

Abstract
Insulin resistance is the best predictor for the development of diabetes in offspring of type 2 diabetic patients, but the mechanism responsible for it remains unknown. Recent studies have demonstrated increased intramyocellular lipid, decreased mitochondrial ATP synthesis, and decreased mitochondrial density in the muscle of lean, insulin-resistant offspring of type 2 diabetic patients. These data suggest an important role for mitochondrial dysfunction in the pathogenesis of type 2 diabetes. To further explore this hypothesis, we assessed rates of substrate oxidation in the muscle of these same individuals using 13C magnetic resonance spectroscopy (MRS). Young, lean, insulin-resistant offspring of type 2 diabetic patients and insulin-sensitive control subjects underwent 13C MRS studies to noninvasively assess rates of substrate oxidation in muscle by monitoring the incorporation of 13C label into C4 glutamate during a [2-13C]acetate infusion. Using this approach, we found that rates of muscle mitochondrial substrate oxidation were decreased by 30% in lean, insulin-resistant offspring (59.8 ± 5.1 nmol · g−1 · min−1, P = 0.02) compared with insulin-sensitive control subjects (96.1 ± 16.3 nmol · g−1 · min−1). These data support the hypothesis that insulin resistance in skeletal muscle of insulin-resistant offspring is associated with dysregulation of intramyocellular fatty acid metabolism, possibly because of an inherited defect in the activity of mitochondrial oxidative phosphorylation.
Offspring of type 2 diabetic patients have a significantly increased risk of developing diabetes during their lifetime (1). The factors that contribute to this inherited risk are unknown, but insulin resistance is the best predictor for future development of the disease (1). In several populations, intramuscular triglyceride, measured by biopsy (2), and intramyocellular lipid (IMCL), measured by 1H magnetic resonance spectroscopy (MRS) techniques (3–6), have been shown to be positively correlated with insulin resistance in skeletal muscle. A mechanism has been proposed whereby accumulation of intracellular fatty acid metabolites, such as diacylglycerol, impairs the insulin signaling cascade via activation of novel protein kinase Cs (θ, βII, and δ) and subsequent phosphorylation of critical serine residues of the insulin receptor substrate-1 (IRS-1) (7–11). Upon insulin stimulation, IRS-1–associated phosphatidylinositol 3-kinase activation is diminished, resulting in decreased insulin-stimulated activation of glucose transport. At present, what factors contribute to the increased accumulation of intracellular lipid in these individuals is unknown.
Previous studies of insulin-resistant offspring have revealed elevated plasma fatty acid concentrations (12) and increased IMCL (5,6), suggesting dysregulation of lipid metabolism in this group of subjects. Recent studies by our group have demonstrated increased IMCL associated with decreased rates of mitochondrial ATP synthesis, assessed by 31P MRS, in the muscle of lean, insulin-resistant offspring of type 2 diabetic patients (13). These data suggest a potentially important role for mitochondrial dysfunction in the pathogenesis of type 2 diabetes. This association is supported by evidence for reduced mitochondrial density, determined by electron microscopy, and impaired insulin signaling in the same cohort of subjects (14). Currently, however, there is no direct evidence of decreased rates of mitochondrial substrate oxidation and therefore fatty acid oxidation in the muscle of insulin-resistant offspring.
To address this issue, we applied a novel 13C MRS method, developed in our laboratory, to directly measure rates of substrate oxidation in the muscle of these insulin-resistant individuals (15,16). The rate of oxidation via the tricarboxylic acid (TCA) cycle was determined noninvasively for each individual using 13C MRS to monitor the incorporation of 13C label into the muscle glutamate pool at the C4 position during an intravenous infusion of [2-13C]acetate (Fig. 1). Computer modeling of the enrichments of plasma [2-13C]acetate and muscle [4-13C]glutamate yielded the TCA cycle flux and provides a direct measure of muscle mitochondrial substrate oxidation rates.
FIG. 1.

Schematic of the TCA cycle, demonstrating incorporation of 13C label from plasma [2-13C]acetate into the muscle [4-13C]glutamate pool. *The carbon position labeled with 13C. Pyr, pyruvate; FFA, free fatty acids; AcCoA, acetyl-CoA; αKG, α-ketoglutarate. A single turn of the TCA cycle is shown; a second turn of the cycle forms [2-13C]glutamate and [3-13C]glutamate.
RESEARCH DESIGN AND METHODS
Twelve insulin-resistant offspring and seven insulin-sensitive subjects were recruited as described previously (13). Briefly, all subjects were selected to be in excellent health, lean, nonsmoking, and taking no medications and to have had a birth weight >2.3 kg (5 lb) and a sedentary lifestyle, as defined by an activity index questionnaire (17). Insulin-resistant offspring were classified as having an insulin sensitivity index (ISI) (18) <4.0 after a 2-h oral glucose-tolerance test (75-g oral glucose load), plus either one parent with type 2 diabetes or a grandparent and one other second-degree relative with type 2 diabetes. Insulin-sensitive control subjects were classified by an ISI >6.3, with or without a family history of type 2 diabetes. Thresholds for classification of insulin sensitivity by ISI corresponded closely to the lowest (<3.7) and highest quartiles (>6.1) of ISI determined in a prior cross-sectional study of 482 healthy, lean, sedentary, nonsmoking individuals (19). Written, informed consent was obtained from each subject after the purpose, nature, and complications of the studies were explained and the protocol was approved by the Yale Human Investigation Committee.
Diet and study preparation
For 3 days before the 13C MRS studies, subjects consumed a weight-maintenance diet containing at least 150 g carbohydrate/day and were instructed not to perform any exercise other than normal walking. The evening before the MRS studies, subjects were admitted to the Yale–New Haven Hospital General Clinical Research Center and fasted overnight, with free access to drinking water.
13C MRS
After an overnight 12-h fast, subjects were transported to the Yale Magnetic Resonance Research Center via wheelchair and positioned supine in the bore of a superconducting magnetic resonance magnet. Experiments were performed on either a 2.1T (Magnex Instruments, Oxford, U.K.) or 4T system (Bruker Biospin, Ettlingen, Germany).
The muscles of the right calf were positioned within a home-built magnetic resonance probe assembly over a 9-cm–diameter 13C surface coil with twin, orthogonal 13-cm 1H quadrature coils for imaging, shimming, and decoupling. After tuning, transverse, gradient-echo, scout images of the calf were acquired to ensure correct positioning and to define a volume for localized shimming using the FASTMAP procedure (20). Typical 1H line widths within the volume of interest were 12 and 13 Hz for the 2.1T and 4T systems, respectively.
13C magnetic resonance spectra on the 2.1T system were acquired using a nonlocalized sequence with NOE (nuclear overhauser enhancement), WALTZ16 decoupling, and a repetition time (TR) of 1.4 s (16). The 13C lipid peak at 34.4 ppm overlaps with the muscle C4-glutamate peak and was suppressed by T1-selective nulling after an adiabatic inversion pulse. Temporal resolution was 10 min, corresponding to 424 averages. 13C magnetic resonance spectra on the 4T system were acquired using a localized 1H-13C polarization-transfer sequence (21) with WALTZ16 decoupling. Selection of a 90-cm3 volume within the calf muscles was achieved using two-dimensional adiabatic outer-volume suppression; IMCL was suppressed using T1-selective nulling, as above. Temporal resolution was 5 min, corresponding to 160 averages at a TR of 1.7 s.
13C magnetic resonance spectra were acquired for 20 min before and during a 120-min infusion of 99% enriched [2-13C]acetate (350 mmol/l sodium salt) at a rate of 3.0 mg · kg−1 · min−1. Plasma samples were obtained at 10-min intervals throughout the study for the measurement of plasma acetate concentration and fractional enrichment by gas chromatography/mass spectrometry (16).
13C MRS data processing
13C free induction decays (FIDs) were processed using XWINNMR version 6.5 (Bruker Biospin); FIDs were zero-filled to 32K points and multiplied by an exponential factor, corresponding to 5 Hz, before Fourier transformation. Reference spectra were manually phased (0 and 1st order), and these phase correction parameters were applied to all subsequent spectra; baselines were corrected by fitting to a 1st-order polynomial. Absolute enrichment of the C2-glutamate peak was determined by integration (±0.5 ppm) relative to the natural abundance enrichment (1.1%) of the reference spectra. Natural abundance C4-glutamate is undetectable in human muscle in vivo because of overlapping lipid resonances. To determine the time course of 13C incorporation into C4-glutamate during the [2-13C]acetate infusion, difference spectra for each time point were obtained by subtracting averaged reference spectra, and the increment in the C4-glutamate peak was determined by integration (±0.5 ppm). Approximate maximal enrichment at C4-glutamate was calculated from that of C2-glutamate, assuming 5% dilution of the C2 pool because of anaplerosis.
CWave modeling
TCA cycle flux (VTCA) for each individual was determined by computer modeling of the incorporation of 13C label from plasma [2-13C]acetate into the muscle [4-13C]glutamate pool, using CWave software (16,22,23). The CWave model consists of isotopic mass balance equations that describe the metabolic fate of the plasma [2-13C]acetate (see below). Upon entry into the myocyte, [2-13C]acetate is converted (Vac) into [2-13C]acetyl CoA, which enters the TCA cycle by condensing with oxaloacetate to form [4-13C]citrate. Entry of unlabeled substrates into the TCA cycle via acetyl CoA or anaplerosis was incorporated into the model as a separate reaction (VPDH+FA). The position of the 13C label is conserved through the initial steps of the TCA cycle, labeling α-ketoglutarate at the C4 position. Glutamate and α-ketoglutarate are in rapid exchange, and equilibration results in the formation of [4-13C]glutamate. As the TCA cycle progresses, the 13C label becomes scrambled between the C2 and C3 positions because of the symmetry of the succinate molecule; a second turn of the TCA cycle yields [2-13C]glutamate and [3-13C]glutamate. CWave determines VTCA as the rate of total carbon flow from acetyl CoA to α-ketoglutarate using a nonlinear least-squares algorithm to fit the curve of C4-glutamate enrichment, assuming a value for the rate of α-ketoglutarate–glutamate exchange that was determined in previous studies. In this model, VTCA is equal to the sum of Vac + VPDH+FA but is independent of the absolute fractional enrichment at C4-glutamate (Vac/VPDH+FA). The intracellular concentration of glutamate was previously measured by muscle biopsy and was found to be 2.41 mmol/l (16); the ratio of natural abundance C2-glutamate/C2-creatine was the same between the groups, and there was no significant difference in the fractional enrichment at C2-glutamate at the end of the [2-13C]acetate infusion, indicating that the intracellular concentration of glutamate was unchanged in insulin-resistant offspring. The rate of exchange between α-ketoglutarate and glutamate (Vx) was determined in prior studies (16,24) and was fixed at 150 nmol · g−1 muscle · min−1, which is significantly faster than the TCA cycle flux.
Isotopic mass balance equations
Mass balance equations are as follows.
Isotope balance equations are as follows.
Muscle metabolite concentrations are as follows: acetyl CoA, 0.05 μmol/g; citrate, 0.2 μmol/g; glutamate, 2.41 μmol/g; and α-ketoglutarate, 0.05 μmol/g.
1H MRS
On a separate occasion, IMCL content of the soleus muscle was assessed in each subject by 1H MRS. Spectra were obtained using either a PRESS (2.1T system) or a STEAM (4T system) sequence and quantified as described previously (25).
Statistical analysis
All data are expressed as means ± SE. Statistical analyses were performed using InStat3 software (GraphPad Software). Statistically significant differences between insulin-sensitive subjects and insulin-resistant offspring were detected using an unpaired Student’s t test. A P value of <0.05 was considered statistically significant.
RESULTS
Subject characteristics
Characteristics of the insulin-sensitive subjects and insulin-resistant offspring are shown in Table 1. The two groups were similar in age, height, weight, and activity, although BMI was slightly higher in the insulin-resistant offspring. Fasting plasma concentrations of glucose and insulin were higher in the insulin-resistant offspring than the insulin-sensitive control subjects. As reported previously (13), all subjects had normal glucose tolerance; however, insulin release in response to the glucose load of the oral glucose tolerance test was greater and IMCL content of the soleus muscle was increased by ~70% (P = 0.03) in insulin-resistant offspring compared with insulin-sensitive control subjects (Table 1). Two of the insulin-sensitive subjects had a family history of diabetes.
TABLE 1.
Characteristics of insulin-sensitive control subjects and insulin-resistant offspring
| Insulin-sensitive control | Insulin-resistant offspring | |
|---|---|---|
| n | 7 | 12 |
| Men/women | 2/5 | 3/9 |
| Age (years) | 26 ± 2 | 26 ± 2 |
| Height (m) | 1.70 ± 0.04 | 1.67 ± 0.03 |
| Weight (kg) | 61 ± 4 | 66 ± 3 |
| BMI (kg/m2) | 20.9 ± 0.7 | 23.7 ± 0.6* |
| Activity index | 2.4 ± 0.4 | 2.5 ± 0.1 |
| Fasting plasma glucose (mg/dl) | 81.3 ± 1.6 | 93.2 ± 3.0† |
| Fasting plasma insulin (μU/ml) | 7.0 ± 1.2 | 12.7 ± 0.7† |
| ISI | 10.3 ± 1.9 | 2.7 ± 0.3† |
| IMCL (%) | 0.53 ± 0.1 | 0.90 ± 0.11† |
Data are means ± SE.
P < 0.01;
P < 0.05.
13C MRS
The concentration and enrichment of plasma acetate were elevated within 5 min upon commencing the infusion of [2-13C]acetate and were stable for the remainder of the experiment for all subjects. Steady-state values for the offspring and control groups are shown in Table 2. Enrichment of the muscle [4-13C]glutamate pool after infusion of [2-13C]acetate, expressed as the mean of all subjects for each subject group, is shown in Fig. 2. The time course of enrichment in muscle [4-13C]glutamate was slower in the insulin-resistant offspring compared with the insulin-sensitive controls with divergence in the enrichments of [4-13C]glutamate between the two groups apparent after only 10 min of infusion of [2-13C]acetate. However, there was no difference in the maximal enrichment of muscle [2-13C]glutamate attained during the infusion or the calculated maximum enrichment at [4-13C]glutamate between the groups (Table 2). When the curves of muscle [4-13C]glutamate incorporation were modeled for each individual using CWave software, the calculated rate of muscle TCA cycle flux (Fig. 3) was reduced by 30% (P = 0.02) in the insulin-resistant offspring (59.8 ± 5.1 nmol · g−1 · min−1) compared with the insulin-sensitive controls (96.1 ± 16.3 nmol · g−1 · min−1). The contribution of acetate to the overall oxidation of substrates by the TCA cycle (Vac/VTCA) was identical in both groups (Table 2). There was no effect of BMI or sex on the rate of TCA cycle flux within either the insulin-resistant or -sensitive groups or for both groups combined.
TABLE 2.
Plasma and muscle metabolite data during the 120-min [2-13C]acetate infusion
| Insulin-sensitive control | Insulin-resistant offspring | |
|---|---|---|
| Plasma [acetate] (mmol/l) | 0.93 ± 0.12 | 0.93 ± 0.05 |
| Plasma [2-13C]acetate APE (%) | 80.8 ± 2.45 | 87.42 ± 0.80* |
| Muscle C2-glutamate APE (%) | 1.20 ± 0.39 | 1.04 ± 0.20 |
| Muscle C4-glutamate APE (%)† | 2.52 ± 0.83 | 2.13 ± 0.43 |
| VTCA (nmol · g−1 · min−1) | 96.1 ± 16.3 | 59.8 ± 5.1‡ |
| Vac/VTCA (%) | 3.4 ± 1.1 | 2.9 ± 0.6 |
Data are means ± SE. APE, atom percent excess.
P < 0.01;
calculated from C2-glutamate assuming 5% anaplerosis;
P < 0.05.
FIG. 2.
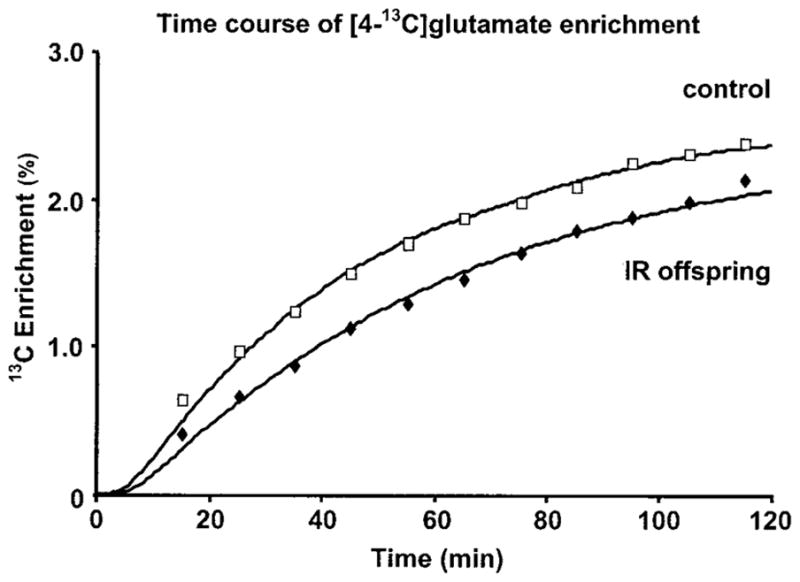
Time course of enrichment (averaged data) of the muscle [4-13C]glutamate pool, measured by 13C MRS, during an infusion of [2-13C]acetate in insulin-resistant offspring of type 2 diabetic patients (◆, n = 12) and insulin-sensitive control subjects (□, n = 7). The curves of incorporation for each individual were computer modeled using CWave software to generate an estimate of TCA cycle flux using a nonlinear least-squares fitting algorithm. A CWave fit of the averaged data for each group is shown and yielded identical TCA cycle fluxes as the data for each individual.
FIG. 3.
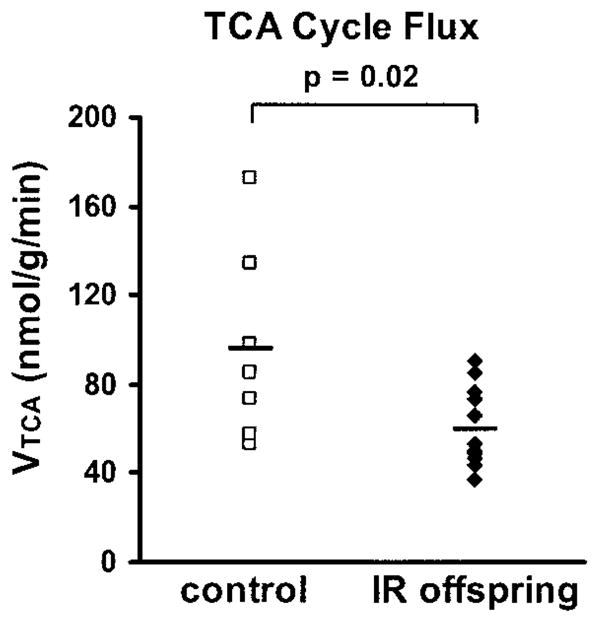
TCA cycle flux (nmol · g−1 muscle · min−1) calculated on an individual-by-individual basis in insulin-resistant offspring of type 2 diabetic patients (◆, n = 12) and insulin-sensitive control subjects (□, n = 7). The average rate of TCA cycle flux for each group is shown by the black bar.
To obtain the most sensitive time course of C4-glutamate enrichment and therefore the most accurate estimate of VTCA, the 13C MRS methods used in this study were optimized to detect C4-glutamate. Although C2-glutamate could also be observed, the rapid time resolution did not permit accurate time courses of C2-glutamate enrichment to be measured on an individual basis. However, to examine whether the rate of anaplerotic flux may have influenced the calculated rate of TCA cycle flux and to test the validity of our assumption that Vx >VTCA, we also assessed datasets of C2-glutamate, C4-glutamate, and plasma acetate enrichment created by averaging the data obtained from each individual for both groups. These data were analyzed using a model that incorporated an anaplerotic flux via pyruvate carboxylase (Vana) and fitted the target data of C2-glutamate enrichment and C4-glutamate incorporation. Setting Vana at a fixed rate corresponding to 5, 10, or 20% of VTCA did not significantly change the calculated VTCA and confirmed that Vx is faster than VTCA.
DISCUSSION
Insulin-resistant offspring of type 2 diabetic patients have an increased lifetime risk of developing diabetes, suggesting that they possess an inherited metabolic defect that predisposes them to develop insulin resistance and eventually diabetes. However, little is known about what this inherited metabolic defect may be. We have previously demonstrated that young, lean but insulin-resistant offspring of type 2 diabetic patients have impaired insulin-stimulated nonoxidative muscle glucose disposal and increased IMCL content compared with age-, height-, weight-, and activity-matched insulin-sensitive controls (13). Accumulation of intracellular lipid has been well correlated with impaired insulin signaling in both muscle and liver (2–6), and mechanisms for IMCL-induced insulin resistance have been proposed (7–11). Deposition of IMCL could either be due to increased delivery of fatty acids to the muscle and/or to decreased fatty acid oxidation (7). In insulin-resistant offspring, we observed that muscle fatty acid delivery, assessed by whole-body and localized rates of lipolysis, was unaltered compared with insulin-sensitive subjects, suggesting that fatty acid oxidation was compromised in these individuals. Supporting this hypothesis, muscle ATP synthesis, measured by 31P saturation-transfer MRS, was decreased, implicating impaired muscle mitochondrial function (13). However, this is an indirect assessment of mitochondrial oxidative activity, and to date, there is no direct evidence of decreased rates of muscle mitochondrial oxidation and therefore fatty acid oxidation in insulin-resistant offspring.
To address this issue, we used in vivo 13C MRS to directly measure muscle oxidation rates. Computer modeling of the incorporation of 13C label from infused [2-13C]acetate into the muscle [4-13C]glutamate pool yields the rate of flux through the TCA cycle. We found that the rate of 13C incorporation into the C4-glutamate pool was slower in muscle of insulin-resistant offspring compared with controls, and this reflected an ~30% decrease in the basal rate of muscle substrate oxidation. This decrease in muscle TCA cycle flux was associated with an increase in IMCL content of the soleus muscle in the insulin-resistant offspring.
This observation is supported by two recent studies of insulin-resistant offspring from our group. Electron microscopy analysis of muscle biopsies obtained from a similar cohort of young, lean, insulin-resistant offspring revealed that mitochondrial density was decreased by 38% compared with insulin-sensitive control subjects (14). This reduction in mitochondrial number was associated with impaired insulin signaling and decreased expression of the mitochondrial proteins: cytochrome oxidase (COX), pyruvate dehydrogenase (PDH), and succinate dehydrogenase (SDH); these enzymes catalyze key steps of mitochondrial oxidative metabolism at the level of the electron transport chain (COX) and the TCA cycle (SDH and PDH). Furthermore, insulin-stimulated rates of ATP synthesis were decreased in insulin-resistant offspring (26), suggesting that the mitochondria of these individuals have an impaired capability to respond to insulin-stimulated energy demands of the muscle.
Studies of the genotypes of different diabetic cohorts have implicated the involvement of peroxisome proliferator–activated receptor-γ coactivator-1α (PGC-1α), a nuclear transcription factor responsible for the regulation of genes involved in mitochondrial oxidation and biogenesis (27), in the diabetic phenotype (28–32). We previously assessed mRNA and protein content of PGC-1α, PGC-1β, and several downstream targets (nuclear respiratory factor-1 and -2 and mitochondrial transcription factor A) in our cohort of insulin-resistant offspring and did not observe any significant alterations in the expression of these factors known to regulate mitochondrial biogenesis (14). Differences in the characteristics of the subjects selected for these studies may explain the discrepancies in the data. Taken together, these studies imply that any defect in the regulation of mitochondrial biogenesis and mitochondrial activity in insulin-resistant offspring occurs either downstream of PGC-1α or by other unknown factors.
A recent study has also investigated lipid oxidation in offspring of type 2 diabetic patients in comparison with control subjects with no family history of diabetes (33). Initial analyses of these data demonstrated impaired insulin sensitivity associated with elevated IMCL in offspring, but detected no difference in rates of fasting whole-body lipid oxidation, calculated from indirect calorimetry. These findings would appear to be contradictory to the data presented in this study; however, when the data from the offspring group was segregated into quartiles based on lipid oxidation, they detected a significant association between whole-body lipid oxidation rate and insulin sensitivity. In contrast, this correlation was not observed in the control group. It is worth noting that in the study by Lattuada et al. (33), lipid oxidation was assessed indirectly and reflected whole-body rates of lipid oxidation and thus may have been too insensitive to detect alterations in the rate of muscle lipid oxidation between offspring and control subjects. In contrast, the present study directly assessed muscle-specific rates of substrate oxidation via the TCA cycle.
It is also possible that the decrease in muscle TCA cycle flux that we observed in offspring could be distal to a primary impairment in fatty acid oxidation or may occur as a consequence, rather than a cause, of insulin resistance. However, the gene array studies in subjects with family history of diabetes show little or no effect on expression of enzymes involved in β-oxidation (30,31). Although Patti et al. (31) did observe decreased expression of a few enzymes of lipid metabolism in diabetic patients, only monoglyceride lipase was significantly affected in offspring. Similarly, although Mootha et al. (30) observed a coordinated decrease in expression of their “OXPHOS” group of enzymes, they found no detectable effect in enzymes involved in lipid oxidation or metabolism. These studies suggest that impaired lipid oxidation was not the cause of decreased TCA cycle flux in offspring. Mitochondrial function could potentially become impaired as a consequence of insulin resistance, and without a longitudinal study of at-risk individuals, it is impossible to discern the primary cause of the decrease in TCA cycle flux. However, we have purposely recruited young, lean, and healthy individuals to eliminate confounding factors such as age, obesity, and diabetes in an attempt to isolate the earliest changes in metabolism associated with insulin resistance. Given that the deficit in muscle TCA cycle flux that we have measured in these subjects occurred before impaired glucose tolerance, this suggests that defective muscle mitochondrial function is an early factor in the pathogenesis of type 2 diabetes.
Taken together, these data support the hypothesis that insulin-resistant offspring of type 2 diabetic patients have an inherited defect in muscle mitochondrial oxidative phosphorylation activity that may be attributed to reduced mitochondrial content. This leads to decreased lipid oxidation, which predisposes these individuals to the accumulation of intramyocellular fatty acid metabolites. Although the mechanism of fat-induced insulin resistance has yet to be fully elucidated, it has been postulated that increase in the intracellular concentration of fatty-acyl CoA and diacylglycerol activates a serine/threonine kinase cascade that impairs insulin signaling at the level of IRS-1 and causes insulin resistance in these individuals (7–11,14,34). Interestingly, lean but insulin-resistant elderly subjects also exhibit impaired mitochondrial oxidative phosphorylation that is associated with increased IMCL, elevated intrahepatic lipid content, and insulin resistance (24). In contrast to insulin-resistant offspring, it is likely that an acquired, rather than inherited, defect in mitochondrial activity may lead to the decrement in muscle mitochondrial function associated with aging (Fig. 4). These data support the hypothesis that mitochondrial activity and biogenesis may represent important new pharmacological targets for the prevention and treatment of type 2 diabetes.
FIG. 4.
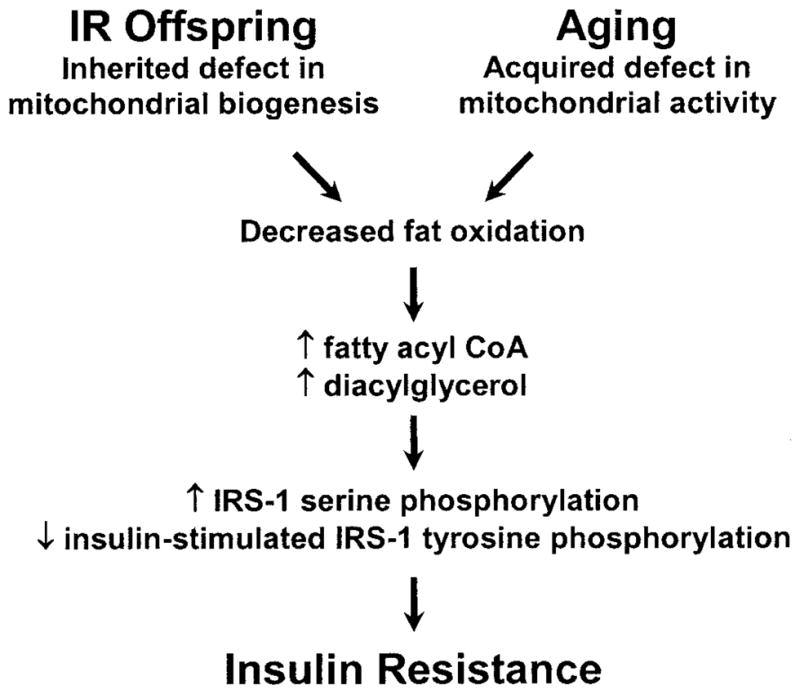
Schematic depicting the central role of the mitochondria and impaired lipid oxidation in causing insulin resistance in offspring of type 2 diabetic patients (IR offspring) and the elderly.
Acknowledgments
G.I.S. has received a Distinguished Clinical Scientist Award from the American Diabetes Association. This work has been supported by National Institutes of Health Grants P01-DK-68229, R01-AG-23686, R01-DK-063192, R01-DK-49230, P30-DK-45735, and M01-RR-00125. CWave was developed by Dr. Graeme Mason at the Yale Magnetic Resonance Research Center.
We thank the subjects for their participation in this study; Dr. James Dziura for statistical assistance; and Yanna Kosover, Anthony Romanelli, Mikhail Smolgovsky, and the staff of the Yale–New Haven Hospital General Clinical Research Center for technical assistance with the studies.
- COX
cytochrome oxidase
- FID
free induction decay
- IMCL
intramyocellular lipid
- IRS-1
insulin receptor substrate-1
- ISI
insulin sensitivity index
- MRS
magnetic resonance spectroscopy
- PDH
pyruvate dehydrogenase
- PGC
peroxisome proliferator–activated receptor-γ coactivator
- SDH
succinate dehydrogenase
- TCA
tricarboxylic acid
References
- 1.Warram JH, Martin BC, Krolewski AS, Soeldner JS, Kahn CR. Slow glucose removal and hyperinsulinemia precede the development of type II diabetes in the offspring of diabetic parents. Ann Intern Med. 1990;113:909–915. doi: 10.7326/0003-4819-113-12-909. [DOI] [PubMed] [Google Scholar]
- 2.Phillips DI, Caddy S, Ilic V, Fielding BA, Frayn KN, Borthwick AC, Taylor R. Intramuscular triglyceride and muscle insulin sensitivity: evidence for a relationship in non-diabetic subjects. Metabolism. 1996;45:947–950. doi: 10.1016/s0026-0495(96)90260-7. [DOI] [PubMed] [Google Scholar]
- 3.Krssak M, Petersen KF, Dresner A, DiPietro L, Vogel SM, Rothman DL, Roden M, Shulman GI. Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: a 1H NMR spectroscopy study. Diabetologia. 1999;42:113–116. doi: 10.1007/s001250051123. [DOI] [PubMed] [Google Scholar]
- 4.Szczepaniak LS, Babcock EE, Schick F, Dobbins RL, Garg A, Burns DK, McGarry JD, Stein DT. Measurement of intracellular triglyceride stores by H spectroscopy: validation in vivo. Am J Physiol Endocrinol Metab. 1999;276:E977–E989. doi: 10.1152/ajpendo.1999.276.5.E977. [DOI] [PubMed] [Google Scholar]
- 5.Perseghin G, Scifo P, De Cobelli F, Pagliato E, Battezzati A, Arcellono C, Vanzulli A, Testolin G, Pozza G, Del Maschio A, Luzi L. Intramyocellular triglyceride content is a determinant of in vivo insulin resistance in humans: a 1H-13C nuclear magnetic resonance spectroscopy assessment in offspring of type 2 diabetic parents. Diabetes. 1999;48:1600–1606. doi: 10.2337/diabetes.48.8.1600. [DOI] [PubMed] [Google Scholar]
- 6.Jacob S, Machann J, Rett K, Brechtel K, Volk A, Renn W, Maerker E, Matthaei S, Schick F, Claussen CD, Häring HU. Association of increased intramyocellular lipid content with insulin resistance in lean nondiabetic offspring of type 2 diabetic subjects. Diabetes. 1999;48:1113–1119. doi: 10.2337/diabetes.48.5.1113. [DOI] [PubMed] [Google Scholar]
- 7.Shulman GI. Cellular mechanisms of insulin resistance. J Clin Invest. 2000;106:171–176. doi: 10.1172/JCI10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Griffin ME, Marcucci MJ, Cline GW, Bell K, Barucci N, Lee D, Goodyear LJ, Kraegen EW, White MF, Shulman GI. Free fatty acid-induced insulin resistance is associated with activation of protein kinase Cθ and alterations in the insulin signaling cascade. Diabetes. 1999;48:1270–1274. doi: 10.2337/diabetes.48.6.1270. [DOI] [PubMed] [Google Scholar]
- 9.Itani SI, Ruderman NB, Schmieder F, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IκB-α. Diabetes. 2002;51:2005–2011. doi: 10.2337/diabetes.51.7.2005. [DOI] [PubMed] [Google Scholar]
- 10.Dresner A, Laurent D, Marcucci M, Griffin ME, Dufour S, Cline GW, Slezak LA, Andersen DK, Hundal RS, Rothman DL, Petersen KF, Shulman GI. Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J Clin Invest. 1999;103:253–259. doi: 10.1172/JCI5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu C, Chen Y, Cline GW, Zhang D, Zong H, Wang Y, Bergeron R, Kim JK, Cushman SW, Cooney GJ, Atcheson B, White MF, Kraegen EW, Shulman GI. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem. 2002;277:50230–50236. doi: 10.1074/jbc.M200958200. [DOI] [PubMed] [Google Scholar]
- 12.Perseghin G, Ghosh S, Gerow K, Shulman GI. Metabolic defects in lean non-diabetic offspring of NIDDM parents: a cross-sectional study. Diabetes. 1997;46:1001–1009. doi: 10.2337/diab.46.6.1001. [DOI] [PubMed] [Google Scholar]
- 13.Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350:664–671. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morino K, Petersen KF, Dufour S, Befroy D, Frattini J, Shatzkes N, Neschen S, White MF, Bilz S, Sono S, Pypaert M, Shulman GI. Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J Clin Invest. 2005;115:3587–3593. doi: 10.1172/JCI25151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jucker BM, Dufour S, Ren J, Cao X, Previs SF, Underhill B, Cadman KS, Shulman GI. Assessment of mitochondrial energy coupling in vivo by 13C/31P NMR. Proc Natl Acad Sci U S A. 2000;100:6880–6884. doi: 10.1073/pnas.120131997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lebon V, Dufour S, Petersen KF, Ren J, Jucker BM, Slezak LA, Cline GW, Rothman DL, Shulman GI. Effect of triiodothyronine on mitochondrial energy coupling in human skeletal muscle. J Clin Invest. 2001;108:733–737. doi: 10.1172/JCI11775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baecke JA, Burema J, Frijters JE. A short questionnaire for the measurement of habitual physical activity in epidemiological studies. Am J Clin Nutr. 1982;36:936–942. doi: 10.1093/ajcn/36.5.936. [DOI] [PubMed] [Google Scholar]
- 18.Matsuda M, DeFronzo RA. Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic insulin clamp. Diabetes Care. 1999;22:1462–1470. doi: 10.2337/diacare.22.9.1462. [DOI] [PubMed] [Google Scholar]
- 19.Petersen KF, Dufour S, Feng J, Befroy D, Dziura J, Man CD, Cobelli C, Shulman GI. Increased prevalence of insulin resistance and nonalcoholic fatty liver disease in Asian-Indian men. Proc Natl Acad Sci U S A. 2006;103:18273–18277. doi: 10.1073/pnas.0608537103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shen J, Rycyna RE, Rothman DL. Improvements on an in vivo automatic shimming method [FASTERMAP] Magn Reson Med. 1997;38:834–839. doi: 10.1002/mrm.1910380521. [DOI] [PubMed] [Google Scholar]
- 21.Doddrell DM, Pegg DT, Bendall MR. Distortionless enhancement of NMR signals by polarization transfer. J Magn Reson. 1982;48:323–327. [Google Scholar]
- 22.Mason GF, Rothman DL, Behar KL, Shulman RG. NMR determination of the TCA cycle rate and alpha/ketoglutarate/glutamate exchange rate in rat brain. J Cereb Blood Flow Metab. 1992;12:434–447. doi: 10.1038/jcbfm.1992.61. [DOI] [PubMed] [Google Scholar]
- 23.Mason GF. Cwave software for the design and analysis of 13C labeling studies performed in-vivo (Abstract) Proc Int Soc Magn Reson Med. 2000;3:1870. [Google Scholar]
- 24.Petersen KF, Befroy D, Dufour S, Dzuira J, Ariyan C, Rothman DL, DiPietro L, Cline GW, Shulman GI. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003;300:1140–1142. doi: 10.1126/science.1082889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mayerson AB, Hundal RS, Dufour S, Lebon V, Befroy D, Cline GW, Enocksson S, Inzucchi SE, Shulman GI, Petersen KF. The effects of rosiglitazone on insulin sensitivity, lipolysis, and hepatic and skeletal muscle triglyceride content in patients with type 2 diabetes. Diabetes. 2002;51:797–802. doi: 10.2337/diabetes.51.3.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Petersen KF, Dufour S, Shulman GI. Decreased insulin-stimulated ATP synthesis and phosphate transport in muscle of insulin-resistant offspring of type 2 diabetic parents. PLoS Med. 2005;2:e233. doi: 10.1371/journal.pmed.0020233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 28.Ek J, Andersen G, Urhammer SA, Gaede PH, Drivsholm T, Borch-Johnsen K, Hansen T, Pedersen O. Mutation analysis of peroxisome proliferator-activated receptor-gamma coactivator-1 (PGC-1) and relationships of identified amino-acid polymorphisms to type II diabetes mellitus. Diabetologia. 2001;44:2220–2226. doi: 10.1007/s001250100032. [DOI] [PubMed] [Google Scholar]
- 29.Muller YL, Bogardus C, Pedersen O, Baier L. A Gly482Ser missense mutation in the peroxisome proliferator–activated receptor-γ coactivator-1 is associated with altered lipid oxidation and early insulin secretion in Pima Indians. Diabetes. 2003;52:895–898. doi: 10.2337/diabetes.52.3.895. [DOI] [PubMed] [Google Scholar]
- 30.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 31.Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone R, Landaker EJ, Goldfine AB, Mun E, DeFronzo R, Finlayson J, Kahn CR, Mandarino LJ. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc Natl Acad Sci U S A. 2003;100:8466–8471. doi: 10.1073/pnas.1032913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pihlajamäki J, Kinnunen M, Ruotsalainen E, Salmenniemi U, Vauhkonen I, Kuulasmaa T, Kainulainen S, Laakso M. Haplotypes of PPARGC1A are associated with glucose tolerance, body mass index and insulin sensitivity in offspring of patients with type 2 diabetes. Diabetologia. 2005;48:1331–1334. doi: 10.1007/s00125-005-1800-9. [DOI] [PubMed] [Google Scholar]
- 33.Lattuada G, Costantino F, Caumo A, Scifo P, Ragogna F, De Cobelli F, Del Maschio A, Luzi L, Perseghin G. Reduced whole-body lipid oxidation is associated with insulin resistance, but not with intramyocellular lipid content in offspring of type 2 diabetic patients. Diabetologia. 2005;48:741–747. doi: 10.1007/s00125-005-1686-6. [DOI] [PubMed] [Google Scholar]
- 34.Morino K, Petersen KF, Shulman GI. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes. 2006;55 (Suppl 2):S9–S15. doi: 10.2337/db06-S002. [DOI] [PMC free article] [PubMed] [Google Scholar]