

Abstract
Trm5 is a eukaryal and archaeal tRNA methyltransferase that catalyzes methyl transfer from S-adenosylmethionine (AdoMet) to the N1 position of G37 directly 3′ to the anticodon. While the biological role of m1G37 in enhancing translational fidelity is well established, the catalytic mechanism of Trm5 has remained obscure. To address the mechanism of Trm5 and more broadly the mechanism of N-methylation to nucleobases, we examined the pH-activity profile of an archaeal Trm5 enzyme, and performed structure-guided mutational analysis. The data reveal a marked dependence of enzyme-catalyzed methyl transfer on hydrogen ion equilibria: the single-turnover rate constant for methylation increases by one order of magnitude from pH 6.0 to reach a plateau at pH 7.0. This suggests a mechanism involving proton transfer from G37 as the key element in catalysis. Consideration of the kinetic data in light of the Trm5–tRNA–AdoMet ternary cocrystal structure, determined in a precatalytic conformation, suggests that proton transfer is associated with an induced fit rearrangement of the complex that precedes formation of the reactive configuration in the active site. Key roles for the conserved R145 side chain in stabilizing a proposed oxyanion at G37-O6, and for E185 as a general base to accept the proton from G37-N1, are suggested based on the mutational analysis.
Keywords: anticodon loop, S-adenosylmethionine (AdoMet), pH-rate profile, active site assembly
INTRODUCTION
The methylation of proteins and nucleic acids at nitrogen plays key roles in regulating gene expression. In proteins, recent work has established the importance of lysine and arginine methylation for controling biological activities (Bedford and Clarke 2009; Ng et al. 2009). Methylation at one or multiple positions of these side chains alters charge distribution, hydrogen bonding, and steric properties, and thus is well suited to regulate the assembly and interactions of macromolecular complexes. In nucleic acids, methylation occurs at nearly all nonglycosidic nitrogens in the canonical nucleobases, including the guanosine N3, which is methylated in wybutosine after an additional ring is added to the original base (Noma et al. 2006). The only positions that have no methylation found so far are the adenosine N3 and N7. Nucleobases with N-methylation are particularly prevalent in structural RNAs, most commonly tRNA and rRNA, accounting for a subset of the over 100 known modified nucleosides (Czerwoniec et al. 2009). However, despite the large variety of chemical and structural contexts in which these RNA N-methylation events occur, and their well-established importance in regulation of protein synthesis (Gustilo et al. 2008), little is known regarding the mechanisms of the enzyme-catalyzed reactions. Only the N-methyltransferases M.EcoRI and M.TaqI, which catalyze formation of m6A in DNA as part of bacterial restriction-modification systems, have been studied in any detail to date (Mashhoon and Reich 1994; Goedecke et al. 2001; Newby et al. 2002).
The m1G37 modification in the tRNA anticodon loop is conserved in all three domains of life (Supplemental Fig. S1), and plays an important role in increasing fidelity of ribosomal decoding in both the cytoplasm and mitochondria (Bjork et al. 1989, 2001; Lee et al. 2007). In methanogens, m1G37 is also essential for tRNA recognition by phosphoseryl- and cysteinyl-tRNA synthetases (Hauenstein and Perona 2008; Hauenstein et al. 2008; Zhang et al. 2008). In all organisms, the synthesis of m1G37 is catalyzed by the AdoMet-dependent tRNA(m1G37) methyltransferase, with S-adenosyl homocysteine (AdoHcy) as the second product. Interestingly, the bacterial enzyme TrmD and archaeal/eukaryal enzyme Trm5 feature distinct tertiary folds in their active-site domains, suggesting that they have evolved separately (Ahn et al. 2003; Elkins et al. 2003; Goto-Ito et al. 2008, 2009). TrmD and Trm5 also recognize different determinants on tRNA: Trm5 requires full integrity of the L-shaped tRNA structure for activity, whereas TrmD requires only the D-stem and anticodon stem–loop motifs in the vertical arm (Christian and Hou 2007).
Trm5 from Methanocaldococcus jannaschii is the lead model system for elucidating the enzyme-catalyzed mechanism of m1G37 formation (Christian et al. 2004, 2006; Christian and Hou 2007). Crystal structures of the enzyme reveal that it is a member of the class I amino-methyltransferase family (Goto-Ito et al. 2008, 2009), which transfers the methyl group from AdoMet to nitrogen in widely different chemical environments. Trm5 features a conserved NLPK motif in the active site, a variant of the consensus NPPY motif in other class I amino-methyl transferases. Kinetic studies show that the rate-limiting step in the catalytic cycle of Trm5 occurs after formation of m1G37 on the enzyme (Christian et al. 2006, 2010), similar to other class I methyltransferases (Flynn et al. 1996; Bhattacharya and Dubey 1999; Vilkaitis et al. 2001). The crystal structure of M. jannaschii Trm5 bound to tRNA and AdoMet reveals a conformation poised for catalysis (Goto-Ito et al. 2009), in which the N1 of G37 is located 2.8 Å from the methyl group of AdoMet. However, although the high pKa (∼9.5) of the guanine N1 suggests that enzyme-catalyzed deprotonation would be necessary for methyl transfer, there is no apparent general base positioned to accept this proton. Thus, the mechanism for methyl transfer remains unclear despite the detailed structural information.
To directly address the role of proton transfer in the Trm5 mechanism, we have determined the pH-dependence of the methylation event as it occurs in the enzyme active site. To our knowledge, this is the first such study for an endocyclic N-methylation reaction on nucleic acids. The data reveal that catalysis by Trm5 is indeed dependent on hydrogen ion equilibria, and suggest that proton transfer from N1 of G37 is necessary to activate the enzyme. Together with the crystal structures of Trm5 and structure-guided mutational analysis, these data place fundamental constraints on the enzyme mechanism and emphasize the importance of an induced-fit process after the initial tRNA binding event to form the reactive configuration required for methyl transfer. Importantly, two strictly conserved residues in the Trm5 family, R145 and E185, have been identified by sequence analysis. The roles of these amino acids in rate enhancement are addressed by structure-guided mutational analysis.
RESULTS AND DISCUSSION
pH-dependent methyl transfer by M. jannaschii Trm5
To measure the pH-dependence of M. jannaschii Trm5 reactivity, we determined the single-turnover rate constant for methyl transfer under conditions of saturating AdoMet and tRNA, with molar excess of enzyme over tRNA. The advantage of single turnover (compared with steady-state) assays is that the observed rate constant (kobs) must correspond to either the chemical step (kchem) or a closely linked induced-fit conformational rearrangement prior to catalysis (kconf) in the following reaction scheme:
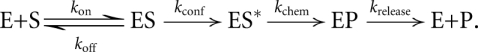 |
Here, ES* represents a Trm5–tRNA–AdoMet ternary complex that has undergone an induced-fit conformational change, so that the reactive substrate moieties are poised for catalysis.
Measurements of the pH-dependence of the single-turnover rate constant were exhaustively controlled, and the quality of the tRNA substrate was rigorously monitored. An in vitro transcript of M. jannaschii tRNACys was refolded by annealing (Supplemental Fig. S1), and its integrity as a Trm5 substrate verified by demonstrating a capacity for methylation to ∼70% levels in extended time courses (e.g., pH 6.0, 8.1, and 9.8, Supplemental Figs. S2a,b). Saturating conditions were established at 7.5 μM Trm5, 0.5 μM tRNACys, and 25 μM [3H]-AdoMet at pH 6.0, 8.1 and 9.8. All reactions were carried out at 55°C with Trm5 present in 15-fold molar excess over tRNA. Further controls established that the order of mixing and the nature of the buffer do not influence the rates. Finally, we also demonstrated that preincubation of Trm5 at pH 6.0, followed by adjustment of the solution to pH 8.0, does not cause irreversible inactivation of the enzyme. Enzyme treated in this manner retained equivalent activity in single-turnover assays.
Time courses of [3H]-methyl group transferred to tRNA were best fit to a single exponential function at all values of pH, yielding kobs. The plot of kobs against pH shows a steep increase in the rate of methyl transfer as the proton concentration is lowered, up to an asymptote at pH 7.0 (Fig. 1A). The logarithmic plot of kobs (log[kobs]) against pH reveals a slope of 0.92 (Fig. 1B), indicating a mechanism involving the deprotonation of one ionizing group. The data are most consistent with the anticipated deprotonation of N1 of G37 and are well fit to the equation:
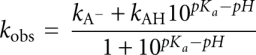 |
where kobs is the observed reaction rate at a specific pH, kAH is the activity of the protonated form of G37 (kAH = 0), kA is the activity of the deionized form of G37, and Ka is the equilibrium constant for the dissociation of the proton (Fersht 1999). An optimal fit of the data (R2 = 0.98) yields pKa = 6.5 ± 0.1, roughly three units below the pKa of the N1 proton of guanosine in solution (Clauwaert and Stockx 1968). Shifts of pKa values of this magnitude and larger have been well documented for other enzymes (Frey and Hegeman 2007). However, it should be noted that a definitive assignment of the pKa to the deprotonation of G37-N1 is not possible based on this experiment alone.
FIGURE 1.

pH-dependent methylation by M. jannaschii Trm5. (A) Plot of the observed rate constant, kobs, against pH. (B) Logarithmic plot of kobs versus pH.
Proposed mechanism of methyl transfer by Trm5
Although the identity of the transferred proton cannot be unambiguously established at present, the dependence of the reaction rate on proton removal is consistent with a mechanism in which the observed rate constant (kobs) corresponds to nucleophilic attack on the methyl group of AdoMet by the deprotonated N1 of G37 in tRNA, with electron transfer to the sulfonium ion (Fig. 2). Proton abstraction from G37 N1 could be facilitated by an enzyme residue functioning as a general base, with a shift of electron density from the N–H bond to the O6 of the guanine ring. The electron sink at O6 is important for promoting the initial proton removal from N1, while a positively charged enzyme side chain could stabilize the increased electron density at O6. Interestingly, interplay between endocyclic position 1 and exocyclic position 6 in purines was previously noted for N6 methylation of adenine, where methylation at N1 occurs first, followed by methyl migration to N6 via the base-catalyzed Dimroth rearrangement, involving ring opening of the adenine base (Engel 1975).
FIGURE 2.

Proposed mechanism for Trm5. Proton abstraction during docking of G37 in the active site by a general base (E185 in M. jannaschii Trm5, upper left) yields deprotonated G37 as visualized in the crystal structure with electron density shifted to O6 (stabilized by a positively charged residue, such as R145 in M. jannaschii Trm5, upper right). Nucleophilic attack on AdoMet by deprotonated N1 then leads to product formation.
The crystal structure of the Trm5–tRNA–AdoMet ternary complex reveals that the methyl group has not been transferred from AdoMet to G37 (Goto-Ito et al. 2009), perhaps because reactivity is greatly reduced at the low temperature of crystallization compared with the optimal 85°C growth temperature of M. jannaschii. For the following reasons, it also appears likely that the crystal lattice has trapped a deprotonated form of G37 with the N1 nitrogen in an sp2-hybridized state, in which the lone pair of electrons is poised to attack the AdoMet methyl group (Fig. 2). First, G37 N1 is located only 2.8–3.0 Å from the electrophilic methyl group of AdoMet, so that an interstitial proton may be sterically excluded. Second, the adjacent positively charged sulfonium group of AdoMet likely also disfavors approach by protonated N1. Finally, despite the demonstrated importance of proton equilibria to catalysis (Fig. 1), the structure does not reveal an appropriately positioned general base that might abstract the N1 proton (see Fig. 3A). This consideration suggests that proton abstraction from N1 occurs prior to the configuration observed in the crystal lattice, which represents a state that is clearly poised for catalysis. Thus, proton removal would have occurred during the induced-fit conformational rearrangement leading to juxtaposition of G37 with AdoMet. The implication is that the pH dependence of kobs in single-turnover analysis corresponds to proton transfer during a slower process of induced fit, rather than the bond-breaking and bond-forming steps of methyl transfer (that is, the measured rate constant kobs corresponds to kconf in the above scheme, and not to kchem).
FIGURE 3.

Structure of the M. jannaschii Trm5 active site bound to tRNA and AdoMet. (A) An overall view of the active site, showing all of the residues that have been tested by mutational analysis in this work. (B) Spatial positioning of the catalytic residues R145 and E185 relative to G37 and AdoMet. The position and interactions of the adjacent R186 are also depicted. (C) Detail of the Trm5 active site showing close juxtaposition of G37 N1 with the AdoMet methyl group. Hydrogen bonds are shown as dotted yellow lines. Key enzyme residues mentioned in the main text are shown in green in the enzyme binary complex with sinefungin, and are shown in red in the enzyme ternary complex with AdoMet and tRNA. (D) Trm5 residues that stabilize the backbone groups of G37 and A38 in the anticodon loop of the ternary complex. Figures are drawn with PyMol (PDB 2YX1) corresponding to Trm5–sinefungin and with PyMol (PDB 2ZZN) corresponding to the Trm5–tRNACys–AdoMet ternary complex (Goto-Ito et al. 2009). H-bonds are shown in yellow dots, while the distance between the N1 position of G37 and the methyl group of AdoMet is shown in gray. Superposition of the two structures in (B), (C), and (D) was done by aligning sinefungin in the binary complex with AdoMet in the ternary complex, with an RSMD of 0.47 Å on the basis of the superposition of α-carbon atoms.
Structure-guided mutational analysis of Trm5
As a test of this proposed mechanism, we used structure-guided mutational analysis of M. jannaschii Trm5 to elucidate the identities of amino acids that may directly facilitate methyl transfer. Mutant enzymes were constructed and analyzed by single-turnover assays with saturating AdoMet and increasing molar ratio of enzyme to tRNA to determine the Kd for tRNA and the saturating rate constant kobs (summarized in Table 1). Importantly, all of the mutational effects were associated with alteration of kobs, rather than Kd, indicating that the driving force for activity and specificity is associated with the first-order induced fit and chemical steps of the reaction scheme, rather than the binding of tRNA. Mutational effects were interpreted by examining the relevant functional groups in the enzyme binary complex with the AdoMet analog sinefungin (Goto-Ito et al. 2008) and in the ternary complex with AdoMet and tRNA, where the base of G37 becomes unstacked from its neighbors in the anticodon loop and flips into the active site (Fig. 3A; Goto-Ito et al. 2009). Comparison of binary and ternary complex structures was used to gain insight into how insertion of the guanine base into the active site affects the positioning of functional groups.
TABLE 1.
Kinetic parameters with respect to tRNA for M. jannaschii Trm5

Structure-based sequence alignments show that the only two strictly conserved residues among Trm5 enzymes are R145 and E185 (Supplemental Fig. S3A,B). Comparison of the binary and ternary Trm5 complexes shows that R145 is located on a mobile surface loop that markedly changes conformation upon tRNA binding. In the tRNA-bound enzyme, R145 orients its guanidinium group toward the Hoogsteen face of the flipped-out G37 base, allowing donation of two H-bonds to the O6 and N7 groups (Fig. 3B). The interaction between conserved R145 and O6 of G37 supports the proposed mechanism (Fig. 2), suggesting that the function of this positively charged residue is to stabilize the negative charge on O6 in the initial step. The R145A mutation reduces kobs by 20-fold, demonstrating the importance of this arginine in facilitating rate enhancement.
Unlike R145, the position and conformation of E185 within the core catalytic domain are not greatly altered upon binding of tRNA G37 in the active site (Fig. 3B). This strictly conserved glutamate does not directly bind either substrate, but is located immediately N-terminal to R186, which does reorient in the ternary complex to stabilize AdoMet binding by forming a charged hydrogen bond with the carboxylate group of the methyl donor. The position of E185 adjacent to the active site, together with its strict conservation in all known Trm5 enzymes and the solution pKa of 4.3 for the side-chain carboxylate, suggest that E185 may function as the general base for removal of the G37-N1 proton, and that proton abstraction catalyzed by this residue occurs together with the induced-fit conformational change leading to the fully assembled active site that is visualized in the crystal structure. The properties of the E185A and E185Q mutants support this hypothesis, because the single-turnover kobs measured for these enzymes is decreased by 300- and 60-fold, respectively, compared with the wild-type (WT) Trm5 (Table 1). The more than threefold improved activity of E185Q compared with E185A suggests that E185 is crucial both for general base catalysis and for the conformational change that precedes catalysis. In contrast, E185D, which retains the capacity to function as a general base (pKa = 3.7), is reduced only 2.4-fold in kobs compared with WT Trm5. Similar to WT Trm5, analysis of the pH-activity dependence of the E185D mutant also reveals a plot of log(kobs) against pH with a slope of 1.0 and a pKa of 6.6 ± 0.2 (Supplemental Fig. S4). WT and E185D Trm5 each therefore appear to function by the same mechanism. Further, the high kobs retained by E185D supports the notion that this parameter corresponds to an induced-fit conformational change, because the flexibility required for motion of the protein and RNA groups relative to each other should allow retraction of the carboxylate by the length of a carbon–carbon bond, while still permitting the juxtaposition required for proton transfer.
Induced fit for active-site assembly
To further test whether R145 or E185 function in active-site assembly, we evaluated their roles in AdoMet binding using pre-steady-state burst kinetics, as described previously (Christian et al. 2010). In this assay, the kinetics of m1G37 synthesis is monitored under conditions of saturating tRNA and molar excess of AdoMet over enzyme, to permit one round of methyl transfer on the enzyme, followed by steady-state turnovers. WT Trm5 exhibits a burst of rapid product synthesis in the first turnover, followed by a slower and linear phase reflecting the rate-limiting product release step. Analysis of the amplitude of the burst phase as a function of AdoMet concentration then allows estimation of the binding affinity of Trm5 to AdoMet (Table 2). While WT Trm5 exhibits Kd (AdoMet) of 0.4 ± 0.1 μM, identical to the value reported previously (Christian et al. 2010), the R145A and E185D mutations elevate Kd (AdoMet) by 10-fold and 11-fold, respectively (Table 2). Since neither R145 nor E185 directly bind AdoMet, these data suggest that each of these catalytically crucial residues are also involved in active-site assembly. Coupling of AdoMet binding to catalysis is also revealed by the properties of the R186A mutant, for which kobs is decreased by 12-fold (Table 1; Fig. 3B). Notably, instead of R186 in archaeal Trm5, sequence homology analysis reveals that eukaryotic organisms have conserved H186 (Supplemental Fig. S3A,B). While H186 in principle could act as a general base for proton abstraction of G37, it is unlikely to perform this function in the presence of a functional E185, due to the strict conservation of the latter residue and the much stronger effect on kobs brought about by its mutation (>300-fold by E185A; Table 1). The role of H186 in eukaryotic Trm5 enzymes is presently unknown.
TABLE 2.
Kinetic parameters with respect to AdoMet for M. jannaschii Trm5

To provide a more complete assessment of catalytic function, we also examined the roles of other highly conserved residues in the active site (Table 1; Fig. 3). The NLPK loop is of central interest, because of its conserved role in stabilizing AdoMet among class I amino-methyl transferases. In the ternary Trm5–tRNACys–AdoMet complex, the side-chain amide of N265 accepts a hydrogen bond from the exocyclic 2-NH2 of G37, while P267 stacks on the guanine ring (Fig. 3C). Although both residues move slightly away from AdoMet upon G37 binding, mutations of N265 had only modest effects on kobs (0.8-, 6-, and 12-fold reduction for N265H, N265Q, and N265A, respectively), whereas mutations of P267 had much more severe effects (120-fold reduction for P267A, P267A/N265Q, and P267A/N265H). The near-WT activity of N265H, which may retain a hydrogen-bond acceptor at an equivalent spatial position, reveals the importance of the hydrogen bond with G37-N2. However, the stronger effect of the mutations containing P267A suggests a key role for this proline in coordinating the active site rearrangement. Y177 stabilizes G37 by stacking on the opposing side relative to P267 (Fig. 3C,D). The Y177A mutation produces a 60-fold decrease in kobs, while the Y177F mutant retains near-WT activity. These findings demonstrate that the hydrogen bond formed by the Y177 hydroxyl group with the 2′-OH of the G37 ribose is not crucial to rate enhancement. We also found that the N265A and Y177A mutations increase Kd for AdoMet by 2.5-fold and 12-fold, respectively (Table 2). The effect of the Y177A mutation on AdoMet binding again reveals structural coupling between the tRNA-G37 and AdoMet binding sites.
The side-chain carboxylate of D223 contacts both the 2′- and 3′-OH groups of the adenosine ribose of AdoMet, and this residue also remains relatively unchanged in its spatial position upon tRNA binding (Fig. 3C). Removal of the carboxylic side chain of D223 resulted in either a severe loss in kobs (D223A, 170-fold) or complete loss of activity (D223N and D223L, greater than 1,200-fold), whereas activity was preserved in D223E (Table 1). These data are consistent with conservation of D223 in the Trm5 family, since the only other naturally occurring alternative is the substitution of aspartate with glutamate. The D223A mutation, which destabilizes the ribose moiety of AdoMet, also increased the Kd for AdoMet by 22-fold to 8.8 ± 1.3 μM. While the properties of the D223 mutants, considered alone, suggest the possibility that this residue might function as the catalytic base, we favor E185 in this role for two reasons. First, as described above, E185 is appropriately positioned to accept a proton from G37-N1 during the induced-fit transition, while D223 binds a distal portion of AdoMet and is not located near the reactive moieties. Second, D223 is conserved as an AdoMet-binding residue in many class I methyltransferases that function to catalyze methyl transfer at a variety of chemically diverse positions on the nucleobases. A role in acid-base catalysis for Trm5 thus appears less plausible.
Several of the mutations studied here have been previously examined by steady-state analysis (Christian et al. 2006). In all cases, the mutational effects on kcat were significantly smaller than the corresponding effects on kobs. For example, the R145A mutation reduced kcat by 2.5-fold (vs. kobs by 20-fold); the Y177A mutation reduced kcat by 6.2-fold (vs. kobs by 60-fold); the D223A mutation reduced kcat by 25-fold (vs. kobs by 170-fold); the N265A mutation reduced kcat by 6.2-fold (vs. kobs by 12-fold); and the P267A mutation reduced kcat by 83-fold (vs. kobs by 120-fold). In each case, the kcat value is also smaller in magnitude than the respectively measured kobs value, suggesting that for these mutants the kcat corresponds to a step after methyl transfer, as demonstrated for the WT enzyme (Christian et al. 2010). The smaller kcat effect compared with the kobs effect thus suggests a lesser role of the specific residue in product release. All of the mutations for which steady-state kinetics were measured also display a larger effect on Km for tRNA (6–12-fold), than on the tRNA binding affinity (more than twofold). This is consistent with product release being the rate-limiting step, because Km represents a complex combination of rate and equilibrium constants in this case. Interpretation of the pre–steady-state data is more straightforward, because the fundamental rate and equilibrium constants are directly measured, giving unambiguous insight into the effects of mutations.
We also used single-turnover assays to assess the roles of several positively charged residues that interact with or lie adjacent to the tRNA anticodon loop backbone near the active site. For example, K318 forms a salt bridge to the phosphate of A38, R181 forms a salt bridge to the phosphate at U39 and to the D-arm backbone at positions C25/G26), and the side chain of K137 is reoriented toward solvent upon tRNA binding (Fig. 3D). Alanine substitutions of these three residues were created and mutant enzymes were evaluated for their performance in tRNA binding and methyl transfer kinetics. Among these mutants, the largest effect was observed for R181A, for which kobs is reduced by 20-fold, suggesting that the bridging interaction between the anticodon loop and the coaxially stacked D-arm is important for catalysis, probably by facilitating the induced-fit rearrangement. K137A is reduced in kobs by fourfold, suggesting a smaller role in the induced-fit process. Surprisingly, the K318A mutation had no effect on kobs despite its interaction with an adjacent phosphate that might have been expected to participate in the flipping of the G37 base into the active site.
CONCLUSION
This work addresses how Trm5 catalyzes deprotonation of the N1 of G37 to enable methyl transfer from AdoMet. In principle, deprotonation of N1 might be envisioned to occur simply from its approach toward the positively charged sulfonium group of AdoMet, without invoking the need for a general base. In this case, the role of the enzyme would be entirely to stabilize the bound substrates and to juxtapose the deprotonated nitrogen and the electrophilic methyl group. However, based on the strong mutational effect of the strictly conserved E185 and R145 residues on kobs for methyl transfer, the data presented here support the reaction mechanism for methyl transfer outlined in Figure 2, in which E185 acts as the general base that abstracts the proton from N1 of G37, while R145 lowers the transition-state free energy by interacting with the incipient negative charge on O6. This proposed mechanism now provides the basis for further experiments to clarify specific details. For example, the use of guanine base analogs will be useful in more definitively establishing whether the measured pKa of 6.5 ± 0.1 associated with pH-dependent methyl transfer (Fig. 1) indeed corresponds to the downward shift in pKa for the N1 of G37, as we have hypothesized.
An important finding is that, while neither E185 nor R145 form a direct interaction with the methyl donor in the ternary complex, both residues enable AdoMet binding, suggesting that both play a further key role in promoting active-site assembly. Active-site organization is also facilitated by coordinated conformational changes of the residues that organize binding to the G37 base (i.e., Y177, N265, and P267) and those that stabilize AdoMet (i.e., R186, N265, and D223). Additional residues (i.e., K318 and R181) appear to be involved in shaping the tRNA backbone in the D-anticodon arm, so as to facilitate stable insertion of G37 into the active site. The involvement of diverse enzyme side chains in the active site suggests that the induced fit rearrangement of the enzyme–tRNA–AdoMet complex is elaborate, coupling both short- and long-range interactions to arrive at the reactive configuration of the complex. The assignment of the proton transfer to the induced-fit rearrangement makes it particularly challenging to identify the residues or motifs that are the key determinants in the process. Further work to develop a fluorescence approach to directly monitor the induced-fit process will be necessary and beneficial to gain insight into this process.
The large catalytic defect of the E185A mutant, demonstrated by a reduction of kobs by greater than 300-fold, reflects loss of both the catalytic carboxylate group and most of the aliphatic portion of the side chain. Because the E185Q mutant remains severely defective with a loss of 60-fold in kobs compared with the WT enzyme, it appears that acid-base catalysis plays a major role in rate enhancement. This notion is distinct from methylation at DNA adenine-N6 by M.TaqI, which is instead critically dependent on substrate organization for rate enhancement (Newby et al. 2002). An additional distinction is that, while deprotonation of G37-N1 by Trm5 occurs early, and the nucleophilic attack on AdoMet is by nitrogen in the sp2 hybridization state (Fig. 2), deprotonation by M.TaqI occurs only after the nitrogen has acquired significant sp3 character (Goedecke et al. 2001; Newby et al. 2002), due to the much higher pKa (∼20) of adenine N6. Consistent with this analysis, a pH kinetic analysis of the m6A DNA methylase M.EcoRI showed only a linear fourfold change in kobs over a wide pH range (Mashhoon and Reich 1994). Based on these two examples, it appears that enzymatic methylation at exocyclic adenine N6 may depend less on acid-base chemistry for rate acceleration than methylation at endocyclic guanine N1. Further studies of additional methltransferases will be necessary to test this prediction.
This work provides a benchmark that will be useful as a model for parallel investigations of the proton transfer mechanisms of other amino methyl transferases that operate on structural RNAs. An important example is the m1G37 synthesis reaction catalyzed by the bacteria-specific TrmD enzyme, which shares no structural homology with Trm5 and so must develop a distinct stereochemical pathway for the same chemical reaction. Other examples include amino methyl transfer reactions that target chemically distinct positions to synthesize m1A, m6A, m2G, m22G, m7G, and m4C, all of which are widely present in nucleic acids and contribute to the central process of gene expression.
MATERIALS AND METHODS
Expression and purification of M. jannaschii Trm5 and tRNACys
M. jannaschii Trm5 with a C-terminal His-tag sequence was expressed in the vector pET-22b in Escherichia coli BL21(DE3)/RIL cells. Protein expression was induced by addition of 0.3 mM IPTG to the growing cultures at A600 between 0.4 and 0.6. Trm5-containing cells were lysed by sonication, and the enzyme purified by heating of the cell extract to 75°C for 20 min to precipitate native proteins, followed by metal-affinity (Talon, ClonTech) and Q Sepharose chromatography steps (Christian et al. 2006). Active-site titration of the purified enzyme based on the amplitude in the burst assay revealed ∼50% active fraction (Christian et al. 2010), which was used to correct the enzyme concentration as determined from the Bradford assay. To prepare tRNA for kinetic analysis, the M. jannaschii tRNACys gene was transcribed from a duplex DNA template constructed from complementary synthetic oligonucleotides containing a 10-bp overlapping region, which were then extended using the Klenow fragment of E. coli DNA polymerase (Sherlin et al. 2001). Transcription reactions were carried out as previously described for synthesis of M. mazei tRNACys transcripts, except that 2′-O methyl sugar modifications at the two 5′ nucleotides of the noncoding strand were omitted (Hauenstein et al. 2008). RNA was recovered by ethanol precipitation, resuspended in 10 mM Tris-HCl (pH 7.5), and heated to 85°C for 3 min. MgCl2 was then added to 2 mM final concentration, and the reactions were slow cooled to room temperature to maximize refolding.
Single-turnover methylation assays
Methylation at the N1 position of G37 in M. jannaschii tRNACys transcripts was assayed at 55°C, the highest temperature compatible with retention of structure in the RNA (optimal growth temperature of M. jannaschii at 85°C). Determination of kinetic parameters Kd (tRNA) and kobs by single-turnover kinetic assays, and determination of the kinetic Kd (AdoMet) by presteady-state assays were as described (Christian et al. 2010), using 3H-labeled S-AdoMet (67.3 Ci/mmol; Perkin-Elmer). Reactions were initiated by addition of tRNA. Aliquots taken at specific time points were precipitated with 5% (w/v) trichloroacetic acid on filter pads, washed extensively, and air dried prior to scintillation counting. Data were background-subtracted and corrected for a quenching efficiency of ∼60%. Curve fitting to the equation for a single ionizing system (see Results and Discussion) was performed with Kaleidagraph.
Measurement of pH-dependent methyl transfer activity
Buffers used in the pH-dependent measurements were as follows: sodium cacodylate (pH 6.0); MES (pH 6.0, 6.1, 6.2, 6.4, 6.5, 6.6); MOPS (pH 6.6, 6.7, 6.9, 7.1); glycyl glycine (pH 7.1, 7.3, 7.5, 8.1, 8.6, 8.7); glycine (pH 8.7, 9.3, 9.8). Each buffer was made as a 5× solution (0.5 M buffer, 0.5 M KCl, 30 mM MgCl2, 0.5 mM EDTA), mixed with a 5× solution B (20 mM DTT and 0.12 mg/mL BSA), and diluted to 1× to check the pH. If the pH was lower than the normal value, drops of 5 M KOH were added to the 5× solution such that the 1× solution became properly adjusted. This adjustment was particularly important for pH values of 6.6 and lower, because measurements of activity without the adjustment failed to reveal the full activity at low pH values. Uncorrected measurements at low pH values led to the previous suggestion that a slope of two characterizes the log kobs versus pH plot (Hou and Perona 2010). No differences in rate were observed for reactions run at pH values where two different buffers were used. Reactions at pH lower than 6.6 were monitored by hand sampling, while those at higher pH values were monitored using a rapid quench apparatus (Kintek RQF-3). For single turnover condition, M. jannaschii Trm5 enzyme was used at 7.5 μM (15-fold molar excess of tRNA) as the active concentration and was added to one syringe at 55°C containing a final concentration of 0.1 M buffer, 0.1 M KCl, 6 mM MgCl2, 0.1 mM EDTA, 4 mM DTT, and 0.024 mg/mL bovine serum albumin (BSA). Upon rapid mixing with 0.5 μM tRNACys (previously heat-cooled and annealed) and 25 μM AdoMet in the same buffer from the second syringe at 55°C, the time course of the reaction was monitored. Substrate saturation at pH 6.0, 8.1, and 9.8 was shown by measuring kobs in reactions for which the enzyme and tRNA concentrations were each reduced by a factor of 2, while maintaining enzyme concentrations in 15-fold molar excess. Control reactions for order of addition were carried out at pH 6, 8, and 10; no significant differences in measured rates were observed. The consistency in the measured kobs across all three pH values establishes that both the tRNA and AdoMet substrates are stable at all relevant pH values. The resistance of the tRNA substrate to alkaline degradation was further confirmed by analysis on a 12% PAGE/7 M urea gel. Standard deviations are plotted in Figure 1.
SUPPLEMENTAL MATERIAL
Supplemental material can be found at http://www.rnajournal.org.
ACKNOWLEDGMENTS
We thank Tom Bruice, Eugene Mueller, and Philip Bevilacqua for helpful discussions, and Eric Wickstrom for assistance in utilizing the 3D projection environment for molecular design and surgical simulation supported by USAMRMC W81XWH-09-1-0577. This work was supported by NIH Grants GM53763 (to J.J.P.) and GM81601 (to Y.M.H.).
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.2376210.
REFERENCES
- Ahn HJ, Kim HW, Yoon HJ, Lee BI, Suh SW, Yang JK 2003. Crystal structure of tRNA(m1G37)methyltransferase: Insights into tRNA recognition. EMBO J 22: 2593–2603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedford MT, Clarke SG 2009. Protein arginine methylation in mammals: Who, what, and why. Mol Cell 33: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya SK, Dubey AK 1999. Kinetic mechanism of cytosine DNA methyltransferase MspI. J Biol Chem 274: 14743–14749 [DOI] [PubMed] [Google Scholar]
- Bjork GR, Wikstrom PM, Bystrom AS 1989. Prevention of translational frameshifting by the modified nucleoside 1-methylguanosine. Science 244: 986–989 [DOI] [PubMed] [Google Scholar]
- Bjork GR, Jacobsson K, Nilsson K, Johansson MJ, Bystrom AS, Persson OP 2001. A primordial tRNA modification required for the evolution of life? EMBO J 20: 231–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian T, Hou YM 2007. Distinct determinants of tRNA recognition by the TrmD and Trm5 methyl transferases. J Mol Biol 373: 623–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian T, Evilia C, Williams S, Hou YM 2004. Distinct origins of tRNA(m1G37) methyltransferase. J Mol Biol 339: 707–719 [DOI] [PubMed] [Google Scholar]
- Christian T, Evilia C, Hou YM 2006. Catalysis by the second class of tRNA(m1G37) methyl transferase requires a conserved proline. Biochemistry 45: 7463–7473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian T, Lahoud G, Liu C, Hou YM 2010. Control of catalytic cycle by a pair of analogous tRNA modification enzymes. J Mol Biol 400: 204–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clauwaert J, Stockx J 1968. Interactions of polynucleotides and thier components. I. Dissociation constants of the bases and their derivatives. Z Naturforsch B 23: 25–30 [PubMed] [Google Scholar]
- Czerwoniec A, Dunin-Horkawicz S, Purta E, Kaminska KH, Kasprzak JM, Bujnicki JM, Grosjean H, Rother K 2009. MODOMICS: A database of RNA modification pathways. 2008 update. Nucleic Acids Res 37: D118–D121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkins PA, Watts JM, Zalacain M, van Thiel A, Vitazka PR, Redlak M, Andraos-Selim C, Rastinejad F, Holmes WM 2003. Insights into catalysis by a knotted TrmD tRNA methyltransferase. J Mol Biol 333: 931–949 [DOI] [PubMed] [Google Scholar]
- Engel JD 1975. Mechanism of the Dimroth rearrangement in adenosine. Biochem Biophys Res Commun 64: 581–586 [DOI] [PubMed] [Google Scholar]
- Fersht AR 1999. Structure and mechanism in protein science. W. H. Freeman and Company, New York [Google Scholar]
- Flynn J, Glickman JF, Reich NO 1996. Murine DNA cytosine-C5 methyltransferase: pre-steady- and steady-state kinetic analysis with regulatory DNA sequences. Biochemistry 35: 7308–7315 [DOI] [PubMed] [Google Scholar]
- Frey PA, Hegeman A 2007. Enzymatic reaction mechanisms. Oxford University Press, New York [Google Scholar]
- Goedecke K, Pignot M, Goody RS, Scheidig AJ, Weinhold E 2001. Structure of the N6-adenine DNA methyltransferase M.TaqI in complex with DNA and a cofactor analog. Nat Struct Biol 8: 121–125 [DOI] [PubMed] [Google Scholar]
- Goto-Ito S, Ito T, Ishii R, Muto Y, Bessho Y, Yokoyama S 2008. Crystal structure of archaeal tRNA(m(1)G37)methyltransferase aTrm5. Proteins 72: 1274–1289 [DOI] [PubMed] [Google Scholar]
- Goto-Ito S, Ito T, Kuratani M, Bessho Y, Yokoyama S 2009. Tertiary structure checkpoint at anticodon loop modification in tRNA functional maturation. Nat Struct Mol Biol 16: 1109–1115 [DOI] [PubMed] [Google Scholar]
- Gustilo EM, Vendeix FA, Agris PF 2008. tRNA's modifications bring order to gene expression. Curr Opin Microbiol 11: 134–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauenstein SI, Perona JJ 2008. Redundant synthesis of cysteinyl-tRNACys in Methanosarcina mazei. J Biol Chem 283: 22007–22017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauenstein SI, Hou YM, Perona JJ 2008. The homotetrameric phosphoseryl-tRNA synthetase from Methanosarcina mazei exhibits half-of-the-sites activity. J Biol Chem 283: 21997–22006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou YM, Perona JJ 2010. Stereochemical mechanisms of tRNA methyltransferases. FEBS Lett 584: 278–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Kramer G, Graham DE, Appling DR 2007. Yeast mitochondrial initiator tRNA is methylated at guanosine 37 by the Trm5-encoded tRNA (guanine-N1-)-methyltransferase. J Biol Chem 282: 27744–27753 [DOI] [PubMed] [Google Scholar]
- Mashhoon N, Reich NO 1994. Investigation of ionizable residues critical for sequence-specific enzymatic DNA modification: Protein modification and steady-state and pre-steady-state kinetic pH analyses of EcoRI DNA methyltransferase. Biochemistry 33: 7113–7119 [DOI] [PubMed] [Google Scholar]
- Newby ZE, Lau EY, Bruice TC 2002. A theoretical examination of the factors controlling the catalytic efficiency of the DNA-(adenine-N6)-methyltransferase from Thermus aquaticus. Proc Natl Acad Sci 99: 7922–7927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SS, Yue WW, Oppermann U, Klose RJ 2009. Dynamic protein methylation in chromatin biology. Cell Mol Life Sci 66: 407–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noma A, Kirino Y, Ikeuchi Y, Suzuki T 2006. Biosynthesis of wybutosine, a hyper-modified nucleoside in eukaryotic phenylalanine tRNA. EMBO J 25: 2142–2154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherlin LD, Bullock TL, Nissan TA, Perona JJ, Lariviere FJ, Uhlenbeck OC, Scaringe SA 2001. Chemical and enzymatic synthesis of tRNAs for high-throughput crystallization. RNA 7: 1671–1678 [PMC free article] [PubMed] [Google Scholar]
- Vilkaitis G, Merkiene E, Serva S, Weinhold E, Klimasauskas S 2001. The mechanism of DNA cytosine-5 methylation. Kinetic and mutational dissection of Hhai methyltransferase. J Biol Chem 276: 20924–20934 [DOI] [PubMed] [Google Scholar]
- Zhang CM, Liu C, Slater S, Hou YM 2008. Aminoacylation of tRNA with phosphoserine for synthesis of cyseinyl-tRNA(Cys). Nat Struct Mol Biol 15: 507–514 [DOI] [PMC free article] [PubMed] [Google Scholar]