

Abstract
Although protein kinase C (PKC) plays an important role in sensitizing prostate cancer cells to apoptosis, and suppression of PKC is able to trigger an apoptotic crisis in cells harboring oncogenic ras, little is known about whether dyregulation of Ras effectors in prostate cancer cells, together with loss of PKC, is synthetically lethal. The current study aims at investigating whether prostate cancer cells with aberrant Ras effector signaling are sensitive to treatment with HMG (a PKC inhibitor) for the induction of apoptosis. We show that prostate cancer DU145 cells expressing a high level of JNK1 become susceptible to apoptosis after treatment with HMG, in which caspase 8 is activated and cytochrome c is released to the cytosol. In contrast, the addition of HMG sensitizes LNCaP or PC3 prostate cancer cells harboring an active Akt to apoptosis, in which ROS is upregulated to induce the UPR and GADD153 expression. The concurrent activation of JNK1 and Akt has an additive effect on apoptosis following PKC suppression. Thus, the data identify Akt and JNK1 as potential targets in prostate cancer cells for PKC inhibition–induced apoptosis.
Keywords: JNK, Akt, caspase, UPR, apoptosis
Introduction
Cell growth and death are carefully orchestrated to guide the development and function of multiple cellular systems. Oncogenic transformation often results from gain of functional changes of signaling pathways. Thus, oncogenic signals are highly dependent upon each other to support the survival of cancer cells.1 Studies demonstrated that oncoproteins not only promote cell growth–related activities but also induce senescence and apoptosis, which are dependent on circumstances, stimuli, or cell types.2 Protein kinase C (PKC) is a family of serine/theronine kinases and consists of more than 10 isoforms that differ in their structures, cellular functions, and tissue distributions.3 PKC α, βI, βII, and γ are the conventional, calcium- and diacylglycerol (DAG)–dependent PKC isoforms, while isozymes of the unconventional PKC subgroup (PKC δ, ε, η, and θ) are independent of calcium for their functions. The atypical PKC isozymes (PKC ζ and λ/ν) require neither DAG nor calcium for being activated. The structural diversity and different tissue distributions render distinct specificities of PKC isozymes that differentially regulate various cellular signaling pathways and further dictate different biological outcomes, including apoptosis. Recently, with the availability of small hairpin RNA (shRNA) and other genetic means to disrupt individual PKC isoforms in vitro and in vivo, studies demonstrated that PKC isoforms are either proapoptotic or antiapoptotic, depending upon cell types, stimuli, or contexts within signaling pathways.4,5
Despite the central involvement in cell growth and differentiation, PKC has been shown to be crucial for cells expressing oncogenic ras to survive, and once PKC was suppressed, these cells underwent apoptosis.6-8 In this PKC suppression–induced apoptotic process, some of the Ras downstream effectors, such as c-Jun kinase (JNK) or PI3K, are the important players for the initiation of apoptosis.7-9
Although the induction of apoptosis in prostate cancer cells has been shown to be through modulating PKC activity or expression,10,11 the underlying mechanisms are still not fully understood. It was reported that hyperactive PKC α in LNCaP cells was critical for phorbol ester PMA–induced apoptotic response.10 However, suppression of PKC α was apoptotic to androgen-independent PC3 cells.12 Studies also demonstrated that the activation of PKC δ in prostate cancer cells caused the autocrine release of death receptor ligands for the execution of cell death program.13 Much attention has been focused on the study of the role of PKC isoforms in the regulation of apoptosis in prostate cancer cells; it is unclear whether other aberrant signaling pathways, such as Ras downstream effector signals, participate in the sensitization of prostate cancer cells to apoptosis.
Some prostate cancer cells, like PC3 cells, harbor a hyperactive Akt that is downstream of Ras/PI3K and due to PTEN mutations.14 PI3K, through activating Akt, is involved in prosurvival activities, and PTEN negatively controls PI3K activity to prevent unlimited growth.15 The role of PTEN in the regulation of prostate cancer becomes more significant from the findings that PTEN heterozygous knockout mice developed prostate tumor.16 Furthermore, increases of Akt activity have been reported to be associated with the development of prostate cancer, through affecting cell growth and angiogenesis.17 Studies also showed that the PI3K/Akt pathway is able to induce apoptosis, depending upon the types of stimuli and circumstances.18 For example, Akt controls the status of several enzymes (such as NADPH oxidase) to promote the generation of ROS, high levels of which elicit apoptosis.19
The ER serves as the site for synthesis, folding, modification, and trafficking of proteins and plays a critical role in the maintenance of homeostasis.20 ER stress has been indicated in a variety of human diseases.21 Pharmacological interference with ER function triggers the accumulation of misfolded proteins, resulting in the adaptive ER stress response program named the unfolded protein response (UPR).22 The UPR attenuates protein translation, induces ER chaperone proteins to alleviate protein aggregation, and activates the proteasome machinery to degrade misfolded proteins. Oncogenic stresses are able to trigger the UPR and further promote transformation, apoptosis, or premature senescence, depending upon the circumstances.23 Studies demonstrated that, under persistent ER stress, the UPR plays a significant role in the initiation of apoptosis.24 ER stress–induced factors, such as GADD153/45, are capable of sensitizing different intracellular targets to execute apoptotic programs. GADD153, an ER protein and member of the CCAAT/enhancer-binding protein (C/EBP) family that is also known as the C/EBP homologous protein 10 (CHOP) transcription factor, was shown to heterodimerize with other C/EBP family members to further induce apoptosis or growth arrest.25 In mouse embryotic fibroblasts, GADD153 elicited programmed cell death through the activation of PUMA or NOXA.26
In the present study, using different prostate cancer cell lines with different activation status or expression levels of Ras downstream effectors, we tested whether these aberrant intracellular pathways, together with the suppression of PKC, are synthetically lethal. Our data suggest that Akt and JNK are targets in prostate cancer cells for the induction of apoptosis triggered by PKC inhibition.
Results
Induction of apoptosis in prostate cancer cells after downregulation of PKC
Studies have demonstrated that human or mouse fibroblasts or lymphocytes harboring a mutated ras, but not cells with a normal ras, are susceptible to apoptosis once PKC is suppressed.6-8,27 In the process, multiple Ras downstream effectors are involved in transmitting apoptotic signaling. Because Ras-related pathways are aberrant in some prostate cancer cell lines, we tested whether these cells were susceptible to apoptosis upon PKC suppression. Human primary prostate epithelial PrEC and immortalized HPV7 as well as prostate cancer LNCaP, PC3, and DU145 cells with or without transiently expressing v-Ha-ras were treated with HMG (1.0 µM) for 24 hours, and a DNA fragmentation (Fig. 1A) or annexin V (Fig. 1B) was conducted. About 37% to 40% of all cell lines overexpressing v-ras underwent apoptosis after the treatment with HMG, indicating that persistent activation of Ras, together with suppression of PKC, sensitizes prostate epithelial or cancer cells to apoptosis. Interestingly, 18% to 20% of DU145 or PC3 cells and more than 10% of LNCaP cells were apoptotic upon the same treatment. In comparison, apoptosis did not occur in either prostate epithelial PrEC or immortalized prostate HPV7 cells after the addition of HMG, indicating that suppression of PKC is not apoptotic to these control cells, as observed in other types of normal cells.6-8,27 The results suggest that hyperactive Akt or JNK1 has a lethal interaction with loss of PKC. To confirm the effect of v-ras infection, Ras activity in the cells was assayed by the Active Ras Pull-Down and Detection kit (Thermo Scientific) (Suppl. Fig. S1A). Ras activity was very low in these prostate cells but highly elevated after the infection with v-ras. We also verified the expression of PKC and inhibitory effect of HMG on this kinase in these prostate cells. A very similar level of PKC in each cell line was revealed by anti-pan-PKC antibody (Suppl. Fig. S1B). The negative effect of HMG on PKC was analyzed by treating the prostate cells with PMA in the presence or absence of HMG and then being assayed with the PKC activity kit (Millipore) (Suppl. Fig. S1C).
Figure 1.
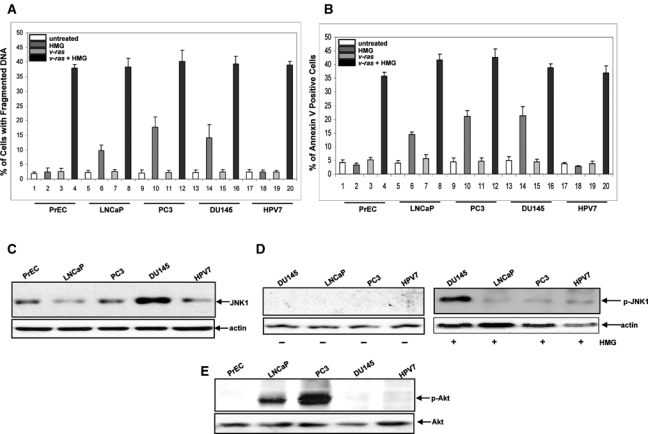
Induction of apoptosis in human prostate epithelial and cancer cells upon HMG treatment. (A and B) Human prostate epithelial PrEC or cancer LNCaP, PC3, DU145, and HPV7 cells, with or without expressing v-Ha-ras, were treated with HMG (1.0 µM) for 24 hours; DNA fragmentation (A) and annexin V (B) assays were performed. Error bar represents the standard deviation (SD) over 5 independent experiments (n = 5, P < 0.05). (C) The expression of JNK1 in the cells was analyzed by immunoblotting. The even loading of total proteins was normalized by actin. (D) The phosphorylation status of JNK1 in untreated or HMG-treated cells was tested using the antiphosphorylated JNK1 antibody. The even loading of total proteins was normalized by actin. (E) The active status of Akt in the cells was immunoblotted with the antiphosphorylated Akt antibody, and the even loading of total proteins was normalized by Akt.
Since studies have demonstrated that JNK and PI3K/Akt are 2 major players in the induction of apoptosis triggered by the suppression of PKC in cells harboring oncogenic ras and some prostate cancer cell lines were moderately susceptible to apoptosis induced by HMG shown in Figure 1A, we examined the expression or activation status of JNK1 and Akt in the prostate cells by immunoblotting. A high level of JNK1 was detected in DU145 cells (Fig. 1C). This stress-related kinase was not phosphorylated in DU145 as well as other cell lines (Fig. 1D, left panel). A slight increase of phosphorylated JNK1 was detected in LNCaP, PC3, and HPV7 cells after the treatment with HMG (Fig. 1D, right panel). In comparison, JNK1 was phosphorylated in DU145 cells after the addition of HMG, which might be due to the upregulation of the baseline activity of this molecule.
It is well known that PTEN is either mutated or deleted in prostate cancer cells, resulting in the upregulation of PI3K/Akt signaling.14 The activation status of Akt in the cells was then examined using the antiphosphorylated Akt antibody (Fig. 1E). The antibody detected a high level of the phosphorylated Akt in PC3 cells and a moderate amount of the active kinase in LNCaP cells.
PKC inhibition, via JNK1, sensitizes DU145 cells to apoptosis
JNK1 was shown to induce apoptosis in cells expressing v-ras through the activation of caspases.28 Since the addition of HMG was able to elicit a moderate magnitude of apoptosis in DU145 cells and to activate JNK, we examined whether caspase 8, the initiator of caspase cascade, was activated under the current experimental settings, using immunoblotting (Fig. 2A, left panels). After PKC inhibition, the cleaved, active caspase 8 was revealed by the antibody only in DU145 cells. Because caspase 8–initiated caspase cascade is able to trigger the release of mitochondrial cytochrome c, the cytosol fractions were isolated and then immunoblotted with cytochrome c antibody (Fig. 2A, right panels). The cytosolic cytochrome c was absent in all untreated cells, and the protein was detected by the antibody only in the cytosol fraction isolated from HMG-treated DU145 cells. The data indicate that caspase signaling was activated in DU145 cells upon PKC suppression.
Figure 2.
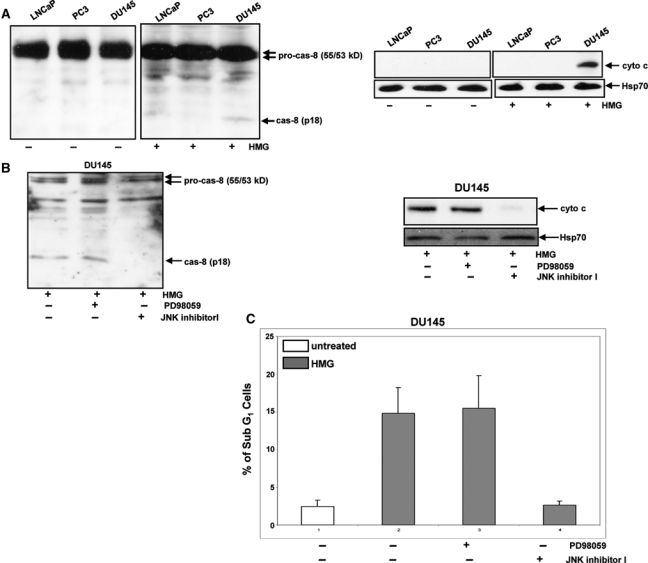
Activation of caspase 8 and cytochrome c after PKC suppression in DU145 cells. (A) After being treated with HMG (1.0 µM) for 24 hours, the cleaved fragment of caspase 8 (left panels) and cytochrome c in the cytosol fraction (right panels) were immunoblotted with the corresponding antibodies. Equal loading of total proteins was verified by procaspase 8 or heat shock protein 70 (Hsp70). (B and C) DU145 cells were treated with either PD98059 (5.0 µM) or JNK inhibitor I (5.0 µM) prior to HMG treatment. Subsequently, the cleaved caspase 8 and cytosolic cytochrome c were tested by immunoblotting, and the percentage of DNA fragmentation was measured by a flow cytometer. The error bars are the standard deviation (SD) over 5 independent experiments (n = 5, P < 0.05).
We further examined the requirement of JNK1 in this process; PD98059 (an inhibitor for ERK1/2 that serves as a control here) or JNK inhibitor was used prior to the treatment with HMG in DU145 cells (Fig. 2B). The cleaved caspase 8 was present in HMG-treated DU145 cells after the suppression of ERK1/2 but not JNK1 (Fig. 2B, left panel). Consistently, the release of cytochrome c to the cytosol in HMG-treated DU145 cells was blocked by JNK1 inhibitor but not PD98059 (Fig. 2B, right panels). DNA fragmentation assay was also conducted for confirmation purposes (Fig. 2C). The addition of JNK1 inhibitor, but not PD98059, blocked this apoptosis process that occurred in HMG-treated DU145 cells. The results from annexin V apoptotic analysis were consistent with those from DNA fragmentation assay (data not shown).
To further determine the role of JNK in the induction of Ras-related apoptosis, JNK1 was transiently infected into LNCaP or PC3 cells. Subsequently, the cytosolic release of cytochrome c in HMG-treated LNCaP or PC3 cells overexpressing JNK1 was examined (Fig. 3A). After the ectopic expression of JNK1, cytochrome c was present in the cytosol fraction in both HMG-treated cells, which was absent in untreated cells. The occurrence of apoptosis in LNCaP/JNK1 or PC3/JNK1 cells was also analyzed by DNA fragmentation assay (Fig. 3B). LNCaP and PC3 cells, after overexpression of JNK1, became more susceptible to HMG-mediated apoptosis. A similar result was obtained from annexin V assay (data not shown). Overall, the data suggested that JNK1 is a crucial factor for sensitizing prostate cells to apoptosis triggered by HMG.
Figure 3.
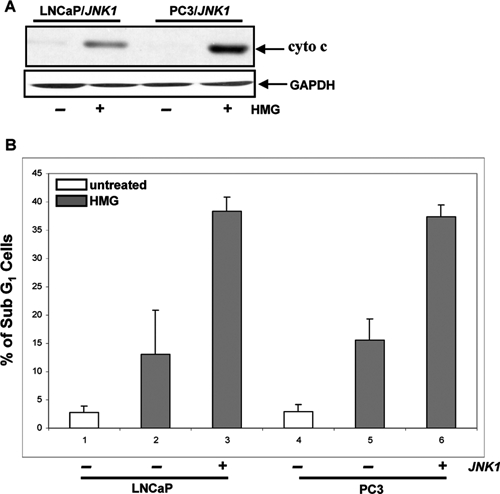
Overexpression of JNK1 sensitizes LNCaP and PC3 cells to apoptosis. (A) A JNK1 expression vector was transiently transfected into LNCaP or PC3 cells. After the treatment with HMG, the cytosolic fractions were isolated from the cells and immunoblotted for cytochrome c expression. (B) The cells with or without ectopically expressing JNK1 were treated with HMG and subsequently analyzed by DNA fragmentation assay. Error bar represents the standard deviation (SD) over 5 independent experiments (n = 5, P < 0.05).
ROS is upregulated, and the UPR is activated in LNCaP and PC3 cells in response to PKC inhibition
Ras promotes cell proliferation or transformation through regulating ROS production.7 PKC suppression has been shown to sensitize murine fibroblasts expressing oncogenic ras to apoptosis, through the upregulation of ROS mediated by the PI3K/Akt pathway.29 Since Akt was active in some prostate cancer cells, ROS in the cells was measured with or without treatment with HMG (Fig. 4A). The level of ROS in untreated prostate cancer LNCaP and PC3 cells that express an active Akt was higher than that in HPV7 and DU145 cells. The introduction of v-ras into HPV7 cells upregulated the amount of ROS. However, the addition of HMG further augmented ROS in LNCaP, PC3, and HPV7/v-ras cells. Next, the expression of ROS modulator HO-1 (heme oxygenase) after PKC suppression was tested using immunoblotting (Fig. 4B). HO-1 was slightly elevated in untreated HPV7/ras cells, which might be induced by hyperactive Ras. The addition of HMG was dramatically upregulated HO-1 in HPV7/ras cells and moderately in PC3 or LNCaP cells, which was blocked by the addition of KP372-1 (data not shown). The data suggested that the equilibrium of the intracellular redox state in cells expressing active v-ras or Akt is perturbed by HMG.
Figure 4.

Perturbation of the redox state in LNCaP and PC3 cells upon PKC suppression. (A) Following being treated with HMG, the cells were stained with DCF and analyzed for ROS levels using a flow cytometer. Error bar represents the standard deviation (SD) over 5 independent experiments (n = 5, P < 0.05). (B and C) The expression of HO-1 (B) or GADD153 and BiP (C) in the cells, after PKC suppression, was tested by immunoblotting. Equal-loading total proteins were verified by actin expression.
Aberrant Akt activation has been shown to cause ER stress and activates the UPR that then switches on the apoptotic machinery.30 GADD153 has been shown to be often upregulated during ER stress and UPR activation and to function as a crucial factor in the induction of stress-induced apoptosis.25 To determine the expression of GADD153 in prostate cancer cells in response to HMG treatment, immunoblotting analysis was performed (Fig. 4C, upper 2 panels). A high level of GADD153 in LNCaP or PC3 cells was detected by the antibody, which was absent in DU145 cells following the treatment. Next, the expression of ER stress–related protein BiP was examined following the treatment with HMG. Consistently, the level of this ER protein was increased in LNCaP or PC3 cells but not in DU145 cells (Fig. 4C, lower 2 panels). Furthermore, the induction of GADD153 and BiP in LNCaP or PC3 cells was abrogated by the treatment with KP372-1 or infected with dn-Akt (data not shown), which support the notion that Akt is responsible for the activation of the UPR.
Akt is required for the induction of apoptosis in GO6976-treated LNCaP or PC3 cells
LNCaP or PC3 cells express an active Akt and are susceptible to apoptosis in response to PKC suppression. This led us to investigate the role of Akt in the initiation of apoptosis. KP372-1 (an Akt inhibitor) and dn-Akt (a dominant-negative Akt inserted in a retroviral vector tagged with HA) were employed. The negative effect of KP372-1 on Akt activation (Fig. 5A) and overexpression of dn-Akt in the prostate cancer cells (Suppl. Fig. S2A) were analyzed by immunoblotting. Subsequently, the susceptibility of HMG-treated LNCaP, PC3, and DU145 cells to apoptosis was tested following suppressing Akt signaling pathway by KP372-1 or dn-Akt, ROS production by NAC (a ROS inhibitor), or GADD153 by GADD153 antisense oligos (Fig. 5B). These chemical or genetic inhibitors blocked the apoptotic process in LNCaP and PC3 cells and played no role in DU145 cells. To further define the effect of Akt on the onset of apoptosis induced by PKC suppression, a retroviral vector inserted with wt-Akt and tagged with HA was transiently introduced into the cells. The expression of HA was analyzed by immunoblotting (Suppl. Fig. S2B). Following PKC inhibition, overexpressed Akt had no further influence on the induction of apoptosis in LNCaP or PC3 cells, indicating that the endogenous, active Akt had maximal effect on this cell death process (Fig. 5C). In contrast, the overexpression of Akt dramatically augmented the magnitude of apoptosis in DU145 cells. The addition of JNK inhibitor I had no effect on apoptosis triggered by HMG in LNCaP or PC3 cells overexpressing wt-Akt but partially blocked the apoptotic process in HMG-treated DU145 cells ectopically expressing wt-Akt. Similar data were obtained from annexin V assay (data not shown). The data suggest that JNK and Akt may act additively to induce a full magnitude of apoptosis in response to HMG treatment.
Figure 5.
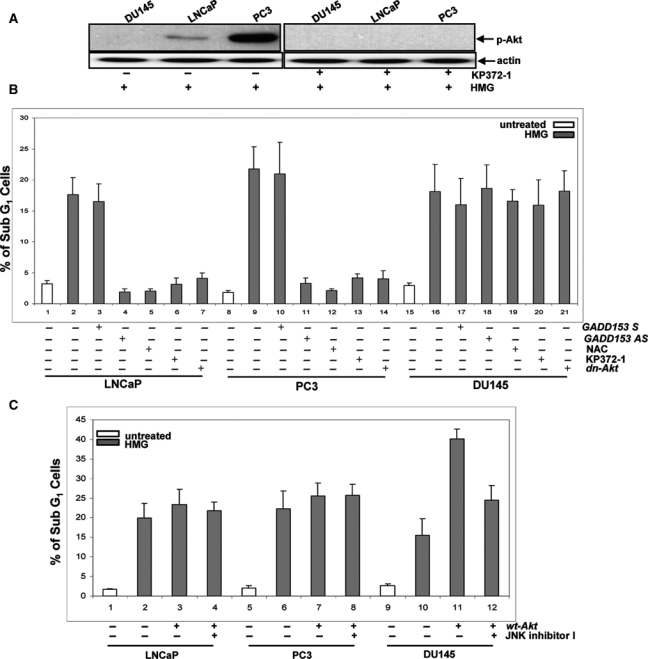
Akt activation is required for HMG-induced apoptosis in prostate cancer cells. (A) In the presence or absence of KP372-1 (1.0 µM), the levels of phosphorylated Akt in HMG-treated LNCaP, PC3, or DU145 cells were examined by immunoblotting. (B) HMG-treated or untreated LNCaP, PC3, or DU145 cells with or without expressing dn-Akt were cultured in the growth medium containing GADD153 antisense, sense oligos, NCA (5.0 µM), or KP372-1. Subsequently, the percentage of the cells with fragmented DNA was measured. Error bar represents the standard deviation (SD) over 5 independent experiments (n = 5, P < 0.05). (C) The prostate cancer cells were transiently infected with wt-Akt or treated with JNK1 inhibitor I. Following the treatment with HMG, the percentages of DNA fragmentation in the cells were measured. Error bar represents the SD over 5 independent experiments (n = 5, P < 0.05).
Differential induction of apoptosis in xenografted prostate cancer tumors
To further test the contribution of Ras downstream effectors to the induction of apoptosis triggered by HMG, xenograft assay was performed. After the inoculation with prostate cancer DU145 and PC3 cells into nude mice subcutaneously, HMG or the combination of HMG with other inhibitors was administrated. When the tumors became detectable 1 week after the inoculation, their diameters were measured every 4 days (Fig. 6A). The growth of the xenografted DU145 tumors was dramatically reduced by HMG injection. When coinjected with JNK inhibitor, HMG became inefficient to block the growth of the tumors, which did not occur after the combination of HMG with KP372-1 (Fig. 6A, left panel). A similar phenomenon was observed in xenografted PC3 tumors coinjected with HMG plus KP372-1, but not with JNK inhibitor (Fig. 6A, right panel). HMG was still able to suppress the growth of the tumors from either DU145 or PC3 cells when coinjected with PD98059 (a MAPK inhibitor) (data not shown). After the animals were sacrificed at 16 days after the inoculation, the tumors were isolated and stained with a TUNEL staining kit (BioVision) (Fig. 6B). Consistently, after the injection of HMG, high numbers of the cells in xenografted DU145 or PC3 tumors were stained positively with TUNEL reagents, which was absent in untreated tumors. However, the coinjection of HMG with JNK inhibitor blocked the occurrence of apoptosis in the xenografted DU145 tumor. The same phenomenon was seen in xenografted PC3 tumor cotreated with HMG plus KP372-1. Most cells in the tumors from either the DU145 or PC3 cell line were stained positively with TUNEL reagents after coinjected HMG with PD98059 (data not shown). Overall, both in vivo and in vitro data suggest that a block of PKC is able to sensitize prostate cancer cells with aberrant Akt or JNK signaling to apoptosis.
Figure 6.
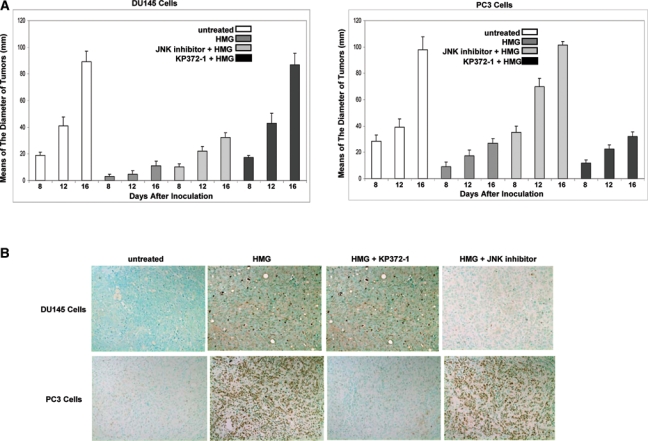
Attenuation of xenografted DU145 or PC3 tumor growth in nude mice upon HMG injection. (A) DU145 or PC3 cells were inoculated subcutaneously into nude mice. A group of 6 mice were injected with HMG or its combination with other inhibitors. The diameters of the tumors were measured starting on the eighth day after the inoculation, which was repeated every 4 days. Error bar represents the standard deviation (SD) over 6 mice (n = 6, P < 0.05). (B) Sixteen days after the inoculation, the animals were sacrificed, and the slides mounted with tumor tissue were stained with TUNEL agents.
Discussion
Emerging evidence shows that PKC not only involves prostate cancer tumorigenesis but also induction of apoptosis.10,11,31 Different PKC isoforms employ different mechanisms to sensitize prostate cancer cells to apoptosis.10,12,13,31 In addition, studies demonstrated that loss of PKC, together with mutated ras, has been shown to trigger a synthetic lethal interaction in various types of cells, in which several kinases regulated by Ras were involved.4,8,32 Since some of Ras downstream effector pathways play important roles in prostate cancer development, we in this study explored the possibility of the sensitization of prostate cancer cells to apoptosis by the suppression of PKC. JNK1 was shown to be overexpressed in DU145 cells. Akt was moderately phosphorylated in LNCaP cells and highly activated in PC3 cells. After the suppression of PKC, DU145 cells underwent apoptosis, in which JNK1 is activated, resulting in caspase 8 cleavages and cytochrome c release. However, LNCaP and PC3 cells expressing an active Akt were also in synergy with the inhibition of PKC for the induction of apoptosis, in which the intracellular redox equilibration was perturbed, leading to persistent activation of the UPR and subsequent induction of GADD153. Thus, the present study suggests that Akt or JNK1 cooperates with PKC to maintain homeostasis for the survival of prostate cancer cells. Once the balance is disrupted, the intrinsic apoptotic machinery is being switched on.
Under normal growth conditions, activation of Ras triggers a wide spectrum of cellular responses, leading to transformation.33 These responses persistently maintained by oncogenic ras are often key events for the promotion of various types of human malignancy. In recent years, it has drawn great attention for the therapeutic potential by the capability of oncogenes, such as ras or myc, to induce apoptosis.2 Through binding to multiple effector proteins, Ras triggers a series of protein phosphorylation chain reactions to regulate growth-related activities.33 Ras also utilizes different downstream effectors for the induction of apoptosis.2,34 After PKC suppression, JNK1 was shown to promote the formation of the death-inducing complex.9 However, the activation of JNK alone is only partially responsible for the activation of the cell death program. In our current study, using different prostate cancer cell lines, we assessed the contribution of signaling pathways downstream of Ras to the induction of apoptosis. The study disclosed a regulatory network for apoptosis triggered by PKC inhibition in prostate cancer. It appears that Akt or JNK1, under the condition of loss of PKC, is able to sensitize prostate cancer cells to apoptosis. In this apoptotic process, Akt signals ROS to activate the UPR-mediated cell death program. In the JNK1-participated apoptotic process, caspase chain reaction appeared to be a key player.
JNK, as one of the Ras downstream effectors, phosphorylates the transcriptional factor c-Jun and further participates in the regulation of cell growth, stress-related activities, or apoptosis.35 The increase of JNK1 activity is observed in TNF- or Fas-induced apoptosis. In this apoptotic process, the association of the death receptor with the ligand initiated the formation of the death-induced signaling complex and subsequently activated JNK1, leading to caspase 8–mediated cascade. A similar action of JNK1 appeared in apoptosis induced by PKC suppression in the cells expressing mutated ras.9,32 Studies also demonstrated that JNK1 was able to inactivate the ASK1 inhibitor, thioredoxin, which in turn mobilized ASK1 signaling to upregulate ROS for the induction of apoptosis.36 In the current experimental setting, JNK1 activation played no role in ROS accumulation. However, the baseline phosphorylation of JNK1 appears to be much higher in DU145 cells in response to PKC inhibition than that in LNCaP or PC3 cells that express a normal level of JNK1. As a result of the increase of JNK1 activity, caspase 8 cleavage is initiated, and subsequently, cytochrome c is released from the mitochondrial to the cytosol.
ROS is an important intracellular signal transducer of growth factors.37 In response to abnormal ligation of growth factor receptors, a moderate increase of intracellular ROS was induced in cells expressing oncogenic ras, which facilitates the process of cellular transformation through altering the structure of the cytoskeleton and further inducing a transformed phenotype in a Rac-dependent fashion.37 In PC12 cells, Ras was suggested to play a crucial role in the upregulation of ROS production upon epidermal growth factor stimulation.38 Studies also demonstrated that oncogenes perturb the equilibrium of intracellular redox state and cause DNA single-strand breaks, which in turn disrupted genetic integrity.39 Despite regulating cell proliferation and transformation, the increase of ROS production was also shown to play an obligatory role in the induction of apoptosis induced by various apoptotic stimuli.40 In NFκB-regulated programmed cell death, ROS accumulation precedes mitochondrial depolarization and caspase activation.41 In this study, we demonstrated that the level of ROS was moderately increased in PC3 cells, which may be due to high demand for the maintenance of the growth for tumor cells. Notably, the inhibition of PKC further augments the level of Akt phosphorylation in PC3 cells, which is coincided with a further increase of the ROS level. It is possible that PKC and Akt function in an opposite way to balance the intracellular redox state. After lifting the regulation rendered by PKC, activated Akt disrupts the equilibrium of the intracellular redox state, which contributes to the induction of apoptosis.
Alternations in proteins and lipid metabolism can cause ER stress and the accumulation of unfolded proteins.24 Dysregulation of ER stress programs often induces persistent activation of the UPR that elicits cytoprotective or cytotoxic reactions. GADD153, as a transcriptional factor, often induced ER stress and plays a crucial role in ER stress–induced apoptosis.24 We showed here that ER stress chaperons were induced in response to PKC suppression in prostate cancer cells expressing an active Akt. The experiments with the antisense oligos suggested that GADD153 functions downstream of Akt and participates in the execution of apoptosis. In this apoptotic process, persistent UPR activation appears to be a major player. It is likely that upon PKC inhibition, genetic or epigenetic mechanisms in prostate cancer cells harboring an active Akt surpass the buffering capacity of the ER and in turn trigger the cell death program. In addition, our study suggests that the ER is able to function as a sensor to detect changes in the cellular microenvironment.
Taken together, the results of our study demonstrate that as Ras downstream effectors, JNK1 and Akt are able to sensitize prostate cancer cells to apoptosis when endogenous PKC is suppressed. However, the concurrent activation of JNK1 and Akt triggers a full execution of the cell death program in the cells after the inhibition of PKC. Since the aberrant activation of growth-related signaling pathways often is an important factor in prostate cancer development, our study supports the notion that these signaling molecules can also serve as intracellular targets for sensitizing prostate cancer to apoptosis and further for developing new therapeutics. However, a better understanding of the underlying mechanisms in prostate cancer cells for the induction of apoptosis triggered by PKC suppression is undoubtedly required.
Materials and Methods
Cell cultures and reagents
Human prostate cancer (LNCaP, PC3, and DU145) and immortalized HVP7 cells were obtained from Dr. Z. Luo (Boston University School of Medicine) and cultured in RPMI1640 medium containing 10% fetal calf serum. PrEC cells (human primary prostate epithelial cells) were grown in Prostate Epithelial Cell Basal Medium (Cambrex Bio Science, Baltimore, MD). The v-Ha-ras, JNK, or active Akt are inserted in a retroviral vector. All the control cells were infected with the vector alone.
For the suppression of GADD153, 30 µM of sense (as control) or antisense oligos were added into cell cultures for 48 hours, and subsequently, the cells were refed with media containing a half dose of the oligos every 4 days. Antibodies used in the study were from Santa Cruz Biotechnology (Santa Cruz, CA), except anti-Ras antibody (Cell Signaling Technology, Danvers, MA).
HMG is a PKC inhibitor that specifically suppresses the activity of the phorbol ester–dependent PKC isoforms (Calbiochem, San Diego, CA). JNK inhibitor I, KP372-1, and PD98059 were also purchased from Calbiochem. The inhibitors were dissolved in DMSO, the volume of which was proved to have no effect on cells.9
DNA fragmentation analysis
A flow cytometric analysis was performed using a FACScan (Becton Dickinson, Mountain View, CA). The data analysis was performed using the Cell-Fit software program (Becton Dickinson). Following treatments, cells were harvested and fixed in 70% cold ethanol. Cells then were stained with 0.1 mg/mL propidium iodide containing 1.5 µg/mL RNase. Following incubation at room temperature for 2 to 4 hours, DNA contents of cells were measured by a FACScan machine (Becton Dickinson) and evaluated with BD FACStation software CellQuest.
Immunoblotting analysis
After treatment, cells were washed in 1x PBS and then lysed in the lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% Triton X-114, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, containing 1 µM phenylmethylsulfonyl fluoride, 1 µg/mL aprotinin, 1 µg/mL leupeptin, 1 µg/mL pepstatin A) on ice for 30 minutes.37 The samples were separated on a 10% SDS-PAGE gel and subsequently transferred to a nitrocellulose membrane. Each experiment was performed at least 3 times for confirmation purposes.
Measurement of ROS
Cells, after the treatments, were washed with ice-cold PBS and resuspended in 5 µg/mL of 2´, 7´-dichlorodihydrofluorescein diacetate (DCF) (Molecular Probes, Eugene, OR). Samples were incubated for 10 minutes at room temperature and analyzed immediately by a flow cytometer.39
Preparation of cytosol fraction
After treatments, cells (1 × 108) were washed twice with 1x PBS and resuspended in 1 mL of 1% Triton X-114 lysis buffer. The cell suspensions were transferred to a 1-mL syringe and sheared by being passed 40 times through a 25-gauge needle. The lysates were centrifuged at 280x g for 10 minutes, and the supernatant was centrifuged at 16,000x g for 30 minutes. The supernatant was collected as the cytosolic fraction.
Xenograft analysis
Male nude mice (NCRNU-M-M, CrTac: Ncr-Foxn 1 nu) (Taconic, Hudson, NY) of ages 4 to 6 weeks were used. Human prostate cancer cells (1 × 106 cells) in 100 µL of 1x PBS were inoculated into nude mice (6 mice per treatment). HMG (30 mg/kg), JNK inhibitor (20 mg/kg), or KP372-1 (20 mg/kg) were administrated at day 0 and then given every 2 days. The size of the tumors was measured routinely. In addition, the water consumption and body weight of the mice were measured daily to monitor the general conditions of the mice. The mice were eventually sacrificed, and the tumors were isolated for further analyses.
Immunohistochemistry analysis
Isolated tumors were fixed in 4% of paraformaldehyde (pH 7.4). The histological specimens were sectioned and analyzed with TUNEL assay.
Statistics
Means and standard deviations of the results of the experiments were computed. Standard deviations are displayed as error bars in the figures.
Supplementary Material
Acknowledgments
The authors thank Dr. Z. Luo (Boston University School of Medicine, Boston, MA) for the reagents.
Footnotes
The study was supported by United States Department of Defense (DOD) (W81XWH-04-1-0246) and National Institutes of Health (NIH) (RO1CA100498) grants.
The author(s) declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
Supplementary material for this article is available on the Genes & Cancer website at http://ganc.sagepub.com/supplemental.
References
- 1. Ding HF, Fisher DE. Induction of apoptosis in cancer: new therapeutic opportunities. Ann Med. 2002;34:451-69 [DOI] [PubMed] [Google Scholar]
- 2. Downward J. Ras signalling and apoptosis. Curr Opin Genet Dev. 1998;8:49-54 [DOI] [PubMed] [Google Scholar]
- 3. Parker PJ, Murray-Rust J. PKC at a glance. J Cell Sci. 2004;117:131-2 [DOI] [PubMed] [Google Scholar]
- 4. Chen CY, Liou J, Forman LW, Faller DV. Differential regulation of discrete apoptotic pathways by Ras. J Biol Chem. 1998;273:16700-9 [DOI] [PubMed] [Google Scholar]
- 5. Mandil R, Ashkenazi E, Blass M, et al. Protein kinase C alpha and protein kinase C delta play opposite roles in the proliferation and apoptosis of glioma cells. Cancer Res. 2001;61:4612-9 [PubMed] [Google Scholar]
- 6. Petti C, Molla A, Vegetti C, Ferrone S, Anichini A, Sensi M. Coexpression of NRAS(Q61R) and BRAF(V600E) in human melanoma cells activates senescence and increases susceptibility to cell-mediated cytotoxicity. Cancer Res. 2006;66:6503-11 [DOI] [PubMed] [Google Scholar]
- 7. Guo JJ, Zhu TB, Luo LY, Huang Y, Sunkavalli RG, Chen CY. PI3K acts in synergy with loss of PKC to elicit apoptosis via the UPR. J Cell Biochem. 2009;107:76-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhu T, Tsuji T, Chen C. Roles of PKC isoforms in the induction of apoptosis elicited by aberrant Ras. Oncogene. 2010;29:1050-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhu T, Chen L, Du W, Tsuji T, Chen CY. Synthetic lethality induced by loss of PKCdelta and mutated Ras. Genes Cancer. 2010;1:142-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Powell CT, Brittis NJ, Stec D, Hug H, Heston WDW, Fair WR. Persistent membrane translocation of protein kinase C alpha during 12-O-tetradecanoylphorbol-13-acetate-induced apoptosis of LNCaP human prostate cancer cells. Cell Growth Differ. 1996;7:419-28 [PubMed] [Google Scholar]
- 11. Gonzalez-Guerrico AM, Meshki J, Xiao LQ, Benavides F, Conti CJ, Kazanietz MG. Molecular mechanisms of protein kinase C-induced apoptosis in prostate cancer cells. J Biochem Mol Biol. 2005;38:639-45 [DOI] [PubMed] [Google Scholar]
- 12. Lamm MLG, Long DD, Goodwin SM, Lee C. Transforming growth factor-beta 1 inhibits membrane association of protein kinase C alpha in a human prostate cancer cell line, PC3. Endocrinology. 1997;138:4657-64 [DOI] [PubMed] [Google Scholar]
- 13. Fujii T, Garcia-Bermejo ML, Bernabo JL, et al. Involvement of protein kinase C delta (PKC delta) in phorbol ester-induced apoptosis in LNCaP prostate cancer cells: lack of proteolytic cleavage of PKC delta. J Biol Chem. 2000;275:7574-82 [DOI] [PubMed] [Google Scholar]
- 14. Li J, Yen C, Liaw D, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943-7 [DOI] [PubMed] [Google Scholar]
- 15. Kandel ES, Hay N. The regulation and activities of the multifunctional serine/threonine kinase Akt/PKB. Exp Cell Res. 1999;253:210-29 [DOI] [PubMed] [Google Scholar]
- 16. Suzuki A, de la Pompa JL, Stambolic V, et al. High cancer susceptibility and embryonic lethality associated with mutation of the PTEN tumor suppressor gene in mice. Curr Biol. 1998;8:1169-78 [DOI] [PubMed] [Google Scholar]
- 17. Jiang BH, Zheng JZ, Aoki M, Vogt PK. Phosphatidylinositol 3-kinase signaling mediates angiogenesis and expression of vascular endothelial growth factor in endothelial cells. Proc Natl Acad Sci U S A. 2000;97:1749-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rosner D, Stoneman V, Littlewood T, et al. Interferon-gamma induces fas trafficking and sensitization to apoptosis in vascular smooth muscle cells via a PI3K-and akt-dependent mechanism. Am J Pathol. 2006;168:2054-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Anastasiadis AG, Stisser BC, Ghafar MA, Burchardt M, Buttyan R. Tumor hypoxia and the progression of prostate cancer. Curr Urol Rep. 2002;3:222-8 [DOI] [PubMed] [Google Scholar]
- 20. Aridor M, Balch WE. Integration of endoplasmic reticulum signaling in health and disease. Nat Med. 1999;5:745-51 [DOI] [PubMed] [Google Scholar]
- 21. Federovitch CM, Ron D, Hampton RY. The dynamic ER: experimental approaches and current questions. Curr Opin Cell Biol. 2005;17:409-14 [DOI] [PubMed] [Google Scholar]
- 22. Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739-89 [DOI] [PubMed] [Google Scholar]
- 23. Barsyte-Lovejoy D, Mao DY, Penn LZ. c-Myc represses the proximal promoters of GADD45a and GADD153 by a post-RNA polymerase II recruitment mechanism. Oncogene. 2004;23:3481-6 [DOI] [PubMed] [Google Scholar]
- 24. McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bc12 and perturbing the cellular redox state. Mol Cell Biol. 2001;21:1249-59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Maytin EV, Ubeda M, Lin JC, Habener JF. Stress-inducible transcription factor CHOP/gadd153 induces apoptosis in mammalian cells via p38 kinase-dependent and -independent mechanisms. Exp Cell Res. 2001;267:193-204 [DOI] [PubMed] [Google Scholar]
- 26. Li JZ, Lee B, Lee AS. Endoplasmic reticulum stress-induced apoptosis: multiple pathways and activation of p53-up-regulated modulator of apoptosis (PUMA) and NOXA by p53. J Biol Chem. 2006;281:7260-70 [DOI] [PubMed] [Google Scholar]
- 27. Xia SH, Forman LW, Faller DV. Protein kinase C delta is required for survival of cells expressing activated p21(RAS). J Biol Chem. 2007;282:13199-210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen CY, Juo P, Liou JS, et al. The recruitment of Fas-associated death domain/caspase-8 in Ras-induced apoptosis. Cell Growth Differ. 2001;12:297-306 [PubMed] [Google Scholar]
- 29. Ronai Z. Deciphering the mammalian stress response: a stressful task. Oncogene. 1999;18:6084-6 [DOI] [PubMed] [Google Scholar]
- 30. Denoyelle C, Abou-Rjaily G, Bezrookove V, et al. Anti-oncogenic role of the endoplasmic reticulum differentially activated by mutations in the MAPK pathway. Nat Cell Biol. 2006;8:1053-63 [DOI] [PubMed] [Google Scholar]
- 31. Zhao X, Gschwend JE, Powell CT, Foster RG, Day KC, Day ML. Retinoblastoma protein-dependent growth signal conflict and caspase activity are required for protein kinase c-signalled apoptosis of prostate epithelial cells. J Biol Chem. 1997;272:22751-7 [DOI] [PubMed] [Google Scholar]
- 32. Liu HS, Chen CY, Lee CH, Chou YI. Selective activation of oncogenic Ha-ras-induced apoptosis in NIH/3T3 cells. Br J Cancer. 1998;77:1777-86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Barbacid M. Ras genes. Annu Rev Biochem. 1987;56:779-827 [DOI] [PubMed] [Google Scholar]
- 34. Ayllon V, Rebollo A. Ras-induced cellular events [review]. Mol Membr Biol. 2000;17:65-73 [DOI] [PubMed] [Google Scholar]
- 35. Franzoso G, Zazzeroni F, Papa S. JNK: a killer on a transcriptional leash. Cell Death Differ. 2003;10:13-5 [DOI] [PubMed] [Google Scholar]
- 36. Yamamoto K, Ichijo H, Korsmeyer SJ. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M. Mol Cell Biol. 1999;19:8469-78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liou JS, Chen CY, Chen JS, Faller DV. Oncogenic Ras mediates apoptosis in response to protein kinase C inhibition through the generation of reactive oxygen species. J Biol Chem. 2000;275:39001-11 [DOI] [PubMed] [Google Scholar]
- 38. Mills EM, Takeda K, Yu ZX, et al. Nerve growth factor treatment prevents the increase in superoxide produced by epidermal growth factor in PC12 cells. J Biol Chem. 1998;273:22165-8 [DOI] [PubMed] [Google Scholar]
- 39. Guo JJ, Chu M, Abbeyquaye T, Chen CY. Persistent nicotine treatment potentiates amplification of the dihydrofolate reductase gene in rat lung epithelial cells as a consequence of Ras activation. J Biol Chem. 2005;280:30422-31 [DOI] [PubMed] [Google Scholar]
- 40. Anderson KM, Ou D, Wu YB, Jajeh A, Harris JE. Induction of type 1 programmed cell death in U937 cells by the antioxidant, butylated hydroxy-toluene or the free radical spin trap, NTBN. Leuk Res. 1999;23:665-73 [DOI] [PubMed] [Google Scholar]
- 41. Khwaja A, Tatton L. Resistance to the cytotoxic effects of tumor necrosis factor alpha can be overcome by inhibition of a FADD/caspase-dependent signaling pathway. J Biol Chem. 1999;274:36817-23 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.