

Abstract
The effects of type 1 diabetes on the contributions of net hepatic glycogenolysis and gluconeogenesis to glucose production (GP) at rest and during moderate (MOD) and high (HI) intensity running were examined in healthy control (n = 6) and type 1 diabetic (n = 5) subjects matched for age, weight, and maximum aerobic capacity by combined noninvasive measurements of hepatic glycogen content using 13C nuclear magnetic resonance spectroscopy and determination of GP using [6,6-2H2]glucose. In the control subjects, GP increased in proportion to the intensity of the exercise [at rest (REST), 14.3 ± 0.5; MOD, 18.1 ± 0.9; HI, 28.8 ± 1.3 μmol/(kg-min); P = 0.001, three-way comparison], and this was accounted for by an increase in the percent contribution of net hepatic glycogenolysis to GP (REST, 32 ± 1%; MOD, 49 ± 5%; HI, 57 ± 5%; P = 0.006). In the diabetic subjects, resting rates of GP were 60% higher than those in the control subjects (P < 0.0001) and increased in proportion to the workload. In contrast, the contributions of net hepatic glycogenolysis to GP were consistently lower than those in the control subjects (REST, 20 ± 6%; MOD, 32 ± 13%; HI, 32 ± 3%; P = 0.006 vs. control), and the exaggerated rates of GP could be entirely accounted for by increased rates of gluconeogenesis. In conclusion, 1) increases in GP in healthy control subjects with exercise intensity can be entirely attributed to increases in net hepatic glycogenolysis. 2) In contrast, moderately controlled type 1 diabetic subjects exhibit increased rates of GP both at rest and during exercise, which can be entirely accounted for by increased gluconeogenesis.
Although it is well established that short-term exercise is accompanied by large increases in glucose production (GP) to meet the metabolic demands of the working muscle, surprisingly little is known about the regulatory mechanisms responsible for this adaptation (1–8). Increases in GP could occur by increases in net hepatic glycogenolysis and/or increases in gluconeogenesis. Previous attempts to measure these processes have been hampered by the limitations of the techniques.
Wahren et al. (9) calculated GP and gluconeogenic precursor uptake across the splanchnic bed in combination with estimates of splanchnic blood flow and concluded that exercise-induced stimulation of glycogenolysis fully accounted for the increase in GP. Extensive studies in dogs by Wasserman et al. (6) suggested that gluconeogenesis increased by 2.5-fold from rest to exercise. However, because this increase could not be entirely accounted for by the exercise-induced increment in GP, they concluded that exercise induced both gluconeogenesis and glycogenolysis.
Even less is known about the impact of type 1 diabetes on these processes. Most studies have found that patients with moderately or poorly controlled type 1 diabetes have increased resting rates of GP and reduced hepatic glycogen concentrations in the fasting state as well as after mixed meal intake, and their hormonal milieu is often similar to that in a stress situation, with absolute or relative hyperglucagonemia and elevated plasma catecholamine concentrations (4, 10–12). To date, no studies have examined the relative contributions of net hepatic glycogenolysis and gluconeogenesis during exercise in these subjects.
The purpose of the present study was to examine the mechanisms responsible for the increases in GP during exercise in healthy individuals and patients with moderately controlled type 1 diabetes.
To do this we used 13C magnetic resonance spectroscopy (MRS) to directly monitor hepatic glycogen concentrations during 200 min of rest (REST) and after a 50-min bout of moderate [MOD; 35% of maximum aerobic capacity (VO2max)] or high (HI; 70% of VO2max) intensity exercise. By combining these measurements with magnetic resonance imaging (MRI) to measure liver volume, the time-dependent change in the rate of net hepatic glycogenolysis could be calculated. The rates of GP was measured with [6,6-2H2] glucose, and the rate of gluconeogenesis was calculated as the difference between GP and net hepatic glycogenolysis (13–15).
Subjects and Methods
Subjects
Six nonsmoking, healthy, control subjects [four men and two women; age, 23 ± 3 yr; body weight, 69 ± 5 kg; body mass index, 22 ± 1 kg/m2; VO2max, 51 ± 3 ml/(kg-min); hemoglobin A1c, 4.4 ± 0.1%] and five healthy subjects with type 1 diabetes in moderate glycemic control were studied [three men and two women; age, 29 ± 3 yr; body weight, 71 ± 5 kg; body mass index, 24 ± 1 kg/m2; VO2max, 44 ± 4 ml/(kg–min; P = 0.186 vs. control); hemoglobin A1c, 7.9 ± 0.4% (P < 0.05 vs. control)]. The mean duration of diabetes was 6 yr. All participants ran approximately 4–6 miles/wk. After determination of VO2max as described below, each subject participated in two experiments over a period of 6–8 wk. The experiments each lasted 250 min and consisted of a 200-min baseline, resting period (REST), followed by 50-min treadmill running at moderate (35% of VO2max; MOD) or high intensity (70% of VO2max; HI).
For 3 d before each of the studies, the subjects were instructed to eat an isocaloric diet (35–40 kcal/kg; 60% carbohydrate, 20% protein, and 20% fat) and to abstain from exercise, caffeine, and alcohol. On the third day patients were admitted to the Yale-New Haven Hospital General Clinical Research Center between 1700–2000 h; dinner was eaten at 1800 h at home or in the General Clinical Research Center. At 2100 h, they were fed a standard meal (~600 kcal with ~60% carbohydrates, 30% fat, and 10% protein) to ensure loading of hepatic glycogen stores before the exercise. The subjects fasted overnight, but had free access to drinking water until the end of the study the following day. The diabetic patients took the last sc dose of NPH insulin at bedtime before the overnight fast, and insulin was withheld the morning of the study to simulate what the patients typically do when they exercise in the morning. Written consent was obtained from each subject after the purpose, nature, and potential complications of the studies were explained. The protocol was approved by the Yale University Human Investigation Committee.
Study protocol
At 0600 h, iv catheters were inserted into an antecubital vein in each arm and kept patent with 0.9% saline. The subjects were transported in a wheelchair to the Magnetic Resonance Center, where the liver volume was measured at 0700 h in a 1.5-Tesla magnet (Signa 3, General Electric, Milwaukee, WI) using multiecho axial scanning (TE20/80, TR2000). Starting at 0800 h after a 10-h fast and completion of the MRI liver volume measurement, liver glycogen concentrations were measured continuously for 200 min at rest with the subjects in the supine position in a 2.1-Tesla magnetic resonance spectrometer (Bruker Biospec Spectrometer, Billerica, MA). After percussion of the borders of the liver, 13C magnetic resonance spectra (MRS) were obtained with a 9-cm circular 13C observation coil and a 12 × 14-cm coplanar butterfly 1H decoupler coil placed rigidly over the lateral aspect of the liver as previously described (16).
At the start of the 13C MRS measurements (−200 min) a primed (corrected for ambient fasting plasma glucose level)-continuous [0.05 mg/(kg-min)] infusion of [6,6-2H2]glucose (Cambridge Isotopes Laboratories, Inc., Andover, MA) was initiated and kept continuously throughout the 250 min of each study (17). During the final 20 min of the 200-min resting periods, blood samples were collected for determination of resting plasma glucose [6,6-2H2]glucose enrichment (m+2). At 0 min the MRS measurements were stopped, and the subjects were taken to an adjacent exercise room. After a 2-min warm-up on the treadmill (Series 2000 Treadmill, Marquette, WI), the subjects began the 50 min of running exercise at the predetermined intensity (35% or 70% of VO2max). The exercise intensity was monitored and verified by measuring rates of oxygen uptake 10–20 and 40–50 min into the exercise. The subjects were encouraged to drink water, and the room temperature was kept constant at 20–22 C.
During the last 30 min of running, blood was collected every 10 min for determination of plasma, insulin, glucagon, nonesterified fatty acids (FA), lactate, cortisol, and catecholamine concentrations and every 5 min for determination of plasma glucose concentrations and plasma [6,6-2H2]glucose enrichment. The last blood sample was collected when the subjects were placed back into the MRS spectrometer, and the final 13C MRS measurements of hepatic glycogen concentration were performed over 10 min. For each subject the order of the MOD and HI studies was selected at random.
Testing of VO2max
Seven to 10 d before the first study, the VO2max of each subject was tested on the running treadmill. After a 5 min warm-up the initial treadmill speed of 2.5 km/h was increased by 1.0 km/h every minute, and the initial incline of 0° was increased by 2° every minute until exhaustion. Oxygen uptake and CO2 production were measured by indirect calorimetry using breath by breath analysis of expired air and measurement of minute ventilation (Vmax 29 Metabolic Monitor, Sensormedics, Yorba Linda, CA). Maximum oxygen uptake was established when two of the following criteria were met: 1) oxygen consumption reached a plateau with increasing workload, 2) the heart rate was greater than the age-predicted maximum value, and 3) the respiratory quotient exceeded 1.1.
MRI measurement of liver volume
On each of the 3 study days the liver volume was measured at 0700 h by clinical imaging on a 1.5-Tesla magnet (Signa II, General Electric, Milwaukee, WI) using multiecho axial scanning (TE20/80, TR2000) as previously described (14). The accuracy of the measurement was assessed with water-filled phantoms of known volume and was determined to be ±5% with a coefficient of variation of ±1%.
13 C MRS of net hepatic glycogen content
13C MRS spectra of the C1 glycosyl unit of hepatic glycogen were obtained at rest during the baseline period from −200 to 0 min and again for 10 min after the completion of the 50-min exercise. Each 10-min block consisted of 4800 scans using a 135° pulse at coil center and a repetition time of 120 msec. 13C MRS spectra were processed with a mild 30-Hz Lorentzian to Gaussian filter and a 500-Hz convolution difference as described (14, 16, 18). After processing, the glycogen line width was 70–90 Hz. The resonance intensity was measured by integrating over a bandwidth of ±120 Hz. The C1 glycosyl units in liver glycogen have been shown to be approximately 100% visible by this method in vivo (19).
The reproducibility of the glycogen concentration measurement was assessed in an earlier study with a coefficient of variation between the pairs of measurements of 7%. The SD in the 13C MRS glycogen concentration measurement due to special noise was ±5% (13).
Gas chromatography-mass spectrometry
Gas chromatography-mass spectrometry analysis of enrichment of [6,6-2H2]glucose in plasma were performed using a Hewlett-Packard 5971A mass selective detector (Wilmington, DE) as previously described (20).
Analyses
Plasma glucose concentrations were measured by the glucose oxidase method using a glucose analyzer (Glucose Analyzer II, Beckman Coulter, Fullerton, CA). Plasma lactate concentrations were measured using a 2300 STAT analyzer (YSI, Inc., Yellow Springs, CA). Plasma FA concentrations were determined using a microfluorimetric method (21). The plasma immunoreactive hormones, insulin, glucagon, and cortisol, were measured using commercially available kits [insulin, LINKO (Diagnostic System Laboratories, Inc., Webster, TX); glucagon, LINKO (ICN Biomedical, Inc., Costa Mesa, CA); cortisol, Diagnostic Product Corp. (Los Angeles, CA)]. Plasma concentrations of epinephrine and norepinephrine were determined using a three-step procedure using HPLC (22).
Continuous indirect calorimetry
To verify that the subjects exercised at the predetermined intensity, oxygen consumption and CO2 production were measured from 10–20 min and from 40–50 min during the 50-min exercise period using breath by breath indirect calorimetry (Vmax 29 Metabolic Monitor, Sensormedics, Yorba Linda, CA). The subjects were familiarized with the running treadmill and the air collection during the initial VO2max test session. A light headband holding the tight-fitting rubber mouthpiece for air collection was placed on the subject’s head, and a nose-clip was used to eliminate air exchange through the nose. After an adjustment period and the initial stabilization period equilibration period (2 min), the subjects began to exercise (23).
Statistical analysis
Data are given as the mean ± SEM. Comparisons within individuals and between groups were made using three-way repeated measures ANOVA with Greenhouse-Geisser corrections.
Calculations
Net hepatic glycogenolysis
Rates of net hepatic glycogenolysis were calculated by finding the best fit of the liver glycogen concentration (millimoles per liter of liver) to a line at the interval from the start of the study to the beginning of the exercise (−200 to 0 min).
Assuming constant rates of net hepatic glycogenolysis from the final resting liver glycogen measurement (0 min) to the start of the exercise (<3 min), liver glycogen concentrations were extrapolated to calculate liver glycogen concentrations at the beginning of the exercise. Likewise, liver glycogen content was extrapolated from the end of the 50-min exercise session to the beginning of the final MRS measurement (<3 min). Because of the short intervals between end of the resting MRS measurements and the beginning of the exercise sessions and vice versa, these adjustments of hepatic glycogen concentrations were minimal (<1%) in both groups. Rates of net hepatic glycogenolysis were calculated by the best fit of the hepatic glycogen concentrations to a line in the interval from at the beginning to the end of the exercise session. The slopes of these lines (resting period and exercise period) were multiplied by the liver volume (liters) and divided by body weight (kilograms) (14). The MRS-determined rate of gluconeogenesis was calculated as the difference between the rates of GP and net hepatic glycogenolysis.
Rates of GP
The resting (REST) rates of GP (micromoles per kilogram of body weight per minute) were calculated by dividing the tracer infusion rate times the tracer enrichment by the average percent enrichment of plasma [6,6-2H2]glucose at steady state and subtracting the tracer infusion rate (16). The (m+2) enrichment was stable at rest, with a mean coefficient of variance of 3% (2). During the exercise studies, GP was calculated using the Steele equations for nonsteady state conditions as modified by Radziuk for nonsteady state conditions adapted for the use of [6,6-2H2]glucose (24, 25).
Results
VO2max
All subjects were considered to be moderately active as assessed by calculation of activity indexes (work, sports, and leisure activity) based on a questionnaire by Baecke et al. (26) and by measuring their VO2max. The VO2max was similar in the two groups: 51 ± 3 ml/(kg-min) (control) and 44 ± 4 ml/(kg-min) (diabetic) as was the workload, expressed as running speed and percent incline during the study [MOD, 3.7 ± 0.4 mph/2.9 ± 1.7% incline (control) vs. 3.5 ± 0.1 mph/1.7 ± 0.8% incline (diabetic); HI, 6.2 ± 1.0 mph/0% incline (control) vs. 5.8 ± 0.4 mph/0% incline (diabetic)].
Plasma concentrations of hormones and metabolites
The mean resting plasma glucose concentration in the control subjects was 5.2 ± 0.1 mmol/liter (94 ± 2 mg/dl) and remained stable during the studies. There was a trend toward an increase in plasma glucose concentrations during HI to 6.4 ± 0.6 mmol/liter (115 ± 11 mg/dl), but this was not significant (P = 0.06 vs. REST; Fig. 1).
Fig. 1.
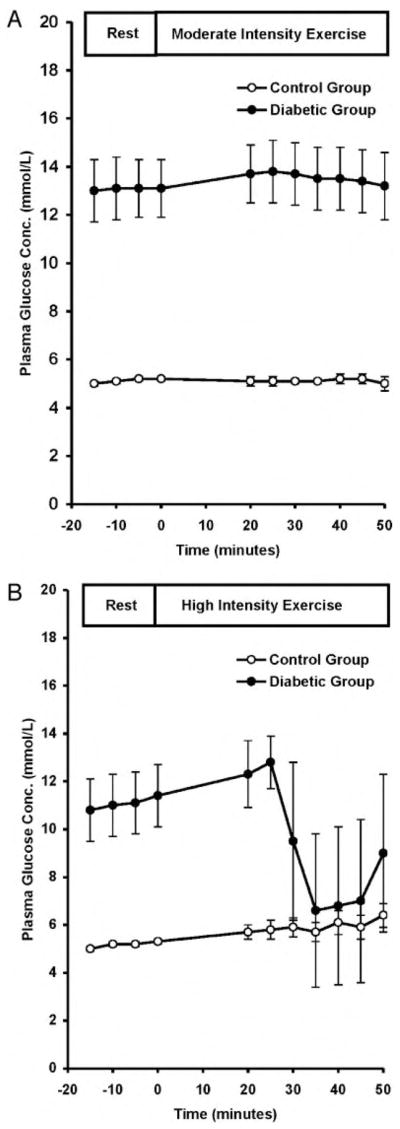
Plasma glucose concentrations at rest and during MOD (A) and HI (B) in control and diabetic subjects. To convert values to milligrams per deciliter, multiply by 0.0551.
The diabetic subjects were hyperglycemic at the start of the studies (P < 0.0001), and plasma glucose levels did not change significantly during the REST and MOD studies (Fig. 1). In contrast, plasma glucose concentrations decreased by about 40% when the exercise intensity was increased to 70% of VO2max [nadir, 6.6 ± 3.2 mmol/liter (119 ± 58 mg/dl); P < 0.005 vs. REST; P < 0.05 vs. control].
In the control and diabetic subjects, plasma concentrations of FA, glycerol, and lactate increased in proportion to the intensity of the exercise, and there were no differences between the groups (data not shown).
Plasma insulin concentrations gradually decreased during both exercise intensities from 66 ± 6 pmol/liter (11 ± 1 μU/ml) to 36 ± 6 pmol/liter (6 ± 1 μU/ml; P < 0.0001) in the control group. Plasma insulin concentrations remained stable at 66 ± 6 pmol/liter (11 ± 1 μU/ml) in the diabetic group in all studies (Fig. 2).
Fig. 2.
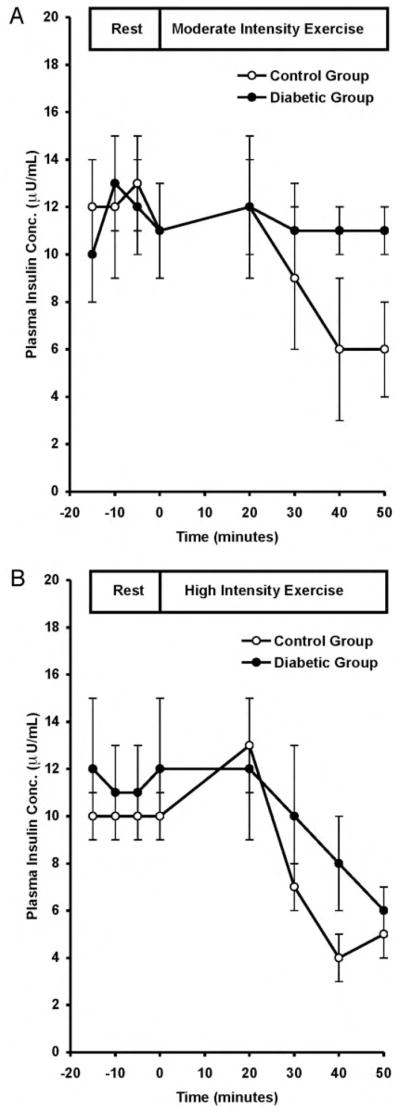
Plasma insulin concentrations at rest and during MOD (A) and HI (B) in control and diabetic subjects. To convert values to microunits per milliliter, divide by 6.
Resting plasma glucagon concentrations were similar in the control and diabetic subjects (51 ± 1 and 49 ± 1 ng/liter; P = 0.70) and did not change with exercise in either group (Fig. 3).
Fig. 3.
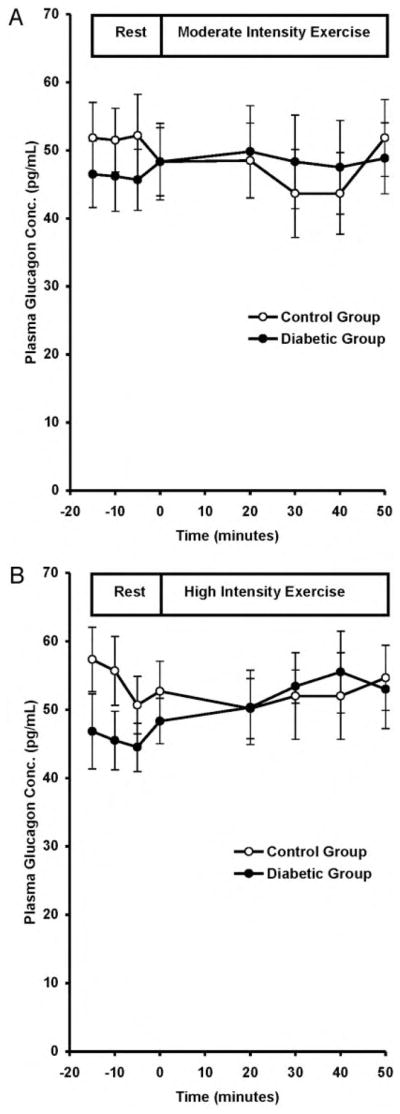
Plasma glucagon concentrations at rest and during MOD (A) and HI (B) in control and diabetic subjects. To convert values to nanograms per liter, divide by 1.
Insulin/glucagon ratio
In the control group, the ratio of plasma insulin to glucagon decreased during exercise in proportion to the workload. During MOD, the insulin/glucagon ratio decreased from 0.24 to 0.14 after 30 min of running (P < 0.05 vs. REST). During HI, the insulin/glucagon ratio decreased to 0.14 after 20 min and continued to decrease to a nadir of 0.08 after 30 min of running (P < 0.001 vs. REST). In the diabetic subjects, the resting insulin/glucagon ratio was 0.24 ± 0.01 (P = 0.205 vs. control) and did not change during MOD, but decreased slightly to 0.19 after 30 min of running at HI and to 0.11 at the end of the 50 min of running (P = 0.06 vs. control).
Catecholamine concentrations
During the MOD period, the plasma epinephrine concentration in the control group gradually increased to a 3-fold peak at 50 min [from 152 ± 11 pmol/liter (28 ± 2 pg/ml) to 720 ± 240 pmol/liter (132 ± 44 pg/ml); P < 0.0001]. This increase was even greater during the HI; plasma epinephrine concentrations immediately rose about 7-fold and remained at this level throughout the 50-min exercise period (P < 0.01). A similar response was observed in fasting concentrations of plasma norepinephrine [966 ± 93 pmol/liter (177 ± 17 pg/ml)], which increased 4-fold with MOD [3,897 ± 229 pmol/liter (714 ± 42 pg/ml); P < 0.01] and 12-fold during HI [12,220 ± 1,255 pmol/liter (2,239 ± 230 pg/ml); P < 0.0001].
In the subjects with type 1 diabetes, resting concentrations of plasma epinephrine and norepinephrine were similar to the control subjects. In contrast to the control subjects, there were no increases in plasma epinephrine concentrations during MOD and HI (Table 1) in the diabetic subjects.
TABLE 1.
Plasma concentrations of epinephrine and norepinephrine in healthy (n = 4) and type 1 diabetic (n = 5) subjects at rest and during 50 min of running exercise at MOD and HI
| Group | Epinephrine (pmol/liter) | Norepinephrine (pmol/liter) |
|---|---|---|
| Rest | ||
| Control subjects | 186 ± 22 | 966 ± 93 |
| Diabetic subjects | 153 ± 11 | 901 ± 49 |
| P value | NS | <0.05 |
| MOD | ||
| Control subjects | 546 ± 76 | 3,897 ± 229 |
| Diabetic subjects | 229 ± 5 | 3,870 ± 33 |
| P value | <0.01 | NS |
| HI | ||
| Control subjects | 1,042 ± 55 | 12,220 ± 1,255 |
| Diabetic subjects | 196 ± 5 | 10,714 ± 1,354 |
| P value | <0.0001 | NS |
To convert picomoles per liter to picograms per milliliter, divide by 5.458. NS, Not significant.
In contrast, plasma norepinephrine concentrations increased in a manner similar to the control subjects (4-fold during MOD and 12-fold during HI; Table 1).
In the resting state, plasma cortisol concentrations were significantly higher in the control group than in the diabetic subjects (29.8 ± 2.4 vs. 12.8 ± 2.9 mmol/liter; P < 0.05). No changes in plasma cortisol concentrations occurred at either exercise intensity in either group.
Hepatic glycogen concentrations and liver volume
In the control subjects, the initial fasting hepatic glycogen concentrations were similar at the beginning of the studies [251 ± 8 mmol/liter liver, and 264 ± 11 mmol/liter liver; mean, 257 ± 7 mmol/liter liver]. Despite similar dietary regimens, hepatic glycogen concentrations were consistently lower in the diabetic subjects than in the control subjects [151 ± 18 mmol/liter liver, and 191 ± 22 mmol/liter liver; mean, 171 ± 15 mmol/liter liver; P < 0.05 vs. control hepatic glycogen concentrations]. There were no differences in liver volumes between the studies or the groups (control subjects, 1.472 ± 0.122 and 1.515 ± 0.128 liters; mean liver volume, 1.494 ± 0.089 liters; diabetic subjects, 1.568 ± 0.071 and 1.536 ± 0.084 liters; mean liver volume, 1.566 ± 0.057 liters; P = 0.323 vs. control).
Rates of net hepatic glycogenolysis
In the control subjects, the rate of net hepatic glycogenolysis increased in direct proportion to the intensity of the exercise from a mean rate of 0.21 ± 0.01 mmol/(liter liver-min) at REST to 0.40 ± 0.03 during MOD and 0.73 ± 0.04 mmol/(liter liver-min) during HI (P < 0.0001; Fig. 4). Despite lower initial hepatic glycogen concentrations in the diabetic subjects, the rate of net hepatic glycogenolysis at REST as well as the exercise-induced increase in the rate of net hepatic glycogenolysis were similar to those in the control subjects [REST, 0.16 ± 0.04 mmol/(liter liver-min); MOD, 0.38 ± 0.14; HI, 0.72 ± 0.11 mmol/(liter liver-min); Fig. 4].
Fig. 4.

Rates of net hepatic glycogenolysis (millimoles per liter of liver per minute) at rest and during MOD and HI in control (□) and diabetic (■) subjects. Cx, Control; DM, type 1 diabetes mellitus.
Rates of GP
Resting rates of GP were similar in the two studies in the control subjects [14.5 ± 0.5 and 14.2 ± 0.6 μmol/(kg-min); mean, 14.3 ± 0.5 μmol/(kg-min); 2.7 ± 0.3 and 2.7 ± 0.2 mg/(kg-min); mean, 2.7 ± 0.1 mg/(kg-min)] and in the diabetic subjects [23.7 ± 3.2 and 21.1 ± 3.6 μmol/(kg-min); mean, 22.4 ± 2.5 μmol/(kg-min); 4.2 ± 0.5 and 3.8 ± 0.5 mg/(kg-min); mean, 4.1 ± 0.4 mg/(kg-min)]. During MOD and HI, the rates of GP in the control subjects increased in proportion to the intensity of the exercise (by 27 ± 8% and 102 ± 11%, respectively; P < 0.001; Fig. 5), and this exercise-induced increase in the rate of GP could be entirely accounted for by the increase in the rates of net hepatic glycogenolysis (P < 0.0001). The rate of gluconeogenesis remained constant during both exercise intensities (Fig. 5).
Fig. 5.

The contributions of net hepatic glycogenolysis and gluconeogenesis to rates of glucose production [μmol/(kg-min)] at rest and during MOD and HI in control (A) and diabetic (B) subjects.
In the diabetic subjects, the resting rate of GP was 51% higher than that in the control subjects (P < 0.0001; Fig. 5) and increased in proportion to the exercise intensity similar to the control subjects (P = 0.132). During MOD and HI, the rates of GP were 60% and 74% higher than in the control subjects (P < 0.01; Fig. 5). The contributions of net hepatic glycogenolysis to GP were consistently low and remained low between 20% at REST and 32% of GP during exercise. In contrast to the control, the exaggerated rates of GP could be entirely accounted for by increased rates of gluconeogenesis (Fig. 5).
Indirect calorimetry
The mean rates of oxygen consumption over the final 10 min of each exercise session increased similarly in both control and diabetic subjects in proportion to the intensity of the physical activity (control subjects: REST, 0.256 ± 0.024 liter/min; MOD, 1.421 ± 0.155 liter/min; HI, 2.570 ± 0.313 liter/min; diabetic subjects: REST, 0.348 ± 0.033 liter/min; MOD, 1.207 ± 0.122 liter/min; HI, 2.385 ± 0.265 liter/min]. No significant change in respiratory quotient values occurred in the control subjects (REST, 0.86 ± 0.02; MOD, 0.86 ± 0.03; HI, 0.88 ± 0.01) or the diabetic subjects (REST, 0.84 ± 0.02; MOD, 0.84 ± 0.02; HI, 0.91 ± 0.03) with increased exercise intensity.
Discussion
In this study we examined the relative contributions of net hepatic glycogenolysis and gluconeogenesis to GP in young, lean, nondiabetic and type 1 diabetic subjects at rest and during moderate and high intensity running exercise.
The major findings are that in nondiabetic humans the rate of GP increased in proportion to the intensity of exercise, which can be entirely accounted for by a matched increase in the rate of net hepatic glycogenolysis. In contrast, the rate of GP was markedly increased in subjects with moderately controlled type 1 diabetes, and this increase could be entirely attributed to the excessive rate of gluconeogenesis both at rest and during exercise.
In the control subjects, mean hepatic glycogen concentrations and the percent contribution of net hepatic glycogenolysis to GP were approximately 20% lower than in our previous studies (257 ± 9 vs. 325 ± 26 mmol/liter liver) (14), which most likely can be attributed to the smaller evening meal given to the subjects in the present study (~600 vs. 1000 kcal) (14). In the diabetic subjects, hepatic glycogen concentrations were 33% lower than in the control subjects, consistent with previous studies (12, 27). This lower hepatic glycogen content in subjects with type 1 diabetes does not appear to be diet related, because all participants ate similar diets for 3 d before each study and were fed a standard meal at bedtime the evening before each study part.
Surprisingly, despite 33% lower initial hepatic glycogen concentrations, the rates of resting net hepatic glycogenolysis were similar to those in the control subjects. However, because the rates of resting GP were increased in the diabetic subjects, net hepatic glycogenolysis accounted for only about 20% of GP. Therefore, increased rates of gluconeogenesis were the major factor responsible for the increased rates of GP in the diabetic subjects at rest. Because rates of GP were 60–74% higher than those in the control subjects during exercise, and rates of net hepatic glycogenolysis were similar to the rates in control subjects, the increased rates of GP in the diabetic subjects both at rest and during exercise could be entirely accounted for by increased rates of gluconeogenesis.
To examine the impact of exercise on liver volume, two healthy subjects underwent MRI of liver volume immediately before and after 50 min of HI treadmill running (65% of VO2max under conditions identical to the original protocol). Liver volume changed less than 12% with 50 min of exercise. This small change in liver volume during exercise would result in a less than 10% underestimation of the rate of net hepatic glycogenolysis to GP during exercise.
Several studies have examined the regulation of blood glucose concentrations and GP in humans during exercise (1, 28–32). Some of the earliest studies to examine GP and gluconeogenesis in humans during exercise were conducted by Felig and Wahren (9, 33). They estimated hepatic gluconeogenesis by measuring blood flow, glucose, and gluconeogenic substrate concentrations across the splanchnic bed during bicycle exercise. Using this method they found that gluconeogenesis contributed only 20–25% to GP at rest, and with exercise gluconeogenesis contributed only 6–11% to GP (9). However, because this method does not take into consideration contributions of gluconeogenic precursors released from the gut, intrahepatic pools of gluconeogenic substrates, and renal gluconeogenesis, this approach might lead to a major underestimation of the contribution of gluconeogenesis to whole body GP (13).
Coggan et al. (3) used the 13C labeling of plasma glucose from an infusion of [13C]bicarbonate to estimate the incorporation of 13C into glucose via pyruvate carboxylase during prolonged high intensity exercise of 2 h at 60% of VO2max and found the contribution of gluconeogenesis to be only about 23% of the GP. However, this method does not adequately account for 13C label dilution in the tricarboxylic acid cycle (34).
These are the first direct measurements of hepatic glycogen metabolism during exercise in type 1 diabetic subjects, and the results demonstrate that the high rate of GP at rest and during exercise is due to a 2- to 2.7-fold increase in gluconeogenesis in subjects with moderately controlled type 1 diabetes. These data are consistent with the observations by Wahren et al. (9, 35) that the relative contribution of gluconeogenesis to GP was increased in subjects with Type 1 diabetes and that the fractional splanchnic extraction of gluconeogenic precursors was 2–3 times higher in type 1 diabetic than in control subjects despite reduced arterial concentrations of alanine, glycine, and threonine.
The hormonal regulatory effects on gluconeogenesis and glycogenolysis during exercise have been studied extensively in animals (36, 37) and humans (10), and there is general agreement that the main factor responsible for the increase in GP during exercise is a decrease in plasma insulin levels and a real or relative increase in plasma glucagon concentrations. Wolfe et al. (10) found that by keeping plasma insulin and glucagon concentrations constant during exercise in humans, plasma glucose concentrations fell, which they ascribed to an increase in peripheral glucose uptake. When plasma glucose concentrations were maintained with a coinfusion of glucose, no increase in GP occurred. The researchers suggested from these observations that a decrease in insulin is necessary to limit peripheral glucose uptake and that an increase in glucagon promotes stimulation of hepatic GP, thus preventing hypoglycemia during exercise. The role of endogenous insulin release in the control of hepatic GP was examined by Wahren et al. (9), who found that although glucose infused during exercise caused approximately 80% suppression of GP in control subjects, it failed to inhibit GP in type 1 diabetic subjects. Based on these findings, the researchers concluded that the ability of glucose to suppress GP is a consequence of glucose-induced endogenous insulin secretion.
We found a decrease in plasma insulin concentrations, which was proportional to the workload in the control subjects. There were no changes in plasma insulin concentrations in the diabetic subjects, which would be anticipated because they were given their NPH insulin at bedtime. No changes in plasma glucagon levels were detected in either group, which resulted in a decrease in the insulin/glucagon ratio in the control subjects and no change in this ratio in the diabetic group during exercise.
The roles of epinephrine and norepinephrine in the regulation of GP during exercise are not clear, although most studies suggest that these hormones play a secondary role to regulate GP during exercise in healthy subjects. In our studies, both epinephrine and norepinephrine concentrations rose dramatically during high intensity running. Howlett et al. (38) infused epinephrine in healthy volunteers during bicycling at 40% of VO2max to mimic the epinephrine levels achieved during bicycling 80% of VO2max. Because they found that GP did not increase during 40% of VO2max with plasma epinephrine levels similar to the exercise at 80% of VO2max, the investigators concluded that epinephrine alone could not account for the increase in GP during high intensity exercise (38).
Our findings suggest that the exercise-induced increase in GP is facilitated by a stimulation of net hepatic glycogenolysis, and this, in turn, is accomplished by a coordinated hormonal response, i.e. a decrease in the insulin/glucagon ratio accompanied by an increase in plasma epinephrine and norepinephrine concentrations (39). Earlier studies in healthy humans have suggested that epinephrine is not a major determinant of GP during exercise (40). By blocking the celiac ganglion, epinephrine secretion was impaired during exercise, but this did not have any effect on the rate of GP, and the results suggest that norepinephrine secretion and/or the central nervous system are the major determinants for the regulation of GP.
The exercise-induced hormonal responses were graded to the exercise intensity. However, the central nervous system and other hormones and substrates, such as glycerol, lactate, and FA, are likely to play a role as well (41–43), although the importance of their respective roles in the regulation of GP during exercise is less clear (40).
In contrast to the exercise-induced increase in plasma epinephrine concentrations in control subjects, plasma epinephrine concentrations did not change during exercise in diabetic subjects. In contrast, plasma norepinephrine concentrations increased in a manner similar to that in control subjects during both exercise intensities. The factors driving the increased gluconeogenesis in these subjects are less clear, but may be partly attributable to a chronically low portal vein insulin/glucagon ratio.
In summary, the contribution of net hepatic glycogenolysis to the rate of whole body GP was directly assessed in healthy volunteers and subjects with moderately controlled type 1 diabetes mellitus using 13C nuclear magnetic resonance spectroscopy at rest and during moderate and high intensity running. We found that increases in GP with exercise can be fully accounted for by increases in the rate of net hepatic glycogenolysis. In contrast, these results suggest that the exaggerated rates of GP in subjects with type 1 diabetes can be attributed to increased rates of gluconeogenesis.
Acknowledgments
We thank Joan Boyer, L.P.N.; Veronika Walton; Laura Burden; Suzanne Vogel, M.P.H.; Donna Caseria, R.D.; James Dziura, Ph.D.; and the staff of the Yale-New Haven Hospital General Clinical Research Center for expert help with the studies.
This work was supported by the Juvenile Diabetes Foundation (Grant 196-092; to K.F.P.) and the USPHS [Grants K23 DK-02347 (to K.F.P.), R01 AG-23686 (to K.F.P.), R01 DK-49230, and M01 RR-00125]. T.B.P. was supported by U.S. Army Contract DAMD 17-96-C-6097.
Abbreviations
- FA
Fatty acid
- GP
glucose production
- HI
high intensity exercise
- MOD
moderate intensity exercise
- MRI
magnetic resonance imaging
- MRS
magnetic resonance spectroscopy
- VO2max
maximum aerobic capacity
References
- 1.Wahren J, Felig P, Ahlborg G, Jorfeldt L. Glucose metabolism during leg exercise in man. J Clin Invest. 1971;50:2715–2725. doi: 10.1172/JCI106772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wahren J. Glucose turnover during exercise in man. Ann NY Acad Sci. 1977;301:45–55. doi: 10.1111/j.1749-6632.1977.tb38184.x. [DOI] [PubMed] [Google Scholar]
- 3.Coggan AR, Swanson SC, Mendenhall LA, Habash DL, Kien CL. Effect of endurance training on hepatic glycogenolysis and gluconeogenesis during prolonged exercise in men. Am J Physiol. 1995;268:E375–E383. doi: 10.1152/ajpendo.1995.268.3.E375. [DOI] [PubMed] [Google Scholar]
- 4.Felig P, Wahren J. Fuel homeostasis in exercise. N Engl J Med. 1975;293:1078–1084. doi: 10.1056/NEJM197511202932107. [DOI] [PubMed] [Google Scholar]
- 5.Wasserman DH, Williams PE, Lacy DB, Green DR, Cherrington AD. Importance of intrahepatic mechanisms to gluconeogenesis from alanine during exercise and recovery. Am J Physiol. 1988;254:E518–E525. doi: 10.1152/ajpendo.1988.254.4.E518. [DOI] [PubMed] [Google Scholar]
- 6.Wasserman DH, Spalding JA, Lacy DB, Colburn CA, Goldstein RE, Cherrington AD. Glucagon is a primary controller of hepatic glycogenolysis and gluconeogenesis during muscular work. Am J Physiol. 1989;257:E108–E117. doi: 10.1152/ajpendo.1989.257.1.E108. [DOI] [PubMed] [Google Scholar]
- 7.Wasserman DH, Mohr T, Kelly P, Lacy DB, Bracy D. Impact of insulin deficiency on glucose fluxes and muscle glucose metabolism during exercise. Diabetes. 1992;41:1229–1238. doi: 10.2337/diab.41.10.1229. [DOI] [PubMed] [Google Scholar]
- 8.Wasserman DH, Johnson JL, Bupp JL, Lacy DB, Bracy DP. Regulation of gluconeogenesis during rest and exercise in the depancreatized dog. Am J Physiol. 1993;265:E51–E60. doi: 10.1152/ajpendo.1993.265.1.E51. [DOI] [PubMed] [Google Scholar]
- 9.Wahren J, Felig P, Cerasi E, Luft R. Splanchnic and peripheral glucose and amino acid metabolism in diabetes mellitus. J Clin Invest. 1972;51:1870–1878. doi: 10.1172/JCI106989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wolfe RR, Nadel ER, Shaw JH, Stephenson LA, Wolfe MH. Role of changes in insulin and glucagon in glucose homeostasis in exercise. J Clin Invest. 1986;77:900–907. doi: 10.1172/JCI112388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galassetti P, Tate D, Neill RA, Morrey S, Davis SN. Effect of gender on counterregulatory responses to euglycemic exercise in type 1 diabetes. J Clin Endocrinol Metab. 2002;87:5144–5150. doi: 10.1210/jc.2002-020757. [DOI] [PubMed] [Google Scholar]
- 12.Hwang JH, Perseghin G, Rothman DL, Cline GW, Magnusson I, Petersen KF, Shulman GI. Impaired net hepatic glycogen synthesis in insulin-dependent diabetic subjects during mixed meal ingestion. A 13C nuclear magnetic resonance spectroscopy study. J Clin Invest. 1995;95:783–787. doi: 10.1172/JCI117727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rothman DL, Magnusson I, Katz LD, Shulman RG, Shulman GI. Quantitation of hepatic glycogenolysis and gluconeogenesis in fasting humans with 13C NMR. Science. 1991;254:573–576. doi: 10.1126/science.1948033. [DOI] [PubMed] [Google Scholar]
- 14.Petersen KF, Price T, Cline GW, Rothman DL, Shulman GI. Contribution of net hepatic glycogenolysis to glucose production during the early post-prandial period. Am J Physiol. 1996;270:E186–E191. doi: 10.1152/ajpendo.1996.270.1.E186. [DOI] [PubMed] [Google Scholar]
- 15.Petersen KF, Krssak M, Navarro V, Chandramouli V, Hundal R, Schumann WC, Landau BR, Shulman GI. Contributions of net hepatic glycogenolysis and gluconeogenesis to glucose production in cirrhosis. Am J Physiol. 1999;276:E529–E535. doi: 10.1152/ajpendo.1999.276.3.E529. [DOI] [PubMed] [Google Scholar]
- 16.Petersen KF, Laurent D, Rothman DL, Cline GW, Shulman GI. Mechanism by which glucose and insulin inhibit net hepatic glycogenolysis in humans. J Clin Invest. 1998;101:1203–1209. doi: 10.1172/JCI579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maggs DG, Buchanan TA, Burant CF, Cline G, Gumbiner B, Hsueh WA, Inzucchi S, Kelley D, Nolan J, Olefsky JM, Polonsky KS, Silver D, Valiquett TR, Shulman GI. Metabolic effects of troglitazone monotherapy in type 2 diabetes mellitus. A randomized, double-blind, placebo-controlled trial. Ann Intern Med. 1998;128:176–185. doi: 10.7326/0003-4819-128-3-199802010-00002. [DOI] [PubMed] [Google Scholar]
- 18.Shulman GI, Rothman DL, Jue T, Stein P, DeFronzo RA, Shulman RG. Quantitation of muscle glycogen synthesis in normal subjects and subjects with non-insulin-dependent diabetes by 13C nuclear magnetic resonance spectroscopy. N Engl J Med. 1990;322:223–228. doi: 10.1056/NEJM199001253220403. [DOI] [PubMed] [Google Scholar]
- 19.Gruetter R, Magnusson I, Rothman DL, Avison MJ, Shulman RG, Shulman GI. Validation of 13C NMR measurements of liver glycogen in vivo. Magn Reson Med. 1994;31:583–588. doi: 10.1002/mrm.1910310602. [DOI] [PubMed] [Google Scholar]
- 20.Mayerson AB, Hundal RS, Dufour S, Lebon V, Befroy D, Cline GW, Enocksson S, Inzucchi SE, Shulman GI, Petersen KF. The effects of rosiglitazone on insulin sensitivity, lipolysis, and hepatic and skeletal muscle triglyceride content in patients with type 2 diabetes. Diabetes. 2002;51:797–802. doi: 10.2337/diabetes.51.3.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miles J, Glasscock R, Aikens J, Gerich J, Haymond M. A microfluorometric method for the determination of free fatty acids in plasma. J Lipid Res. 1983;24:96–99. [PubMed] [Google Scholar]
- 22.Laurent D, Schneider KE, Prusaczyk WK, Franklin C, Vogel SM, Krssak M, Petersen KF, Goforth HW, Shulman GI. Effects of caffeine on muscle glycogen utilization and the neuroendocrine axis during exercise. J Clin Endocrinol Metab. 2000;85:2170–2175. doi: 10.1210/jcem.85.6.6655. [DOI] [PubMed] [Google Scholar]
- 23.Lusk G Animal calorimetry. Analysis of the oxidation of mixtures of carbohydrates and fat: a correction. J Biol Chem. 1924;59:41–42. [Google Scholar]
- 24.Radziuk J, Norwich KH, Vranic M. Experimental validation of measurements of glucose turnover in nonsteady state. Am J Physiol. 1978;234:E84–E93. doi: 10.1152/ajpendo.1978.234.1.E84. [DOI] [PubMed] [Google Scholar]
- 25.Petersen KF, Sullivan JT. Effects of a novel glucagon receptor antagonist (Bay 27–9955) on glucagon-stimulated glucose production in humans. Diabetologia. 2001;44:2018–2024. doi: 10.1007/s001250100006. [DOI] [PubMed] [Google Scholar]
- 26.Baecke JA, Burema J, Frijters JE. A short questionnaire for the measurement of habitual physical activity in epidemiological studies. Am J Clin Nutr. 1982;36:936–942. doi: 10.1093/ajcn/36.5.936. [DOI] [PubMed] [Google Scholar]
- 27.Bischoff MG, Bernroider E, Krssak M, Krebs M, Stingl H, Nowotny P, Yu C, Shulman GI, Waldhäusl W, Roden M. Hepatic glycogen metabolism in type 1 diabetes after long-term near normoglycemia. Diabetes. 2002;51:49–54. doi: 10.2337/diabetes.51.1.49. [DOI] [PubMed] [Google Scholar]
- 28.Felig P, Wahren J. Role of insulin and glucagon in the regulation of hepatic glucose production during exercise. Diabetes. 1979;28(Suppl 1):71–75. doi: 10.2337/diab.28.1.s71. [DOI] [PubMed] [Google Scholar]
- 29.Vranic M, Berger M. Exercise and diabetes mellitus. Diabetes. 1979;28:147–163. doi: 10.2337/diab.28.2.147. [DOI] [PubMed] [Google Scholar]
- 30.Vranic M. Banting Lecture: glucose turnover. A key to understanding the pathogenesis of diabetes (indirect effects of insulin) Diabetes. 1992;41:1188–1206. doi: 10.2337/diab.41.9.1188. [DOI] [PubMed] [Google Scholar]
- 31.Kjaer M, Kiens B, Hargreaves M, Richter EA. Influence of active muscle mass on glucose homeostasis during exercise in humans. J Appl Physiol. 1991;71:552–557. doi: 10.1152/jappl.1991.71.2.552. [DOI] [PubMed] [Google Scholar]
- 32.Richter EA, Turcotte L, Hespel P, Kiens B. Metabolic responses to exercise. Effects of endurance training and implications for diabetes. Diabetes Care. 1992;15:1767–1776. doi: 10.2337/diacare.15.11.1767. [DOI] [PubMed] [Google Scholar]
- 33.Felig P, Wahren J. Amino acid metabolism in exercising man. J Clin Invest. 1971;50:2703–2714. doi: 10.1172/JCI106771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Katz J. Determination of gluconeogenesis in vivo with 14C-labeled substrates. Am J Physiol. 1985;248:R391–R399. doi: 10.1152/ajpregu.1985.248.4.R391. [DOI] [PubMed] [Google Scholar]
- 35.Wahren J, Hagenfeldt L, Felig P. Splanchnic and leg exchange of glucose, amino acids, and free fatty acids during exercise in diabetes mellitus. J Clin Invest. 1975;55:1303–1314. doi: 10.1172/JCI108050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wasserman DH, Lickley HL, Vranic M. Interactions between glucagon and other counterregulatory hormones during normoglycemic and hypoglycemic exercise in dogs. J Clin Invest. 1984;74:1404–1413. doi: 10.1172/JCI111551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cherrington AD, Williams PE, Shulman GI, Lacy WW. Differential time course of glucagon’s effect on glycogenolysis and gluconeogenesis in the conscious dog. Diabetes. 1981;30:180–187. doi: 10.2337/diab.30.3.180. [DOI] [PubMed] [Google Scholar]
- 38.Howlett K, Febbraio M, Hargreaves M. Glucose production during strenuous exercise in humans: role of epinephrine. Am J Physiol. 1999;276:E1130–E1135. doi: 10.1152/ajpendo.1999.276.6.E1130. [DOI] [PubMed] [Google Scholar]
- 39.Sherwin RS, Fisher M, Hendler R, Felig P. Hyperglucagonemia and blood glucose regulation in normal, obese and diabetic subjects. N Engl J Med. 1976;294:455–461. doi: 10.1056/NEJM197602262940901. [DOI] [PubMed] [Google Scholar]
- 40.Kjaer M, Engfred K, Fernandes A, Secher NH, Galbo H. Regulation of hepatic glucose production during exercise in humans: role of sympatho-adrenergic activity. Am J Physiol. 1993;265:E275–E283. doi: 10.1152/ajpendo.1993.265.2.E275. [DOI] [PubMed] [Google Scholar]
- 41.Christensen NJ. Abnormally high plasma catecholamines at rest and during exercise in ketotic juvenile diabetics. Scand J Clin Lab Invest. 1970;26:343–344. doi: 10.3109/00365517009046243. [DOI] [PubMed] [Google Scholar]
- 42.Christensen NJ, Galbo H, Hansen JF, Hesse B, Richter EA, Trap-Jensen J. Catecholamines and exercise. Diabetes. 1979;28(Suppl 1):58–62. doi: 10.2337/diab.28.1.s58. [DOI] [PubMed] [Google Scholar]
- 43.Christensen NJ, Galbo H. Sympathetic nervous activity during exercise. Annu Rev Physiol. 1983;45:139–153. doi: 10.1146/annurev.ph.45.030183.001035. [DOI] [PubMed] [Google Scholar]