


Abstract
Background:
Current studies suggest an interaction between vascular mechanisms and neurodegenerative processes that leads to late-onset Alzheimer disease (AD). We tested whether AD pathology was associated with white matter hyperintensities (WMH) or cerebral infarcts in the oldest old individuals.
Methods:
Brains from 132 subjects over 85 years old, who came to autopsy from the Vantaa 85+ population-based cohort, were scanned by postmortem MRI and examined for neuropathologic changes. Coronal images were analyzed to determine the degree of frontal and parietal periventricular WMH (PVWMH) and deep WMH (DWMH) and cerebral infarcts. Neuropathologic variables included Consortium to Establish a Registry for Alzheimer's Disease scores for neuritic plaques and Braak staging among subjects in 5 groups: normal aging (NA), borderline with insufficient AD pathology, AD, AD plus other pathology, and other primary degenerative diseases.
Results:
Frontal DWMH were detected in >50% of the sample. Both frontal PVWMH and DWMH were significantly more extensive in the AD group compared to the NA group or the NA and borderline groups combined. Frontal PVWMH and DWMH were also associated with increased Braak staging (p = 0.03) and the neuritic plaque load (p = 0.01). Further analysis revealed there were a greater number of cerebral infarcts associated with frontal DWMH (p = 0.03) but not with frontal PVWMH.
Conclusions:
Our study showed an association between neurofibrillary pathology and frontal PVWMH and DWMH (rather than parietal), as a surrogate of small vessel disease, particularly in very old community-dwelling individuals.
GLOSSARY
- AD
= Alzheimer disease;
- AGD
= argyrophilic grain disease;
- CERAD
= Consortium to Establish a Registry for Alzheimer's Disease;
- CVD
= cerebrovascular disease;
- DLB
= dementia with Lewy bodies;
- DTI
= diffusion tensor imaging;
- DWMH
= deep white matter hyperintensities;
- MR
= magnetic resonance;
- NA
= normal aging;
- PVWMH
= periventricular white matter hyperintensities;
- VaD
= vascular dementia;
- WM
= white matter;
- WMH
= white matter hyperintensities.
Recent studies suggest that cerebrovascular disease (CVD) and vascular risk factors increase risk of Alzheimer disease (AD) and other dementias.1–3 Midlife history of vascular risk factors such as arterial hypertension, dyslipidemia, hyperinsulinemia and type 2 diabetes, obesity, atherosclerosis, and arrhythmias2–6 appears to be associated with brain changes and greater risk of cognitive impairment and dementia in later life. Understanding how these systemic vascular factors precisely modify the brain aging process to cause dementia and possibly increase production of AD-type pathology has considerable implications for diagnosis, prevention, and treatment.
Parallel studies also indicate a substantial clinical and pathologic overlap between AD and vascular dementia (VaD)7–9 with an increasing burden of mixed pathology in the very old.10 Despite these realities, there remains an important debate currently as to whether ischemic cerebrovascular disease contributes directly to the development of Alzheimer pathology7 or whether cerebrovascular disease is merely an independent, comorbid process that increases the likelihood of a dementia diagnosis in patients with asymptomatic low-grade AD pathology.11 The mode or nature of the interaction between these processes resulting in dementia, particularly in the oldest survivors often with mixed pathology,12,13 remains unclear. One approach to reliably explore such interaction is by examining whether graded neurodegenerative pathology is associated with vascular changes, i.e., white matter hyperintensities (WMH) or cerebral infarctions in subjects with and without dementia who are recruited from unselected population-based samples.
In this study, with emphasis on brain pathology in the very old (>85 years of age), we assessed whether there were any relationships between white matter (WM) abnormalities seen on postmortem MRI and macroscopic brain infarcts, and neurodegenerative pathology apparent at autopsy in brains from the Vantaa 85+ population-based cohort study.14
METHODS
Study subjects and brain tissue.
The Vantaa 85+ study comprised assessment of 601 community-dwelling persons, all at least 85 years of age, who were living in the town of Vantaa (Southern Finland) on April 1, 1991.15 During a 10-year follow-up from 1991 to 2001, we accrued 304 autopsies, which all included neuropathologic examination. Of these, 145 brains were scanned by magnetic resonance (MR) between 3 months and 4 years after fixation in phosphate-buffered 10% formalin solution.
Standard protocol approvals, registrations, and patient consents.
This prospective, population-based cohort study has been approved by the ethics committee of the Health Centre of the city of Vantaa and the tissue retention by the National Authority for Medicolegal Affairs, Helsinki, Finland. Individual consents from the subjects were also obtained.
Postmortem MRI and analysis.
For the subsequent analysis, we excluded 13 of the 145 scanned brains because the WM could not be analyzed reliably due to the presence of hemorrhages, tumors, and artifacts. The brains were prepared and MR imaged as described previously with the same scanning protocol.16
The Scheltens rating scale17 for the visual assessment of signal hyperintensities in the MRI was used to rate severity of WM changes. The images with increasing severity of frontal or parietal periventricular WMH (on a 2-point scale) and the frontal, parietal, temporal, and occipital deep WMH (on a 6-point scale) were rated by a skilled reader (E.C.W.v.S., weighted kappa score for interrater reliability 0.85) blinded to other data. Figure 1, A and B, shows the regions of interest and severity WMH with the scores. Individuals with score 1 (none had score 0) for frontal periventricular WMH and subjects with scores 0–3 for frontal deep WMH represented those with mild WM abnormality, while those with more extensive lesions formed the groups of individuals with severe WM abnormality.

Figure 1 Differences in severity of white matter hyperintensities
Individuals with score 1 (none had score 0) for frontal periventricular white matter hyperintensities (A, frontal capping, within circle) and subjects with scores 0–3 for frontal deep white matter hyperintensities (DWMH) (A, punctiform lesions, arrow) represented those with mild white matter (WM) abnormality, while those with more extensive lesions (B), frontal capping extending into the WM (circle right) and confluent DWMH (circle left), formed the groups of individuals with severe WM abnormality.
Neuropathologic data acquisition and analysis.
MR-imaged cerebrums were cut into 1-cm-thick coronal slices and examined for cavitated lesions or solid cerebral infarcts. The macroscopic infarcts were confirmed by subsequent histologic examination. Samples were also obtained from the left middle frontal, superior temporal, and middle temporal gyri, and inferior parietal lobule, according to the Consortium to Establish a Registry for Alzheimer's Disease (CERAD) protocol for the diagnosis of AD,18 to be cut at a thickness of 10 μm for staining with a modified Bielschowsky method for neuritic plaques and neurofibrillary tangles. The maximum density of neuritic plaques was scored according to the CERAD protocol.18 Tangles were counted as described previously.19 To estimate the amyloid-β protein load in the neocortex, the fraction of the cortex covered by methenamine-silver stained plaques was quantified as described previously15 on 8-μm-thick sections cut from the same CERAD blocks (see above).
Tissue blocks for staging of neurofibrillary changes and for detection of the argyrophilic grains were obtained and processed as described previously.14 Braak staging was performed as described previously.19,20 The Braak stage, CERAD score, amyloid burden, and tangle counts were examined independently of each other.
Samples from the right hippocampus and midbrain were embedded in paraffin and cut for the hematoxylin-eosin staining and α-synuclein (Transduction Laboratories, Lexington, KY) immunohistochemistry for the detection of hippocampal sclerosis and Lewy-related pathology.21 All the gross and microscopic examinations were performed blinded to the clinical data and postmortem MRI findings.
Description of neurodegenerative and vascular changes.
Each case (n = 132) was assigned to 1 of the 5 groups defined by the severity of AD or other type of primary neurodegenerative pathology (table 1). The normal aging group (NA; n = 15) included subjects who had Braak stage 0, I, or II, no or sparse neuritic plaques, according to the CERAD protocol, and no other primary neurodegenerative disease. Subjects in the borderline group (n = 34) did not fulfill criteria for NA, AD, or other primary neurodegenerative pathology. The AD group (n = 30) included cases that exhibited Braak stages IV, V, or VI together with a CERAD score of moderate or frequent. Additional AD-related features included neocortical tangle counts and neocortical amyloid-β protein load (table 1). The AD plus other group (n = 18) included cases that met pathologic criteria for both AD and other primary neurodegenerative diseases (table 1). This “other” group (n = 35) had cases defined as dementia with Lewy bodies (DLB, n = 6), DLB with argyrophilic grain disease (AGD) (n = 3), AGD with or without hippocampal sclerosis (HS) (n = 17), HS alone (n = 6), and types of tauopathy (n = 3) (table 1).
Table 1 Age, gender, disease severity, and frontal PVWMH and DWMH by MRI in different pathologic groups
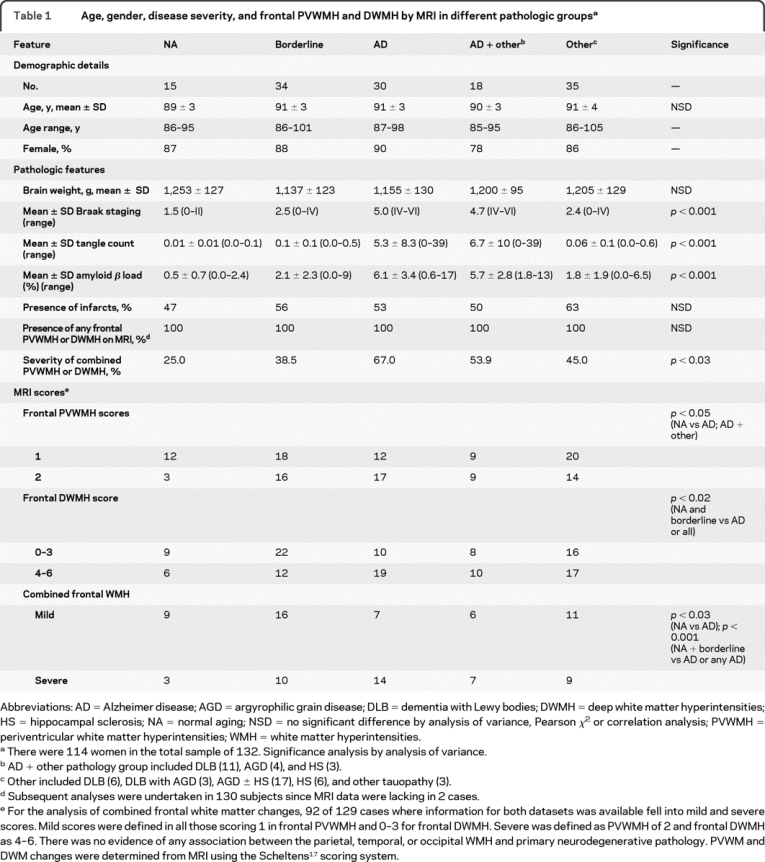
Lewy body–related pathology included subjects who had diffuse neocortical Lewy-related pathology.21 Diagnosis of AGD was based on the presence of a belt-like infiltrate of Gallyas-positive spindle- or comma-shaped neurophilic structures.16 HS was neuropathologically defined as a total or subtotal (few scattered neurons seen) loss of neurons in the CA1 region of the hippocampus and other tauopathy included cases with tangle only dementia and subjects who showed Gallyas-positive ring- or Pick body–like inclusions.16
The subjects were also classified according to the presence of the macroscopic infarcts, their anatomic location (cortex, WM, basal ganglia), size (widest dimension no more than 15 mm, over 15 mm), and number (less than 3, at least 3; table 1).
Statistical analyses.
Standard statistical methods involving nonparametric techniques and linear regression were appropriate for the data analysis (n ≥25). All statistical analysis was performed using the SPSS v.10 software (Chicago, IL). Pearson χ2 and Fisher exact tests, where applicable, were used to compare the proportion of individuals in various groups. Associations between numeric variables were determined using the Spearman rank correlation analysis (coefficient estimate r). Analysis of variance was used to decipher trends in variables and further scrutinized by post hoc tests. Comparisons between categories and numeric variables, e.g., age, measures of pathologic lesions, cortical Aβ, or NFT counts, were assessed by the Mann-Whitney U test. Significance was considered at a p value equal to or less than 0.05.
Each variable for Alzheimer type neuropathology, i.e., CERAD score, Braak stage, neocortical amyloid β load, and neocortical neurofibrillary tangle counts, was analyzed separately. The groups for individuals with different neocortical amyloid β load or neurofibrillary tangle counts were formed by using a rank order. Thus, the 3 groups formed represented those with the lowest amyloid-β load or neurofibrillary tangle counts, the highest amyloid β load or neurofibrillary tangle counts, and those in between. In some of the analyses, we combined the NA and borderline groups and considered them as one. The justification for this was that the NA and borderline groups did not meet any diagnostic criteria for the common primary neurodegenerative disorder.
RESULTS
Table 1 shows the distribution of 132 subjects within the main pathologic diagnostic groups. There were no apparent differences in the mean age or brain weights between the groups. Females were overrepresented (78%–90%) in each of this very old group. As expected, the frequency of clinically diagnosed dementia increased with severity of pathologic criteria which defined each group. The AD and the AD plus other groups exhibited the highest AD-type pathology burden (p = 0.000). There was no significant difference between the groups in the proportion of subjects with at least one macroscopic infarct (table 1).
Evaluation of the successful MRI indicated that some degree of PVWMH or DWMH was present in all (100%) subjects whereas moderate to severe (scale 4–6) frontal DWMH were detected in 64 (50%) of 129 subjects (table 1). Both frontal PVWMH and DWMH were more extensive in the AD group compared to the NA group or compared to the NA and borderline groups together. A similar result was evident when all those with primary neurodegenerative disorder were compared either to the NA group alone or to that combined with the borderline group. We also noted that the combined frontal WM changes were greater in those subjects in the 10th decade and older compared to younger subjects, i.e., aged <90 vs >91 years, irrespective of the presence of any other pathology (p = 0.002, Mann-Whitney U test). There was no evidence of any association between parietal PVWMH and parietal, temporal, and occipital DWMH, and AD pathology (p > 0.05). When we combined the scores for both PVWMH and DWMH and evaluated as mild and severe degrees (table 1), the association between frontal WM changes and AD was clearly evident. There was also a significant association between the NA and borderline groups vs AD and AD plus other pathology groups (table 1).
To delineate the possible association between WMH and AD-type pathology, in subsequent analyses we concentrated on the AD, borderline, and NA groups. Irrespective of this demarcation, all the changes were maintained when the AD + other pathology group were included together in the analysis. Severe frontal PVWMH and DWMH were significantly more common in those with moderate to frequent neuritic plaques than in subjects with lower density of cortical neuritic plaques. The Braak stage for neurofibrillary pathology was also significantly associated with the extent of both frontal PVWMH and DWMH (table 2). The proportion of individuals with extensive lesions increased as the Braak stage increased. However, again there was no relationship between WMH in any of the other lobes and the AD pathology (not shown).
Table 2 Relationship between frontal PVWMH and DWMH and severity of Alzheimer type of pathology
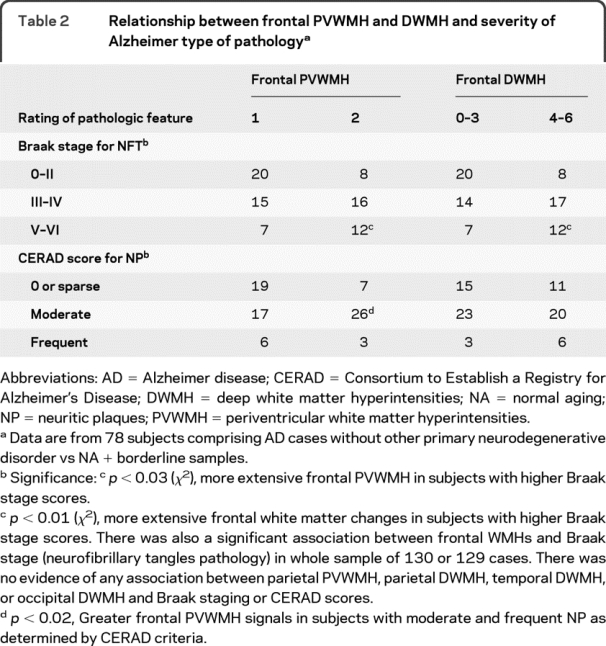
We found no evidence for any association between the frontal WMH and neocortical numerical count for amyloid β deposition or load (tables 1 and 2). There was, however, a weak trend apparent with frontal PVWMH (p = 0.05): the proportion of individuals with extensive PVWMH increased as the amount of cortical amyloid β protein increased (not shown). Not surprisingly, each of the variables for neurofibrillary pathology and amyloid plaque burden for the whole oldest old sample was related to each other (data not shown).
We further tested the whole sample as well as the AD vs the NA + borderline groups for an association between severity of AD type of pathology and presence or absence, location, size, or number of infarcts (figure 2). Uneven numbers of macroscopic infarcts were found in the cortex, WM, and basal ganglia in 56% of the 132 subjects. Smaller infarcts (<15 mm) were the most common type (67%) among cases with AD-type and AD+ other pathology (table e-1 on the Neurology® Web site at www.neurology.org). When the distribution of infarcts was analyzed by location against the pathologic groups, cortical infarcts rather than those in WM or basal ganglia were greater in the borderline, AD, and other pathology groups (p < 0.006). The presence of infarcts was associated with moderate Braak scores (p < 0.02) but not with more severe neurofibrillary pathology, neuritic plaques, or amyloid load (figure 2). Figure 3 shows that there were no relationships between the number of macroscopic cerebral infarcts and frontal PVWMH but more profoundly there were greater numbers of cerebral infarcts in parallel with frontal DWMH (p = 0.03). However, in those cases without macroscopic infarcts, there was a relationship between AD and severity of frontal PVWMH as opposed to DWMH (table e-2). Infarcts in the WM and basal ganglia did not reveal any relationships with any of the pathologic features.
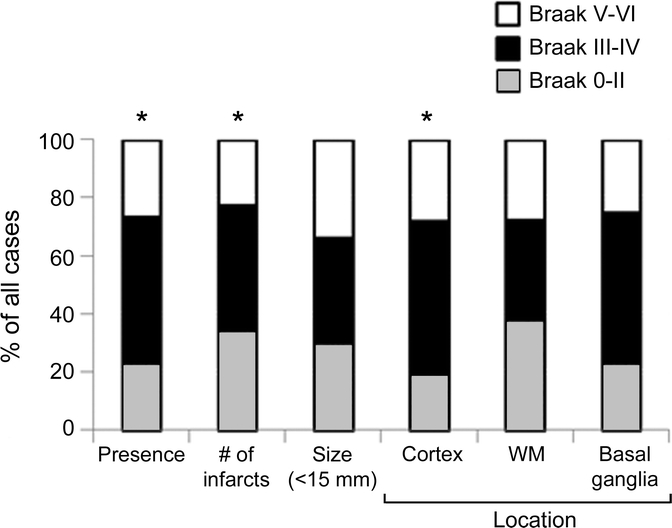
Figure 2 Relationship between Braak neurofibrillary tangles staging and presence, number, size, and location of cerebral infarcts in 85+-year-olds
Histograms show % cases with distribution of Braak staging by size and location of infarcts. Data are from 78 subjects comprising Alzheimer disease (AD) cases without other primary neurodegenerative disorder vs non-AD + borderline samples. *Significance: p < 0.03 (χ2), presence of vascular lesion or infarcts and moderate Braak scores. DWMH = deep white matter hyperintensities; PVWMH = periventricular white matter hyperintensities.
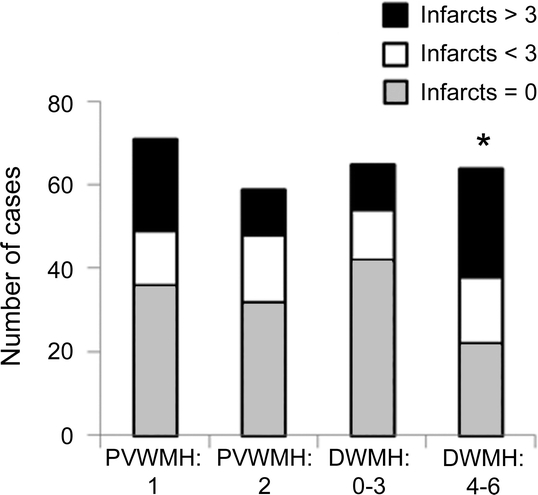
Figure 3 Histograms showing distribution of numbers of infarcts in cases with periventricular white matter hyperintensities (PVWMH) and frontal deep white matter hyperintensities (DWMH)
Data are from 132 subjects comprising Alzheimer disease (AD) cases without other primary neurodegenerative disorder vs NA + borderline samples. *Significance: p < 0.03 (χ2), association of frontal DWMH with infarcts. There was also greater severity of frontal PVWMH signals in subjects with AD who had exhibited no macroinfarcts (p < 0.02; not shown). WM = white matter.
DISCUSSION
Few studies have focused on dissecting the pathologic changes in the oldest or very old (>85 years) within unselected community-dwelling individuals.10 In this study, as in others,22 the frontal lobe revealed the widest interindividual variation in the WM abnormalities. Despite the high age of all these individuals, the burden of frontal WM changes still correlated independently with age. Our results indicate a link between the frontal, but not temporal, parietal, or occipital, WM abnormalities and the presence of neurofibrillary pathology in the oldest old. We observed an association between severity of the frontal PVWM and DWM changes detected by MRI and high Braak stage scores. These results accord with our previous in vivo MRI observations.23,24 The presence and the progression of WM changes are associated with progression of medial temporal atrophy in AD which is also pathologically characterized by a high burden of neurofibrillary tangles.
We did not find an association between the frontal WM changes upon MR and actual neocortical tangle counts. Such a discrepancy between individual counts and the Braak stages may not be surprising, because occasional neocortical tangles can be seen in almost any Braak stage less than V, and thus, a low tangle count does not predict the Braak stage. In addition, the relationship between the Braak stages and WMH was linear, indicating that the WM changes may develop independently or before the neocortical neurofibrillary pathology. There was some evidence that other neocortical tau pathology, the density of the neuritic plaques, is associated with the size of the periventricular WM hyperintensities.
How might the association between frontal WM lesions and neurofibrillary pathology be explained? We offer 3 possible scenarios. First, it relates to the vascular origin of AD-related degenerative changes. It is unlikely that an occlusive disease in large or middle-sized arteries supplying the brain would explain the relationship because macroscopic infarcts did not show linear association with the AD-type pathology. On the other hand, because the extent of cerebral amyloid angiopathy and the presence of microinfarcts were not analyzed, the possibility that pathology within the very small blood vessels of brain presumably in WM would have provided the link between the neurofibrillary pathology and WM changes cannot be excluded. It is already known that WM attenuation increases with older age25 and is frequently observed in neurodegenerative disorders such as AD26 and in clinically diagnosed DLB, though to a lesser extent than in VaD.27,28 Recent diffusion tensor imaging (DTI) studies suggest that the WM microstructure disintegrates with age in particular fiber populations within the prefrontal region and internal capsule.29,30 WM changes frequently though not always coincide with cerebrovascular risk factors such as hypertension, atherosclerosis, and diabetes, and are evident in VaD. The vascular basis of WM change involves arteriosclerosis, microinfarction, perivascular spacing (état criblé), along with myelin loss and axonal swelling.31 Disturbances in CSF production, disruption in blood–brain barrier permeability to macromolecules, and cerebral edema32,33 have been cited as important causes in the development of WM changes. There may also be age-related functional deficits in branches of the perforating arteries.
Furthermore, we previously showed that the lesional or myelin-depleted deep WM is in a chronic hypoxic state as indicated by the hypoxia-related messengers.34 This occurred without relevance to the primary degenerative pathology in the neocortex.26,34 On the other hand, microarrays suggest that WM lesions represent areas with a complex molecular phenotype35; multiple pathways are activated indicating WM changes arise through tissue ischemia but may also reflect the contribution of other factors like blood–brain barrier dysfunction. Thus, it is conceivable that the WM abnormalities instigate the neurofibrillary pathology in the oldest old brains from the Vantaa 85+ cohort involving a small vessel disease component.
In the second scenario, could it be that there is direct interaction between cortical infarcts and neurofibrillary pathology in association cortices and WM changes are coincidental to this? Our results showed that macroscopic cerebral infarcts were present in approximately half of the subjects in each diagnostic group, the cortex being the most common location. This is consistent with the high degree of mixed neurodegenerative and vascular pathologies in community-dwelling elderly subjects.10 We did not detect a relationship between the types or the size of cerebral infarcts and AD-type pathology but there was a clear association between the presence of infarcts and moderate neurofibrillary pathology as assessed by Braak staging in AD cases. Other studies have emphasized that a significant association exists between cortical watershed microinfarcts and AD (32% cases vs 3% controls).31,36 While this association may be related to the known association between cerebral amyloid angiopathy and AD, it is possible that small cortical infarcts affect local neuronal circuits to induce retrograde effects and neurofibrillary pathology in association cortices.
The third scenario entails that WM changes result directly from cortical neurodegeneration and reflect prior gray matter loss. Thus there is the possibility that WM changes reflected on MR are caused by pathologic processes originating within neurons or the neural networks. One simple explanation could be the death or degeneration of neurons, particularly within the limbic region, results in decreased axonal density with loss of myelin in the frontal WM. This is consistent with the DTI-based MR studies suggesting that WM microstructural damage accompanies diffusivity changes and tissue loss within cortical and subcortical gray matter in old age and in different neurodegenerative dementias in the absence of infarcts or cerebrovascular disease.30,37,38
There was also no apparent relationship between the frontal WM changes and the actual burden of amyloid plaques or the neocortical amyloid-β load per area or even the Thal staging39 for amyloid (R. Kalaria, unpublished data). A previous study40 indicated that the frontal astrogliosis and amount of axonal APP, but not intensity of the myelin staining in patients with AD, are closely related to parenchymal amyloid. Therefore, hypothetically, the WM changes detected on MRI may be more related to the presence or absence of myelin than the other markers of WM damage. On the other hand, our study was based on an unselected population, not only on patients with AD.10
ACKNOWLEDGMENT
The authors thank the patients and families for their cooperation in the investigation of this study.
DISCLOSURE
Dr. Polvikoski and Dr. van Straaten report no disclosures. Dr. Barkhof serves on scientific advisory boards for Lundbeck Inc., Bayer Schering Pharma, Sanofi-Aventis, UCB, Novartis, Biogen Idec, BioMS Medical, and Merck Serono; serves on the editorial boards of Brain, the Journal of Neurology, Neurosurgery, and Psychiatry, European Radiology, the Journal of Neurology, and Neuroradiology; has received speaker honoraria from Novartis and Merck Serono; serves as a consultant for Sanofi-Aventis, UCB, Novartis, Biogen Idec, BioMS Medical, and Medicinova, Inc.; and receives research support from the Dutch MS Research Foundation. Dr. Sulkava has received speaker honoraria from Novartis and has received research support from the Academy of Finland. Dr. Aronen, Dr. Niinistö, and Dr. Oinas report no disclosures. Dr. Scheltens serves on scientific advisory boards for Danone, Wyeth/Elan Corporation, and Bristol-Myers Squibb; has received funding for travel from Lundbeck, Inc.; served as an Associate Editor of the Journal of Neurology, Neurosurgery & Psychiatry; serves a Book Review Editor for Alzheimer's Disease and Associated Disorders and on the editorial board of Dementia Geriatric Cognitive Disorders; has received speaker honoraria from Lundbeck Inc. and Danone; and receives research support from Alzheimer Nederland, the Alzheimer Center, and Stichting VUmc fonds. Dr. Erkinjuntti serves on scientific advisory boards for Johnson & Johnson, Pfizer Inc., and SERVIER; and has served on speakers' bureaus for and received speaker honoraria from Janssen and Shire plc. Dr. Kalaria has served on a scientific advisory board for the Alzheimer's Research Trust UK; has received funding for travel from the Marabou Foundation, Sweden; serves on editorial advisory boards for Alzheimer's Disease and Associated Disorders, European Neurology, NeuroReport, and Behavioral and Brain Functions; and has received speaker honoraria from Pfizer Inc.
Address correspondence and reprint requests to Prof. Raj N. Kalaria, Institute for Ageing and Health, Wolfson Research Centre (Neuropathology), Campus for Ageing & Vitality, Newcastle upon Tyne, NE4 5PL, UK r.n.kalaria@ncl.ac.uk
Supplemental data at www.neurology.org
e-Pub ahead of print on November 3, 2010, at www.neurology.org.
Study funding: Work in Newcastle is supported by the Medical Research Council (UK), the Alzheimer's Research Trust (UK), and the National Institutes of Health (NINDS).
Disclosure: Author disclosures are provided at the end of the article.
Received July 19, 2009. Accepted in final form August 4, 2010.
REFERENCES
- 1.Viswanathan A, Rocca WA, Tzourio C. Vascular risk factors and dementia: how to move forward? Neurology 2009;72:368–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Skoog I, Lernfelt B, Landahl S, et al. 15-year longitudinal study of blood pressure and dementia. Lancet 1996;347:1141–1145. [DOI] [PubMed] [Google Scholar]
- 3.Luchsinger J, Mayeux R. Cardiovascular risk factors and Alzheimer's disease. Curr Atheroscler Rep 2004;6:261–266. [DOI] [PubMed] [Google Scholar]
- 4.Tan ZS, Seshadri S, Beiser A, et al. Plasma total cholesterol level as a risk factor for Alzheimer disease: the Framingham Study. Arch Intern Med 2003;163:1053–1057. [DOI] [PubMed] [Google Scholar]
- 5.Biessels GJ, Staekenborg S, Brunner E, Brayne C, Scheltens P. Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol 2006;5:64–74. [DOI] [PubMed] [Google Scholar]
- 6.Pi-Sunyer FX. The obesity epidemic: pathophysiology and consequences of obesity. Obes Res 2002;10(suppl 2):97S–104S. [DOI] [PubMed] [Google Scholar]
- 7.Kalaria RN. The role of cerebral ischemia in Alzheimer's disease. Neurobiol Aging 2000;21:321–330. [DOI] [PubMed] [Google Scholar]
- 8.Lewis H, Beher D, Cookson N, et al. Quantification of Alzheimer pathology in ageing and dementia: age-related accumulation of amyloid-beta(42) peptide in vascular dementia. Neuropathol Appl Neurobiol 2006;32:103–118. [DOI] [PubMed] [Google Scholar]
- 9.Nolan KA, Lino MM, Seligmann AW, Blass JP. Absence of vascular dementia in an autopsy series from a dementia clinic. J Am Geriatr Soc 1998;46:597–604. [DOI] [PubMed] [Google Scholar]
- 10.Pathological correlates of late-onset dementia in a multicentre, community-based population in England and Wales. Lancet 2001;357:169–175. [DOI] [PubMed] [Google Scholar]
- 11.Roman GC, Royall DR. A diagnostic dilemma: is “Alzheimer's dementia” Alzheimer's disease, vascular dementia, or both? Lancet Neurol 2004;3:141. [DOI] [PubMed] [Google Scholar]
- 12.Bacchetta JP, Kovari E, Merlo M, et al. Validation of clinical criteria for possible vascular dementia in the oldest-old. Neurobiol Aging 2007;28:579–585. [DOI] [PubMed] [Google Scholar]
- 13.Delaere P, He Y, Fayet G, Duyckaerts C, Hauw JJ. Beta A4 deposits are constant in the brain of the oldest old: an immunocytochemical study of 20 French centenarians. Neurobiol Aging 1993;14:191–194. [DOI] [PubMed] [Google Scholar]
- 14.Polvikoski T, Sulkava R, Myllykangas L, et al. Prevalence of Alzheimer's disease in very elderly people: a prospective neuropathological study. Neurology 2001;56:1690–1696. [DOI] [PubMed] [Google Scholar]
- 15.Polvikoski T, Sulkava R, Haltia M, et al. Apolipoprotein E, dementia, and cortical deposition of beta-amyloid protein. N Engl J Med 1995;333:1242–1247. [DOI] [PubMed] [Google Scholar]
- 16.Barkhof F, Polvikoski TM, van Straaten EC, et al. The significance of medial temporal lobe atrophy: a postmortem MRI study in the very old. Neurology 2007;69:1521–1527. [DOI] [PubMed] [Google Scholar]
- 17.Scheltens P, Barkhof F, Leys D, et al. A semiquantitative rating scale for the assessment of signal hyperintensities on magnetic resonance imaging. J Neurol Sci 1993;114:7–12. [DOI] [PubMed] [Google Scholar]
- 18.Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD): part II: standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 1991;41:479–486. [DOI] [PubMed] [Google Scholar]
- 19.Myllykangas L, Polvikoski T, Sulkava R, et al. Genetic association of alpha2-macroglobulin with Alzheimer's disease in a Finnish elderly population. Ann Neurol 1999;46:382–390. [PubMed] [Google Scholar]
- 20.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991;82:239–259. [DOI] [PubMed] [Google Scholar]
- 21.McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 2005;65:1863–1872. [DOI] [PubMed] [Google Scholar]
- 22.Capizzano AA, Acion L, Bekinschtein T, et al. White matter hyperintensities are significantly associated with cortical atrophy in Alzheimer's disease. J Neurol Neurosurg Psychiatry 2004;75:822–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Leeuw FE, Barkhof F, Scheltens P. White matter lesions and hippocampal atrophy in Alzheimer's disease. Neurology 2004;62:310–312. [DOI] [PubMed] [Google Scholar]
- 24.de Leeuw FE, Korf E, Barkhof F, Scheltens P. White matter lesions are associated with progression of medial temporal lobe atrophy in Alzheimer disease. Stroke 2006;37:2248–2252. [DOI] [PubMed] [Google Scholar]
- 25.Firbank MJ, Minett T, O'Brien JT. Changes in DWI and MRS associated with white matter hyperintensities in elderly subjects. Neurology 2003;61:950–954. [DOI] [PubMed] [Google Scholar]
- 26.Ihara M, Polvikoski TM, Hall R, et al. Quantification of myelin loss in frontal lobe white matter in vascular dementia, Alzheimer's disease, and dementia with Lewy bodies Acta Neuropathol 2010;119:579–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barber R, Scheltens P, Gholkar A, et al. White matter lesions on magnetic resonance imaging in dementia with Lewy bodies, Alzheimer's disease, vascular dementia, and normal aging. J Neurol Neurosurg Psychiatry 1999;67:66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Erkinjuntti T, Benavente O, Eliasziw M, et al. Diffuse vacuolization (spongiosis) and arteriolosclerosis in the frontal white matter occurs in vascular dementia. Arch Neurol 1996;53:325–332. [DOI] [PubMed] [Google Scholar]
- 29.Bartzokis G, Sultzer D, Lu PH, Nuechterlein KH, Mintz J, Cummings JL. Heterogeneous age-related breakdown of white matter structural integrity: implications for cortical “disconnection” in aging and Alzheimer's disease. Neurobiol Aging 2004;25:843–851. [DOI] [PubMed] [Google Scholar]
- 30.Salat DH, Tuch DS, Greve DN, et al. Age-related alterations in white matter microstructure measured by diffusion tensor imaging. Neurobiol Aging 2005;26:1215–1227. [DOI] [PubMed] [Google Scholar]
- 31.Kalaria RN, Kenny RA, Ballard CG, Perry R, Ince P, Polvikoski T. Towards defining the neuropathological substrates of vascular dementia. J Neurol Sci 2004;226:75–80. [DOI] [PubMed] [Google Scholar]
- 32.Jellinger KA. The enigma of vascular cognitive disorder and vascular dementia. Acta Neuropathol 2007;113:349–388. [DOI] [PubMed] [Google Scholar]
- 33.Pantoni L, Garcia JH. Pathogenesis of leukoaraiosis: a review. Stroke 1997;28:652–659. [DOI] [PubMed] [Google Scholar]
- 34.Fernando MS, Simpson JE, Matthews F, et al. White matter lesions in an unselected cohort of the elderly: molecular pathology suggests origin from chronic hypoperfusion injury. Stroke 2006;37:1391–1398. [DOI] [PubMed] [Google Scholar]
- 35.Simpson JE, Hosny O, Wharton SB, et al. Microarray RNA expression analysis of cerebral white matter lesions reveals changes in multiple functional pathways. Stroke 2009;40:369–375. [DOI] [PubMed] [Google Scholar]
- 36.Suter OC, Sunthorn T, Kraftsik R, et al. Cerebral hypoperfusion generates cortical watershed microinfarcts in Alzheimer disease. Stroke 2002;33:1986–1992. [DOI] [PubMed] [Google Scholar]
- 37.Giorgio A, Santelli L, Tomassini V, et al. Age-related changes in grey and white matter structure throughout adulthood. Neuroimage 2010;51:943–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kantarci K, Avula R, Senjem ML, et al. Dementia with Lewy bodies and Alzheimer disease: neurodegenerative patterns characterized by DTI. Neurology 2010;74:1814–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thal DR, Rub U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 2002;58:1791–1800. [DOI] [PubMed] [Google Scholar]
- 40.Chalmers K, Wilcock G, Love S. Contributors to white matter damage in the frontal lobe in Alzheimer's disease. Neuropathol Appl Neurobiol 2005;31:623–631. [DOI] [PubMed] [Google Scholar]