of the amine group slightly increases to about 8.4 upon binding to the G40A inactive mutant of glmS. These results suggest that GlcN6P acts as a general acid in the self-cleavage of glmS. Upon binding to glmS, GlcN6P can easily release a proton to the 5′-oxygen of G1 during self-cleavage of the backbone phosphodiester bond. However, in the G40A inactive mutant of glmS, the results suggest that the ability of GlcN6P to easily release its proton is diminished, in addition to the possible lack of G40 as an effective base.
Keywords: free energy calculation, RNA self-cleavage, riboswitch, ribozyme, pKa shift
INTRODUCTION
Self-cleaving catalytic RNAs (ribozymes) regulate genes in many organisms through the cleavage of the backbone phosphodiester bond. Well-known self-cleaving ribozymes include the Hammerhead, Hairpin, Hepatitis delta virus (HDV), Varkud satellite (VS), and glmS ribozymes (Bevilacqua and Yajima 2006). GlmS ribozyme is located at the 5′ untranslated region of the mRNA carrying the gene for glucosamine-6-phosphate (GlcN6P) synthase, a member of the amidotransferase family of enzymes, in Gram-positive bacteria (Barrick et al. 2004; Winkler et al. 2004). GlcN6P is the precursor of cell walls of Gram-positive bacteria (Winkler et al. 2004); therefore, glmS ribozyme offers a good target for antibiotic development.
The GlmS ribozyme from Thermoanaerobacter tengcongensis (Fig. 1A) is ∼150 nucleotides (nt) long (Klein et al. 2007b) and self-cleaves its backbone phosphodiester bond between A(-1) and G1 in the presence of GlcN6P. It possesses a “switch-like” activity that is controlled by the concentration of GlcN6P. When the concentration of GlcN6P (Fig. 1B) is high, it activates self-cleavage upon binding to glmS, repressing the gene (Barrick et al. 2004; Winkler et al. 2004). Therefore, the glmS ribozyme is also a metabolite sensing riboswitch (Edwards et al. 2007), and the glmS-GlcN6P complex is stabilized by a bridging Mg2+ that does not participate in the self-cleavage mechanism (Roth et al. 2006). Many of the gene-regulating riboswitches rely on the binding of small metabolites that function as allosteric activators. For example, the expression platform of the SAM-II riboswitch undergoes conformational changes upon binding S-adenosylmethionine (SAM), resulting in stabilization of its pseudoknot structure and termination of translation (Corbino et al. 2005; Gilbert et al. 2008; Kelley and Hamelberg 2010). However, the conformations of the precleavage state, transition state mimic, and post-cleavage state of the glmS riboswitch/ribozyme are very similar, eliminating the possibility that GlcN6P is an allosteric activator (Klein and Ferre-D'Amare 2006; Lim et al. 2006; Cochrane et al. 2007, 2009; Klein et al. 2007b). The exact role of GlcN6P remains unclear. The rate of self-cleavage of glmS is about 106 times slower in the absence of GlcN6P, increasing from <10−5 min−1 to >10 min−1 under physiological conditions upon binding of GlcN6P (McCarthy et al. 2005; Roth et al. 2006; Klein et al. 2007a). On the other hand, glucose-6-phosphate (Glc6P), which has a hydroxyl group instead of an amine group, does not activate self-cleavage of glmS and acts as an inhibitor (McCarthy et al. 2005).
FIGURE 1.

X-ray crystal structure of glmS ribozyme (A) from Thermoanaerobacter tengcongensis complexed with GlcN6P (B), shown in red. Loop and helix segments are labeled according to Klein et al. (2007b; Fig. 1C). glmS self-cleaves its backbone phosphodiester bond between A(-1) and G1, shown in green. G40 is shown in yellow.
Therefore, the presence of the amine functional group is very important for catalysis, and many studies have proposed that GlcN6P could act as a coenzyme in the self-cleavage of glmS through the general acid-base mechanism (Fig. 2; Hampel and Tinsley 2006; Klein and Ferre-D'Amare 2006; Cochrane et al. 2007, 2009). The general acid-base catalytic mechanism has been extensively studied (Thompson and Raines 1994; Breslow and Chapman 1996; Sowa et al. 1997; Bevilacqua 2003; Emilsson et al. 2003; Bevilacqua et al. 2004; Lonnberg and Lonnberg 2005; Bevilacqua and Yajima 2006) and is common in both protein (Thompson and Raines 1994; Breslow and Chapman 1996; Sowa et al. 1997) and nucleic acid chemistry (Nakano et al. 2000; Han and Burke 2005; Guo et al. 2009). In glmS, the proposed cleavage mechanism is similar to that of other self-cleaving ribozymes, in which cleavage is initiated by the binding of GlcN6P. The 2′-hydrogen of the 2′-hydroxyl group of A(-1) is removed by a general base, and an SN2 nucleophilic attack to the scissile phosphorus atom of the backbone phosphate group is carried out by the 2′ O of A(-1), forming a penta-oxygen phosphate transition state (Fig. 2). The 5′-oxygen of G1 accepts a hydrogen from a general acid, breaking the backbone phosphdiester bond between 5′ O of G1 and the scissile P, forming the 5′-hydroxyl and 2′,3′-cyclic phosphate termini (Winkler et al. 2004; Fedor and Williamson 2005). In the presence of GlcN6P, this cleavage reaction happens spontaneously. What is the exact role of GlcN6P? The amine group of GlcN6P could potentially act as a general base or a general acid in the self-cleavage mechanism, since it has the ability to accept and donate a proton under the right condition. However, since the amine group of GlcN6P is far away (>5 Å) from the 2′ OH of A(-1) to effectively act as a base, the remaining possibility is that GlcN6P is the general acid, donating a proton to the 5′ O of G1 during the cleavage of the phosphodiester bond.
FIGURE 2.

The active site and the proposed mechanism for the self-cleavage of the glmS ribozyme. A view of the active site (A) of the glmS ribozyme with A(-1) and G1 shown in green, 2′ O of A(-1) shown as red sphere, the scissile P shown as yellow, and 5′ O of G1 also shown as red sphere. The precleavage (B), transition (C), and post-cleavage (D) states of the cleavage site with the general acid, HA, and base, B− are also shown.
The pKa of the amine group of GlcN6P in solution is about 8.2. Therefore, under physiological conditions, the amine group of GlcN6P cannot easily donate a proton and is somewhat less effective as a general acid. However, the active site environment could optimize the microscopic pKa in order to increase catalytic efficiency, similar to the well-studied HDV. In HDV, the pKa of N3 of C76 is perturbed toward neutrality, and the perturbed C76 has been shown to act as the general acid in the self-cleavage, based on experimental and computational studies (Nakano et al. 2000; Shih and Been 2001; Krasovska et al. 2005). Also, using Raman crystallography, Guo et al. (2009) showed that the pKa of N1 of A38 in the hairpin ribozyme (HPRZ) changes from about 3.7 in solution to about 5.5 in the folded HPRZ, allowing A38 to effectively act as a general acid. Therefore, in order to decipher the role of GlcN6P in the self-cleavage of glmS, we have sought to determine the effect of the active site environment on the binding and pKa of the amine group of GlcN6P using all-atom molecular dynamics simulations and detailed free energy calculations using thermodynamic integration. We have also investigated the effects of the G40A mutation of glmS on the binding and the protonation state preference of the amine group of GlcN6P. The rate (∼10−5 min−1) of self-cleavage of the G40A mutant of glmS in the presence of GlcN6P is similar to that of wild-type glmS in the absence of GlcN6P, even though the configuration of the binding site and the binding orientation of GlcN6P in the wild type and the inactive G40A mutant of glmS are almost identical (Klein et al. 2007a).
RESULTS AND DISCUSSION
The active site environment of glmS alters the pKa of the amine group of GlcN6P
The pKa of the amine group of GlcN6P in water was experimentally measured to be about 8.2 (McCarthy et al. 2005). Based on our free energy simulation results as summarized in Table 1, the pKa of the amine group of GlcN6P decreases by more than 2 and becomes more acidic upon entering the active site of glmS. At around the physiological pH, the amount of protons released upon GlcN6P entering the binding site of glmS can be estimated by the Henderson-Hasselbalch relationship in Equation 1:
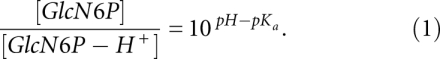 |
TABLE 1.
Relative binding free energies and changes in pKa of GlcN6P

The ratio of [GlcN6P]/[GlcN6P-H+] increases by more than 150 times upon GlcN6P moving from solution to the active site. The results suggest that the significant shift in the protonation state of GlcN6P upon entering the binding site of glmS would provide the proton required for GlcN6P to effectively act as a general acid in the cleavage of the backbone phosphodiester bond, as depicted in Figure 2. Interestingly, glmS is almost completely inactive in the absence of GlcN6P, which suggests that the transition state has a much lower energy in the presence of GlcN6P. Therefore, another possible and important role of GlcN6P in glmS cleavage could be stabilization of the penta-oxygen transition state by the protonated form of GlcN6P upon entering the binding site. Hence, the lack of an effective general acid and the destabilization of the transition state could be reasons why glmS does not self-cleave in the absence of GlcN6P.
On the other hand, the pKa of the amine group of GlcN6P slightly increases by about 0.24 upon binding to the active site of the G40A inactive mutant of glmS. This result suggests that the binding site environment of the G40A inactive mutant makes it harder for GlcN6P to release a proton upon binding and prevents GlcN6P from effectively acting as a general acid in addition to other possible effects due to the mutation, thus maintaining the cleavage rate of the free glmS. For example, the general base that removes the hydrogen from 2′-OH of A(-1) is not known and has been proposed to be G40. Therefore, since the unperturbed pKa of imine group of G is about 9.2 (Saenger 1984; Bevilacqua et al. 2004) and that of A is about 3.5 (Saenger 1984; Bevilacqua et al. 2004), G would be expected to act more effectively as a general base than A at the physiological pH due to the fact that the unperturbed pKa of the 2′-OH group of A(-1) is expected to be greater than 14. Why would the G40A mutant then be completely inactive in the presence of GlcN6P? If G40 is the base and everything remains equal, then the G40A mutant should maintain at least some activity in the presence of GlcN6P (Ferre-D'Amare 2010). However, the G40A mutation alters the active site environment of glmS, which alters the pKa of the amine group of GlcN6P, such that GlcN6P cannot effectively act as a general acid. Furthermore, the active site of the G40A mutant is slightly more electronegative than that of the wild type (as shown below), thus destabilizing the negatively charged penta-oxygen phosphate transition state. Hence, the lack of an effective base and an effective acid and an increase of the energy of the transition state are possibly responsible for the sixth order of magnitude decrease in the cleavage rate of the G40A mutant in the presence of GlcN6P. Therefore, our results suggest that the general acid (the amine group of GlcN6P) has a pKa of about 6 and the general base (if G is the base) has a pKa of about 9.2, similar to the possibility in the hairpin ribozyme, with pKas of 5.4 (A38; the acid) and 9.5 (Guo et al. 2009; Ferre-D'Amare 2010), respectively.
Configuration and the local environment of the binding site of glmS
In order to further understand the implications of the structure of the glmS ribozyme and the effect of the G40A mutation on the environment of the active site, we carried out two 50 nsec of unrestrained molecular dynamics simulations of the wild-type and G40A mutant complexes. It is obvious from the proposed mechanism of self-cleavage of glmS shown in Figure 2 that several atomic distances and orientations of several functional groups are important for activity. The atoms involved and the respective distances and angle are shown in Figure 3.
FIGURE 3.

Normalized probability distribution, p(r), of important atomic distances and angle in the binding site of both the wild-type (blue lines) and G40A inactive mutant (red lines) of the glmS ribozyme. (A) Distance from GlcN6P:N to G1:O5′. (B) Distance from A(-1):O2′ to scissile P (P*). (C) Angle between A(-1):O2′, P*, and G1:O5′. (D) Distance from G40:N1 to A(-1):O2′ (A40:N1 to A[-1]:O2′ in the G40A inactive mutant). The insets in A to D are the atomic view of these distances and angles. The cleavage site A(-1) and G1, the metabolite GlcN6P, and residue 40 are shown and colored by element. The rest of the active site is shown as white surface. The functional essential atoms are labeled (green) within the figure.
The distance between the amine nitrogen of GlcN6P and the 5′-oxygen of G1 is ∼2.8 Å for both the wild type and the G40A mutant (Fig. 3A), with a very narrow distribution. This distance is short enough to allow the amine group of GlcN6P to protonate the leaving group, thus positioning itself to act as a general acid upon entering the active site of glmS. The oxygen of the 2′-OH of A(-1) acts as a nucleophile after its proton has been removed by a general base and attacks the scissile phosphorus atom of the phosphate backbone between A(-1) and G1. The distance between the oxygen of the 2′-OH group of A(-1) and P also has a very narrow distribution and is ∼3.1 Å for both the wild type and the G40A mutant (Fig. 3B). The attacking nucleophile, 2′ O, the scissile phosphorus atom, P, and the leaving 5′ O atom are all in an almost straight line, as can be seen from the distribution of the angle formed by 2′ O, P, and 5′ O (Fig. 3C).
It has been suggested that the general base is G40 (Cochrane et al. 2007, 2009; Klein et al. 2007a), since the imine NH functional group with a pKa of about 9.2 is near the 2′-OH group of A(-1) (Klein and Ferre-D'Amare 2006; Cochrane et al. 2007). Therefore, we have also measured the distances between the oxygen of the 2′-OH group of A(-1) and N1 of G40 in the wild type and A40 in G40A inactive mutant of glmS, in order to investigate if there is a major difference between the wild type and the mutant, since adenine could also act as a general base, under the right conditions (Ditzler et al. 2009). Both distances show a broad distribution with similar range of values between ∼3 and 5 Å. However, the shapes of the distributions are slightly different, as shown in Figure 3D. Obviously, N1 of G40 of the wild type and of A40 in the G40A mutant are both within a reasonable distance from 2′-OH of A(-1) for proton transfer. The above results therefore suggest that the configuration of the active site in the wild type and G40A mutant of glmS are reasonably similar, as was observed from the X-ray crystal structures and concluded from a recent molecular dynamics simulation study (Banas et al. 2010). The active site is already preorganized for self-cleavage in the presence of GlcN6P. Since the G40A mutation does not alter the configuration of the binding site, the lack of self-cleavage could be due to the absence of the proposed general base G40 and due to changes in environment of the binding site that renders GlcN6P ineffective as a general acid.
Likewise, the hydration densities in the active sites of the wild type and G40A inactive mutant of glmS are very similar, as can be seen in Figure 4. Figure 4 shows the hydration occupancy of the active sites of the wild type and the G40A mutant, calculated using the PTRAJ module in AMBER 9 as previously described (Hamelberg et al. 2006), with the dimension of each grid set to (0.5 Å)3. Two water molecules, labeled as WAT1 and WAT2 in Figure 4, located in the active site of glmS are conserved in the G40A mutant and are highly localized during the molecular dynamics simulations. These water molecules have also been suggested to possibly take part in the self-cleavage by participating in the proton relay (Klein and Ferre-D'Amare 2006).
FIGURE 4.

Hydration density of the binding site of the wild type (A) and G40A inactive mutant (B) of the glmS ribozyme. GlcN6P is shown using ball and stick and colored by element. WAT1 and WAT2 are the two highly localized and conserved water molecules in the binding site. Red-colored mesh represents 10 times that of bulk water. Green-colored mesh shows six times that of bulk water. Shown using a green sphere is a Mg2+ ion in the binding site that helps to stabilize the complex.
Obviously, the electrostatic environment of an ionizable group is the dominant factor that would affect its pKa (Jones and Wilson 1981; Potter et al. 1994; Misra and Honig 1995; Lee et al. 2002; Allison and Xin 2006; Tang et al. 2007). A more electropositive environment will most likely stabilize the unprotonated form of the ionizable group, while the more electronegative environment will tend to stabilize the protonated form. Therefore, we have calculated the electrostatic potential of the free wild type and free G40A mutant of glmS by solving the nonlinear Poisson Boltzmann equation using APBS (Baker et al. 2001), in order to study the electrostatic environment of the binding site, as shown in Figure 5. As evident in Figure 5, the active site of the G40A mutant of glmS is slightly more electronegative than the wild type. The amine group of GlcN6P is bound in a pocket formed exclusively by bases from the P2.1 and P2.2 domains (Fig. 1). This pocket has a higher potential in the wild type than that of the G40A mutant. This result suggests that changes in the functional groups as a result of the G40A mutation slightly decrease the electrostatic potential in the active site, thus stabilizing the protonated form of the amine group of GlcN6P and destabilizing the penta-oxygen transition state. The proposed transition state of the self-cleavage of glmS is highly negatively charged; therefore, a more electronegative active site environment will destabilize the transition state and considerably slow down the reaction. One of the major changes due to the G40A mutation is that the central imine (NH) group of G40 with a partial positive charge on the hydrogen becomes an unprotonated nitrogen in the G40A mutant with a partial negative charge. The change in electrostatic environment of the active site of the G40A mutant could explain the slight increase in the pKa of the amine group of GlcN6P. Additionally, we have examined the distance between the nitrogen of the amine group of GlcN6P and N1 of G40 in the wild type and A40 of the G40A inactive mutant of glmS, also shown in Figure 5. This average distance is ∼6.9 Å and is slightly larger for the wild-type glmS than that of the G40A mutant. Clearly, G40 is close enough to the binding site, such that mutating this residue would alter the electrostatic potential around the amine group of GlcN6P.
FIGURE 5.

Electrostatic potential map of the binding site of the wild-type (A) and G40A inactive mutant (B) of the glmS ribozyme. The normalized probability distribution, p(r), of the distance between GlcN6P:N and G40:N1 of the wild-type glmS (blue line) and the distance between GlcN6P:N and A40:N1 of the G40A inactive mutant (red line). (C) The electrostatic maps are colored by potential with a range from −10 kT/e to +10 kT/e. Red color denotes negative potential and blue color denotes positive potential.
Implications of the pKa shift of the amine group of GlcN6P upon binding to glmS
The results suggest that GlcN6P is acting as a general acid in the self-cleavage of glmS, and they are consistent with available experiments. Binding of GlcN6P to glmS is a key step for self-cleavage and is accompanied by decrease in the pKa of the amine group from approximately 8.2 to approximately 6. The free energy differences used to estimate the change in pKa also provide the relative binding affinity between the protonated and deprotonated forms of GlcN6P and glmS. The deprotonated form of GlcN6P has a lower binding free energy than the protonated form (ΔG4 < ΔG2) for wild-type glmS by >2 kcal/mol. These results are consistent with the observation by Klein et al. (2007b) that glmS prefers to bind GlcN6P with the amine group in the deprotonated state. These results are also consistent with the fact that GlcN6P binds very poorly to glmS at low pH of about 5.5 (Klein et al. 2007b; Cochrane et al. 2009) and Glc6P, which lacks the amine group, does not show any pH-dependent binding with glmS (Klein et al. 2007b). Also, it was shown that the cleavage rate of glmS increases with pH, reaching the maximum rate around pH 8 (Winkler et al. 2004; McCarthy et al. 2005; Klein et al. 2007b; Cochrane et al. 2009). Our results suggest that, as the pH is increased from 5, the proton release by the amine group of GlcN6P will progressively become easier, resulting in enhanced binding and increased cleavage rate.
The dependence of the kinetics and catalytic activity of self-cleavage of glmS from Bacillus anthracis on pH has also been investigated by Cochrane et al. (2009). Their plots of 1/Km and kcat/Km suggest the presence of ionizable groups of apparent pKas of 6.3 ± 0.2 and 7.5 ± 0.2, respectively, where Km is the concentration of GlcN6P needed to reach half-maximal cleavage rate and kcat is the maximum cleavage rate. They attributed the pKa of 6.3 ± 0.2 to the phosphate moiety of GlcN6P, since the pKa of the phosphate group in solution is approximately 6.1, and there are no other functional groups in glmS with pKa near 6. The second pKa of 7.5 ± 0.2 was assumed to be that of the amine group of GlcN6P because the pKa of that group in solution is approximately 8.2. However, an alternative explanation could be provided in light of our simulation results and since the above interpretation of the experimental results did not take into account the fact that the pKa of the amine and phosphate groups of GlcN6P could be perturbed by the active site environment of glmS. Our results suggest that the pKa of 6.3 ± 0.2 is due to the amine group of GlcN6P, since the pKa in the active site of glmS is calculated to be around 6, and the pKa of 7.5 ± 0.2 could be the total effect of the amine group and the general base (maybe G40 with unperturbed pKa of ∼9.2), since kobs = f * k1, where f is the fraction of the functional form of general acid and base, and k1 is the rate of bond breaking (Bevilacqua 2003; Smith et al. 2008). Also, the kinetics of self-cleavage of glmS cannot be described with a single pKa, since the plot of pH versus kcat fits very well with a slope of 0.7, instead of 1 (Cochrane et al. 2009).
Titration of the phosphate group was also suggested to be the reason for the pH-dependent binding of GlcN6P to glmS (Cochrane et al. 2009) in the pH range of 5–9, since the pKa of the phosphate group in solution is about 6. However, metal ions, such as Mg2+, near phosphate groups are expected to alter the pKa (Anderson and Record 1995; Misra and Draper 2000). Therefore, it would be difficult to protonate the phosphate group of GlcN6P, which interacts with a bridging Mg2+ in the binding site of glmS that is mainly responsible for the binding of GlcN6P and stability of the complex. Hence, the activity of glmS in the presence of GlcN6P decreases with decreasing concentration of Mg2+ (Winkler et al. 2004), and GlcN, which lacks the phosphate group on position six, is a poorer activator of glmS than GlcN6P due to the fact that it forms a less stable complex with glmS (Winkler et al. 2004; McCarthy et al. 2005; Blount et al. 2006; Jansen et al. 2006; Link et al. 2006; Mayer and Famulok 2006).
Furthermore, the fact that the substrate analog, Glc6P, which has an hydroxyl functional group instead of the amine group, binds to glmS in a pH-independent fashion (Klein et al. 2007b), suggests that the amine group and not the phosphate group is responsible for the pH-dependent effect of GlcN6P binding.
Conclusion
GlmS ribozyme undergoes self-cleavage of the backbone phosphodiester bond between A(-1) and G1 in bacteria and has riboswitch action mediated by the metabolite GlcN6P. The pKa of the amine of GlcN6P is shown to decrease from approximately 8.2 to approximately 6 upon binding to the wild-type glmS, based on free energy calculations and molecular dynamics simulations. The results suggest that GlcN6P can easily release its proton as soon as it enters the binding site of glmS; therefore, GlcN6P is proposed to act as the general acid in the self-cleavage of glmS ribozyme. Unlikely, the wild-type glmS, the pKa of the amine group of GlcN6P is slightly increased upon binding to the G40A inactive mutant of glmS. This slight increase in the pKa of the amine group of GlcN6P renders GlcN6P ineffective as a general acid, since GlcN6P will hold on to its proton. Without a general acid and possibly a general base near the cleavage site, and with a less stable transition state due to the electrostatic environment of the active site, the cleavage rate would remain close to basal level, as experimentally observed. The results also suggest that the amine group of GlcN6P, not the phosphate group, is responsible for the pH-dependent binding to glmS. The deprotonated form of GlcN6P has a higher binding affinity with wild-type glmS than the protonated form, also consistent with experiments. The results of this work therefore reveal a possible interplay of the active site environment of glmS and GlcN6P, of which G40 is an active participant.
Here, we have studied the role of GlcN6P in the self-cleavage of the glmS ribozyme. We conclude that GlcN6P is acting as a general acid in the cleavage of the backbone phosphodiester bond between A(-1) and G1. The general base necessary to initiate self-cleavage by abstracting the proton from the 2′-OH group of A(-1) has been proposed to be G40. However, there are several other possibilities, including a water molecule and one of the nonbridging backbone phosphate oxygen. Several of these possibilities were recently investigated using molecular dynamics simulations, and it was proposed that the phosphate backbone oxygen is the general base (Banas et al. 2010). Additionally, we believe that similar pKa calculations carried out in the present work could be used to shed light on the functional group that is playing the role as the base in the self-cleavage of the glmS ribozyme/riboswitch.
MATERIALS AND METHODS
pKa, the negative log of the dissociation constant of a proton from an acid (Ka), is a measure of the ability of a molecule to act as an acid (HA) at a particular pH. It is related to the free energy change of deprotonation, 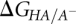 , by
, by
therefore,
where kB is the Boltzmann constant and T is the temperature. When HA moves from one electrostatic environment to another, the pKa could change. A thermodynamic cycle (as shown in Fig. 6) enables one to evaluate the change in free energy, and hence the pKa, in different electrostatic environments. Figure 6 is a schematic representation of the thermodynamic cycle of binding of the protonated and deprotonated forms of GlcN6P to the binding site of glmS ribozyme.
FIGURE 6.

Thermodynamic cycle connecting the deprotonation of GlcN6P in water and in the binding site of the glmS ribozyme.
From the thermodynamic cycle, one can write
where ΔΔG is the relative free energy of binding. The absolute binding free energies, ΔG2 and ΔG4, are difficult to calculate from simulations. However, the difference between the free energies of deprotonation, ΔG1 and ΔG3, can be estimated with some amount of accuracy from free energy calculations by molecular dynamics simulations, assuming that certain contributions, such as bond breaking energies, would cancel out in the difference, ΔG1 − ΔG3. The change in the microscopic pKa, ΔpKa, of the amine group of GlcN6P as it moves from water to the binding site of glmS can therefore be estimated by Equation 5,
Since the microscopic pKa of the amine group of GlcN6P is known, the pKa in the binding site of glmS could therefore be estimated. ΔG1 and ΔG3 in Equation 5 are the free energies of perturbing the partial charges from one protonation state to the other in glmS and solution, respectively, and are calculated using thermodynamics integration (Warshel et al. 1986; Gao et al. 1989; Potter et al. 1994; Misra and Honig 1995; Simonson et al. 2004; Ghosh and Cui 2008) with molecular dynamics simulations.
The Amber 9 suite of programs (Case et al. 2005) was used to carry out all of the molecular dynamics simulations and free energies calculations. The free energy of deprotonating GlcN6P in solution, ΔG3, was calculated by perturbing the partial charges of the protonated GlcN6P to the deprotonated form using thermodynamics integration in explicit TIP3P water (Jorgensen et al. 1983). The potential energy function, V(λ), defining the transformation from the protonated state to the deprotonated state of GlcN6P is controlled by λ, where λ = 0 for the protonated state and λ = 1 for the deprotonated state. The integral over <∂V(λ)/∂λ>λ, from λ = 0–1 gives the free energy change. The integral was calculated using a seven-point Gaussian quadrature at seven discrete λ values: 0.02544, 0.12923, 0.29707, 0.50000, 0.70292, 0.87076, and 0.97455. The partial atomic charges for the protonated and deprotonated form of GlcN6P were derived using the standard two-step RESP method (Bayly et al. 1993) from the electrostatic potential calculated using Gaussian03 (Frisch et al. 2004) at the HF/6-31G* level of theory. The force field parameters used for both forms of GlcN6P were obtained from the generalized AMBER force field (GAFF) (Wang et al. 2004) parameter set.
The free energy, ΔG1, of deprotonating GlcN6P in the binding site of glmS was also calculated. The three-dimensional structure of the complex was taken from the 1.7 Å X-ray crystal structure of the T. tengcongensis glmS ribozyme with Protein Data Bank (PDB) identification no. 2Z75(Klein et al. 2007b). The modified Cornell et al. (1995) force field parameters by Perez et al. (2007) were used to carry out the simulations. The glmS–GlcN6P complex was solvated in an octahedron of TIP3P water, up to 10 Å away from the complex. Ten Mg2+ ions were added to the system, in addition to the nine present in the X-ray crystal structure, in order to get ∼0.03 M of Mg2+. One hundred five Na+ were then added to neutralize the system. During the free energy simulations, the phosphorus atoms of the backbone of glmS were constraint with a very weak force constant of 1 kcal/mol/Å2, so that the three-dimensional structure does not move far away from the crystal structure.
The system was equilibrated by carrying out several rounds of minimization while holding the ribozyme and ligand with a harmonic constraint of force constant of 500, 200, 100, 50, and 25 kcal/mol/Å2 during each run. For each run, a total of 2000 steps of minimization were carried out. After the five rounds of minimization, the system was warmed up from 100K to 300K in 0.5 nsec during a molecular dynamics simulation with a harmonic constraint of force constant of 25 kcal/mol/Å2 applied to the complex using a time step of 1 fsec. At 300K, the system was further equilibrated for 1 nsec with a harmonic constraint of force constant 5 kcal/mol/Å2 applied only to the phosphorus atoms of the phosphate backbone of the ribozyme using a time step of 2 fsec. This equilibrated structure was used both for the free energy calculation described above and the 50-nsec unconstraint molecular dynamics simulation.
All simulations were carried out at a constant temperature of 300K and constant pressure of 1 bar. The temperature was controlled using the Langevin thermostat with a collision frequency of 1 psec−1. A time step of 2 fsec was used to numerically solve Newton's equation of motion. The electrostatic interactions were calculated using particle mesh Ewald (PME) method (Essmann et al. 1995), and all bonds involving H-atoms were constrained using the SHAKE algorithm (Ryckaert et al. 1977). The cutoff for all long-range nonbonded interaction was set to 9 Å. For each λ value during the thermodynamic integration, 1,000,000 steps (2 nsec) of molecular dynamics simulations were performed, and data from the second half were used. The first half was considered to be an equilibration phase. Each simulation was repeated three times with a different initial random seed.
ACKNOWLEDGMENTS
We thank Dr. Adrian Ferré-D'Amaré for helpful comments and discussions. This work was supported in part by research initiation grants from Georgia State University, the Department of Chemistry, and the Georgia Cancer Coalition (GCC) scholar award, and a National Science Foundation CAREER Award. This work was also supported by Georgia State's IBM System p5 supercomputer, acquired through a partnership of the Southeastern Universities Research Association and IBM supporting the SURAgrid initiative.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.2334110.
REFERENCES
- Allison S, Xin Y 2006. Electrokinetic transport of a spherical gel-layer model particle: Inclusion of charge regulation and application to polystyrene sulfonate. J Colloid Interface Sci 299: 977–988 [DOI] [PubMed] [Google Scholar]
- Anderson CF, Record MT 1995. Salt nucleic-acid interactions. Annu Rev Phys Chem 46: 657–700 [DOI] [PubMed] [Google Scholar]
- Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA 2001. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc Natl Acad Sci 98: 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banas P, Walter NG, Sponer J, Otyepka M 2010. Protonation states of the key active site residues and structural dynamics of glmS riboswitch as reveled by molecular dynamics. J Phys Chem B 114: 8701–8712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrick JE, Corbino KA, Winkler WC, Nahvi A, Mandal M, Collins J, Lee M, Roth A, Sudarsan N, Jona I, et al. 2004. New RNA motifs suggest an expanded scope for riboswitches in bacterial genetic control. Proc Natl Acad Sci 101: 6421–6426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayly CI, Cieplak P, Cornell WD, Kollman PA 1993. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges—the Resp model. J Phys Chem 97: 10269–10280 [Google Scholar]
- Bevilacqua PC 2003. Mechanistic considerations for general acid-base catalysis by RNA: Revisiting the mechanism of the hairpin ribozyme. Biochemistry 42: 2259–2265 [DOI] [PubMed] [Google Scholar]
- Bevilacqua PC, Yajima R 2006. Nucleobase catalysis in ribozyme mechanism. Curr Opin Chem Biol 10: 455–464 [DOI] [PubMed] [Google Scholar]
- Bevilacqua PC, Brown TS, Nakano S, Yajima R 2004. Catalytic roles for proton transfer and protonation in ribozymes. Biopolymers 73: 90–109 [DOI] [PubMed] [Google Scholar]
- Blount K, Puskarz I, Penchovsky R, Breaker R 2006. Development and application of a high-throughput assay for glmS riboswitch activators. RNA Biol 3: 77–81 [DOI] [PubMed] [Google Scholar]
- Breslow R, Chapman WH 1996. On the mechanism of action of ribonuclease A: Relevance of enzymatic studies with a p-nitrophenylphosphate ester and a thiophosphate ester. Proc Natl Acad Sci 93: 10018–10021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods RJ 2005. The Amber biomolecular simulation programs. J Comput Chem 26: 1668–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochrane JC, Lipchock SV, Strobel SA 2007. Structural investigation of the GlmS ribozyme bound to its catalytic cofactor. Chem Biol 14: 97–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochrane JC, Lipchock SV, Smith KD, Strobel SA 2009. Structural and chemical basis for glucosamine 6-phosphate binding and activation of the glmS ribozyme. Biochemistry 48: 3239–3246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbino KA, Barrick JE, Lim J, Welz R, Tucker BJ, Puskarz I, Mandal M, Rudnick ND, Breaker RR 2005. Evidence for a second class of S-adenosylmethionine riboswitches and other regulatory RNA motifs in alpha-proteobacteria. Genome Biol 6: R70.71–R70.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA 1995. A second generation force-field for the simulation of proteins, nucleic-acids, and organic-molecules. J Am Chem Soc 117: 5179–5197 [Google Scholar]
- Ditzler MA, Sponer J, Walter NG 2009. Molecular dynamics suggest multifunctionality of an adenine imino group in acid-base catalysis of the hairpin ribozyme. RNA 15: 560–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards TE, Klein DJ, Ferre-D'Amare AR 2007. Riboswitches: Small-molecule recognition by gene regulatory RNAs. Curr Opin Struct Biol 17: 273–279 [DOI] [PubMed] [Google Scholar]
- Emilsson GM, Nakamura S, Roth A, Breaker RR 2003. Ribozyme speed limits. RNA 9: 907–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG 1995. A smooth particle mesh Ewald method. J Chem Phys 103: 8577–8593 [Google Scholar]
- Fedor MJ, Williamson JR 2005. The catalytic diversity of RNAS. Nat Rev Mol Cell Biol 6: 399–412 [DOI] [PubMed] [Google Scholar]
- Ferre-D'Amare AR 2010. The glmS ribozyme: Use of a small molecule coenzyme by a gene-regulatory RNA. Q Rev Biophys 8: 1–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JJA, Vreven T, Kudin KN, Burant JC, et al. 2004. Gaussian 03. Gaussian, Inc, Wallingford, CT [Google Scholar]
- Gao J, Kuczera K, Tidor B, Karplus M 1989. Hidden thermodynamics of mutant proteins: A molecular dynamics analysis. Science 244: 1069–1072 [DOI] [PubMed] [Google Scholar]
- Ghosh N, Cui Q 2008. pKa of residue 66 in Staphylococal nuclease. I. Insights from QM/MM simulations with conventional sampling. J Phys Chem B 112: 8387–8397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert SD, Rambo RP, Van Tyne D, Batey RT 2008. Structure of the SAM-II riboswitch bound to S-adenosylmethionine. Nat Struct Mol Biol 15: 177–182 [DOI] [PubMed] [Google Scholar]
- Guo M, Spitale RC, Volpini R, Krucinska J, Cristalli G, Carey PR, Wedekind JE 2009. Direct Raman measurement of an elevated base pKa in the active site of a small ribozyme in a precatalytic conformation. J Am Chem Soc 131: 12908–12909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamelberg D, Shen TY, and McCammon JA 2006. Insight into the role of hydration on protein dynamics. J Chem Phys 125: 094905 doi: 10.1063/1.2232131 [DOI] [PubMed] [Google Scholar]
- Hampel KJ, Tinsley MM 2006. Evidence for preorganization of the glmS ribozyme ligand binding pocket. Biochemistry 45: 7861–7871 [DOI] [PubMed] [Google Scholar]
- Han J, Burke JM 2005. Model for general acid-base catalysis by the hammerhead ribozyme: pH-activity relationships of G8 and G12 variants at the putative active site. Biochemistry 44: 7864–7870 [DOI] [PubMed] [Google Scholar]
- Jansen JA, McCarthy TJ, Soukup GA, Soukup JK 2006. Backbone and nucleobase contacts to glucosamine-6-phosphate in the glmS ribozyme. Nat Struct Mol Biol 13: 517–523 [DOI] [PubMed] [Google Scholar]
- Jones RL, Wilson WD 1981. Effect of ionic strength on the pKa of ligands bound to DNA. Biopolymers 20: 141–154 [DOI] [PubMed] [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML 1983. Comparison of simple potential functions for simulating liquid water. J Chem Phys 79: 926–935 [Google Scholar]
- Kelley JM, Hamelberg D 2010. Atomistic basis for the on-off signaling mechanism in SAM-II riboswitch. Nucleic Acids Res 38: 1392–1400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein DJ, Ferre-D'Amare AR 2006. Structural basis of glmS ribozyme activation by glucosamine-6-phosphate. Science 313: 1752–1756 [DOI] [PubMed] [Google Scholar]
- Klein DJ, Been MD, Ferre-D'Amare AR 2007a. Essential role of an active-site guanine in glmS ribozyme catalysis. J Am Chem Soc 129: 14858–14859 [DOI] [PubMed] [Google Scholar]
- Klein DJ, Wilkinson SR, Been MD, Ferre-D'Amare AR 2007b. Requirement of helix p2.2 and nucleotide g1 for positioning the cleavage site and cofactor of the glmS ribozyme. J Mol Biol 373: 178–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasovska MV, Sefcikova J, Spackova N, Sponer J, Walter NG 2005. Structural dynamics of precursor and product of the RNA enzyme from the hepatitis delta virus as revealed by molecular dynamics simulations. J Mol Biol 351: 731–748 [DOI] [PubMed] [Google Scholar]
- Lee KK, Fitch CA, Garcia-Moreno B 2002. Distance dependence and salt sensitivity of pairwise, coulombic interactions in a protein. Protein Sci 11: 1004–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J, Grove BC, Roth A, Breaker RR 2006. Characteristics of ligand recognition by a glmS self-cleaving ribozyme. Angew Chem Int Ed 45: 6689–6693 [DOI] [PubMed] [Google Scholar]
- Link KH, Guo LX, Breaker RR 2006. Examination of the structural and functional versatility of glmS ribozymes by using in vitro selection. Nucleic Acids Res 34: 4968–4975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonnberg T, Lonnberg H 2005. Chemical models for ribozyme action. Curr Opin Chem Biol 9: 665–673 [DOI] [PubMed] [Google Scholar]
- Mayer G, Famulok M 2006. High-throughput-compatible assay for glmS riboswitch metabolite dependence. ChemBioChem 7: 602–604 [DOI] [PubMed] [Google Scholar]
- McCarthy TJ, Plog MA, Floy SA, Jansen JA, Soukup JK, Soukup GA 2005. Ligand requirements for glmS ribozyme self-cleavage. Chem Biol 12: 1221–1226 [DOI] [PubMed] [Google Scholar]
- Misra VK, Draper DE 2000. Mg2+ binding to tRNA revisited: The nonlinear Poisson-Boltzmann model. J Mol Biol 299: 813–825 [DOI] [PubMed] [Google Scholar]
- Misra VK, Honig B 1995. On the magnitude of the electrostatic contribution to ligand–DNA interactions. Proc Natl Acad Sci 92: 4691–4695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano S, Chadalavada DM, Bevilacqua PC 2000. General acid-base catalysis in the mechanism of a hepatitis delta virus ribozyme. Science 287: 1493–1497 [DOI] [PubMed] [Google Scholar]
- Perez A, Marchan I, Svozil D, Sponer J, Cheatham TE, Laughton CA, Orozco M 2007. Refinement of the AMBER force field for nucleic acids: Improving the description of α/γ conformers. Biophys J 92: 3817–3829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter MJ, Gilson MK, McCammon JA 1994. small molecule pKa prediction with continuum electrostatics calculations. J Am Chem Soc 116: 10298–10299 [Google Scholar]
- Roth A, Nahvi A, Lee M, Jona I, Breaker RR 2006. Characteristics of the glmS ribozyme suggest only structural roles for divalent metal ions. RNA 12: 607–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckaert J-P, Ciccotti G, Berendsen HJC 1977. Numerical integration of the Cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J Comput Phys 23: 327–341 [Google Scholar]
- Saenger W, ed. 1984. Principles of nucleic acid structure. Spring-Verlag, New York [Google Scholar]
- Shih I, Been MD 2001. Involvement of a cytosine side chain in proton transfer in the rate-determining step of ribozyme self-cleavage. Proc Natl Acad Sci 98: 1489–1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonson T, Carlsson J, Case DA 2004. Proton binding to proteins: pKa calculations with explicit and implicit solvent models. J Am Chem Soc 126: 4167–4180 [DOI] [PubMed] [Google Scholar]
- Smith MD, Mehdizadeh R, Olive JE, Collins RA 2008. The ionic environment determines ribozyme cleavage rate by modulation of nucleobase pKa. RNA 14: 1942–1949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowa GA, Hengge AC, Cleland WW 1997. O-18 isotope effects support a concerted mechanism for ribonuclease A. J Am Chem Soc 119: 2319–2320 [Google Scholar]
- Tang CL, Alexov E, Pyle AM, Honig B 2007. Calculation of pKa in RNA: On the structural origins and functional roles of protonated nucleotides. J Mol Biol 366: 1475–1496 [DOI] [PubMed] [Google Scholar]
- Thompson JE, Raines RT 1994. Value of general acid-base catalysis to ribonuclease-A. J Am Chem Soc 116: 5467–5468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA 2004. Development and testing of a general amber force field. J Comput Chem 25: 1157–1174 [DOI] [PubMed] [Google Scholar]
- Warshel A, Sussman F, King G 1986. Free energy of charges in solvated proteins: Microscopic calculations using a reversible charging process. Biochemistry 25: 8368–8372 [DOI] [PubMed] [Google Scholar]
- Winkler WC, Nahvi A, Roth A, Collins JA, Breaker RR 2004. Control of gene expression by a natural metabolite-responsive ribozyme. Nature 428: 281–286 [DOI] [PubMed] [Google Scholar]