

Abstract
Objective:
To asses the presence of cortical demyelination in brains of patients with neuromyelitis optica (NMO). NMO is an autoimmune inflammatory demyelinating disease that specifically targets aquaporin-4-rich regions of the CNS. Since aquaporin-4 is highly expressed in normal cortex, we anticipated that cortical demyelination may occur in NMO.
Methods:
This is a cross-sectional neuropathologic study performed on archival forebrain and cerebellar tissue sections from 19 autopsied patients with a clinically and/or pathologically confirmed NMO spectrum disorder.
Results:
Detailed immunohistochemical analyses of 19 archival NMO cases revealed preservation of aquaporin-4 in a normal distribution within cerebral and cerebellar cortices, and no evidence of cortical demyelination.
Conclusions:
This study provides a plausible explanation for the absence of a secondary progressive clinical course in NMO and shows that cognitive and cortical neuroimaging abnormalities previously reported in NMO cannot be attributed to cortical demyelination. Lack of cortical demyelination is another characteristic that further distinguishes NMO from MS.
GLOSSARY
- AQP4
= aquaporin-4;
- BBB
= blood-brain barrier;
- IgG
= immunoglobulin G;
- MS
= multiple sclerosis;
- NMO
= neuromyelitis optica;
- NMOSD
= neuromyelitis optica spectrum disorder.
Neuromyelitis optica (NMO) is an autoimmune inflammatory demyelinating relapsing disease of the CNS in which a disease-specific circulating autoantibody (NMO-immunoglobulin G [IgG]) binds to the aquaporin-4 (AQP4) water channel that is concentrated in the astrocytic foot processes.1–3 Increasing evidence supports a primary pathogenic role for this IgG.4–6 Clinical, laboratory, neuroimaging, and pathologic features distinguish NMO from classic multiple sclerosis (MS).7,8
Cognitive deficits and neuroimaging abnormalities have been described in normal-appearing gray matter in NMO.9,10 Since AQP4 is heavily expressed in normal human cortex,7,11 we anticipated finding cortical pathology in NMO. This study describes our findings from a systematic examination of NMO patients' brains for evidence of cortical demyelination.
METHODS
This cross-sectional neuropathologic study was performed on archival forebrain and cerebellar tissue obtained from 19 autopsied patients with a clinically and/or pathologically confirmed NMO spectrum disorder (NMOSD)8: NMO (n = 16) or NMOSD (n = 3; 2 relapsing longitudinally extensive transverse myelitis, 1 relapsing optic neuritis). Autopsies were performed at Mayo Clinic, Rochester, MN, or sent in from other institutions for diagnostic purposes between 1958 and 2009. The study was approved by the Institutional Review Board of Mayo Clinic, Rochester, MN. Clinical follow-up was obtained via review of medical records. Descriptive statistics were used to characterize the population demographics and number of blocks analyzed (table). Median age was 45 years at symptom onset (range 12-80) and 50 years at death (range 16-80). Median disease duration was 36 months (range 8-180). NMO-IgG serostatus was known in 6 cases (positive in 5, including all 3 NMOSD cases). Causes of death were respiratory-related (n = 14), perforated ulcer (n = 1), subarachnoid hemorrhage (n = 1), or unknown (n = 3) (table). We determined the spectrum of cortical demyelinated plaque types according to established criteria.12,13 Plaques were classified as follows: 1) leukocortical demyelinated lesions involving cortical gray matter and white matter at the gray-white junction with sparing of superficial cortical layers; 2) intracortical lesions which were confluent or perivenous demyelinated lesions confined to the cortical gray matter with sparing of superficial cortical and subcortical U fiber layers; or 3) subpial lesions extending variable distances from the cortical pial surface with or without involvement of the underlying white matter. We analyzed 82 tissue blocks containing cerebral or cerebellar cortex for evidence of cortical demyelination, and excluded 6 blocks from 3 NMO cases which contained confounding pathologic changes consistent with ischemic infarcts. We analyzed a median number of 3 blocks per case (range 1-12) from the 76 remaining tissue blocks representing all 19 cases (table).
Table Patient demographics and number and location of cortical blocks analyzed
Specimens were fixed in 10%-15% formalin and embedded in paraffin. Sections were stained with hematoxylin-eosin to demonstrate tissue and cell morphology. Immunohistochemistry was performed without modification using an avidin-biotin or an alkaline phosphatase/antialkaline phosphatase technique as previously described.7,14 Sections were incubated with primary antibodies overnight at 4°C. We used primary antibodies specific for myelin proteolipid protein (PLP, polyclonal; Serotec, Oxford, USA) and AQP4 (C-terminal residues 249-323, affinity purified rabbit polyclonal IgG; Sigma-Aldrich), and as controls, omitted primary antibodies.7
RESULTS
Detailed analysis of the neocortex from all forebrain lobes, archicortex (hippocampus), mesocortex (parahippocampus, cingulate, and insula), and cerebellar cortex revealed no subpial, intracortical, or leukocortical lesions. Myelin was preserved in all cortical layers (figure 1, A, B, E, F, I, and J), in the dentate gyrus and all fields of Ammon's horn (figure 2, A, B, D, F, and H), and in the molecular, infraganglionic, and supraganglionic layers of the cerebellum (n = 9 cerebellar blocks from 9 NMO cases) (figure 2, J, L, and N).

Figure 1 Absence of cortical demyelination and preservation of aquaporin-4 (AQP4) in cerebral cortex of patients with neuromyelitis optica (NMO)
Cortical demyelination is absent and AQP4 is preserved in a normal distribution in the frontal (A-D), insular (E-G), and temporal (I-L) cortices of NMO brains. Cortical areas within rectangles in (A), (E), and (I) are shown at higher magnification in (B), (F), and (J), respectively. Insets (D) and (L) show intense AQP4 immunoreactivity concentrated in the end-feet of protoplasmic astrocytes abutting the blood vessels. Inset (H) shows intense AQP4 immunoreactivity in glia limitans. (A, B, E, F, I, J) Proteolipid protein. (C, D, G, H, K, L) AQP4. (A, I) Scale bar = 1.5 mm. (E) Scale bar = 1 mm. (B, C, F, G, J, K) Scale bar = 500 μm. (D, H, L) Scale bar = 25 μm.

Figure 2 Absence of cortical demyelination and preservation of aquaporin-4 (AQP4) in hippocampus and cerebellar cortex of patients with neuromyelitis optica (NMO)
Cortical demyelination is absent and AQP4 is preserved in a normal distribution in the hippocampus (A-I) and cerebellar cortex of patients with NMO (J-O). Dentate gyrus (B-C), CA4 (D-E), CA3-2 (F-G), and CA1 (H-I) show preservation of myelin (B, D, F, H) and AQP4 (C, E, G, I). The myelin is preserved in the molecular, infraganglionic, and supraganglionic layers of the cerebellum13 (J, L, N) and AQP4 immunoreactivity is most intense in Bergmann glia and astrocytes of the granule cell layer (K, M, O). (A, B, D, F, H, J, L, N) Proteolipid protein. (C, E, G, I, K, M, O) AQP4. (A) Scale bar = 1 mm. (B-I, L, M) Scale bar = 250 μm. (J, K) Scale bar = 500 μm. (N, O) Scale bar = 100 μm. DG = dentate gyrus; CA4 = field 4 of cornu Ammonis; CA3-2 = fields 3 and 2 of cornu Ammonis; CA1 = field 1 of cornu Ammonis.
The cortical distribution of AQP4 as previously described for normal tissues7,11 was preserved in NMO (figure 1, C, G, and J, and figure 2, C, E, G, I, K, M, and O), with the most intense AQP4 immunoreactivity concentrated in the end-feet of protoplasmic astrocytes abutting the blood vessels (figure 1, D and L) at the glia limitans in the cerebral cortex (figure 1H), and in Bergmann glia and astrocytes of the granule cell layer in the cerebellar cortex (figure 2, K, M, and O). Additional cortical neuropathologic findings observed in NMO included astrogliosis (figure 3, B, D, E, and G) mostly involving interlaminar astrocytes (figure 3, B and D) and scattered red and/or pyknotic neurons (figure 3, C, E, and G) within the cerebral but not cerebellar cortex. Myelin and AQP4 immunoreactivity was normal appearing even in cortical regions with profound astrogliosis (figure 3, F and H).
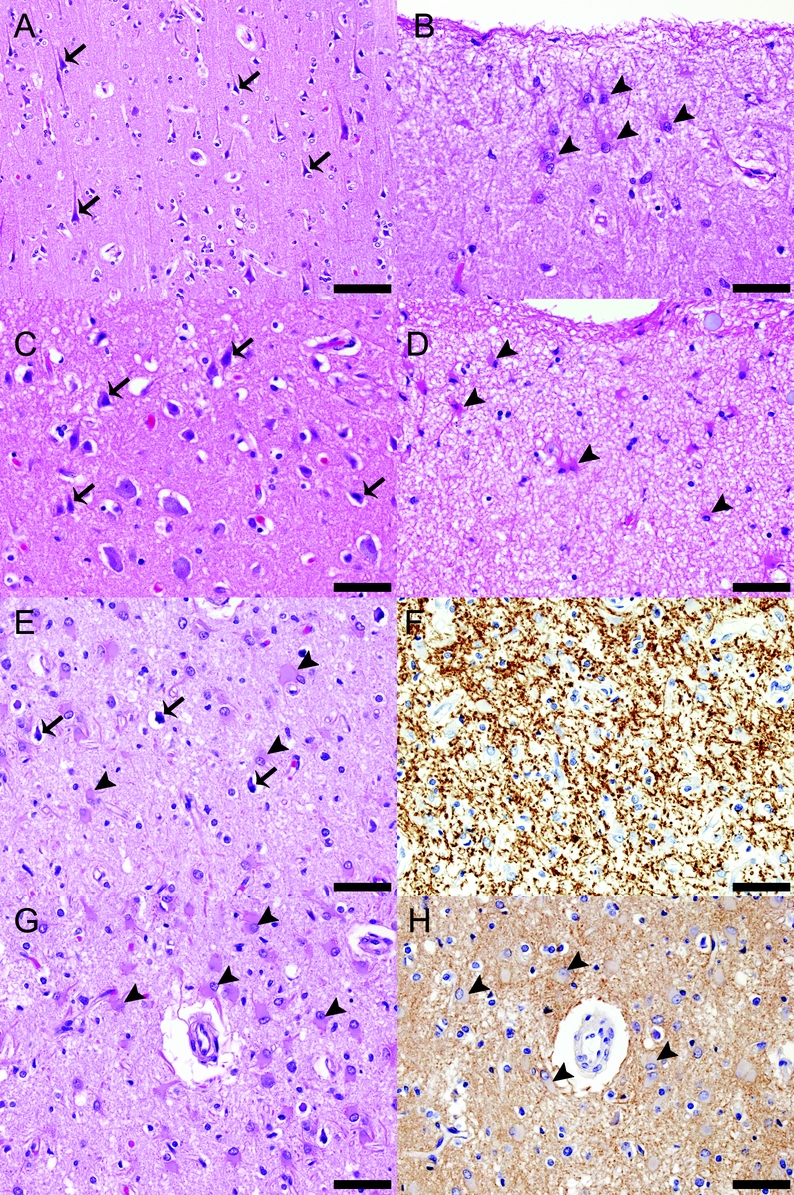
Figure 3 Additional neuropathologic findings in cortices of patients with neuromyelitis optica (NMO)
Pathologic findings in the cortex (illustrated in this figure for 3 individual patients with NMO) included astrogliosis and scattered focal red and/or pyknotic neurons. Cortices from patients 1 (A, B) and 2 (C, D) show numerous red neurons (arrows in [A] and [C], respectively) and astrocytosis involving the interlaminar astrocytes (arrowheads in [B] and [D], respectively). The cortex from patient 3 (E-H) shows pyknotic neurons (arrows in [E]) on a background of profound astrogliosis (arrowheads in [E]) in all layers of the cortex (E, G). Despite these changes, the myelin (F) and aquaporin-4 (AQP4) (H) are preserved. (A-E, G) Hematoxylin-eosin. (F) Proteolipid protein. (H) AQP4. (A) Scale bar = 100 μm. (B-H) Scale bar = 50 μm.
DISCUSSION
Although brain lesions in NMO have been described in AQP4-enriched periventricular white and deep gray matter structures, this is the first study to show the absence of cortical demyelination in NMO, despite AQP4 being heavily concentrated in this region.8,15,16 NMO is caused by a disease-specific serum autoantibody (NMO-IgG) that specifically targets the AQP4 water channel in the astrocytic foot processes.2,3,7 The preservation of AQP4 and myelin in cortical regions may relate to regional differences in blood-brain barrier (BBB) permeability. Functional and structural differences between white and cortical gray matter BBB have been described.17,18 Perivascular astrocytes arranged in rows, cylinder-like segmented, thick and abundant perivascular astrocytic processes, and larger perivascular spaces are characteristic for the white matter and suggest that a single astrocyte has more contact area with the vessel wall, while the absence of perivascular glial rows, narrow perivascular spaces, and thin, sheet-like astrocytic processes that encapsulate blood vessels are characteristic for the cortical gray matter.17,18 These differences are thought to account for selective white matter permeability in brain edema,18 HIV-1 encephalitis,19 and following administration of IL-2,20 although the exact mechanism is unknown. Alternatively, perhaps a differential expression of M1 and M23 isoforms of AQP4 could render the cortex less susceptible to NMO-IgG attack.21
Particularly remarkable is the preservation of myelin in cortical regions that are susceptible to demyelination in MS, namely the cingulate, insular, frontal, and temporal cortices.22,23 Cortical demyelination in chronic MS is extensive. In the progressive stage of the disease, demyelination characteristically affects up to 70% of the cerebral and 90% of the cerebellar cortex.13,24 Despite more severe axonal injury in NMO than MS, paradoxically a secondary progressive clinical course is uncommon in NMO.25 This may relate to the absence of cortical demyelination. The findings we report here further pathogenically distinguish NMO from MS and reinforce the hypothesis that cortical demyelination is an important contributor to progressive neurologic disability in MS.24 Cortical demyelination in MS accumulates with disease duration. Limited cortical demyelination has been reported in acute and early relapsing cases of MS. Nevertheless, preservation of cortical myelin in NMO is unlikely simply due to sampling bias among severe acute fulminant cases, given that 3 of our NMO cases had a disease duration greater than 8 years (table). While one of these cases (no. 15 in the table, 180 months disease duration) had limited sampling (3 blocks), the other 2 (no. 12, 108 months, and no. 13, 96 months disease duration) had extensive sampling (7 and 12 blocks, respectively) (table).
Cognitive deficits have been reported in both MS23 and NMO.9 Neuroimaging and neuropathologic studies suggest brain atrophy26 and cortical demyelination23 as substrates for the cognitive decline observed in patients with MS. However, unlike patients with MS, patients with NMO reportedly do not develop neuroimaging evidence of brain atrophy.10 These neuroimaging findings are compatible with our pathologic observations reporting an absence of cortical demyelinating lesions in NMO brains.10 Furthermore, the reduced magnetization transfer and increased mean diffusivity described in normal-appearing gray matter of NMO brains10 cannot be explained by cortical demyelination. Nevertheless, nonconventional MRI studies suggest that cortical gray matter is damaged in NMO.10 Our findings of prominent astrogliosis and neuronal pathology in NMO cerebral cortex may be the pathologic correlates of these cortical imaging abnormalities. The lack of pyknotic neurons in NMO cerebellar cortex argues against these cortical changes being due to terminal hypoxic events. Although retrograde degeneration of neurons secondary to lesions in the spinal cord, optic nerves, or white matter may contribute to cortical damage, it is pertinent that we did not observe the ballooned or chromatolytic neurons that would be anticipated in a gross retrograde somatic reaction to neuritic injury.
Our finding that AQP4 and myelin are preserved in the cerebral and cerebellar cortices provides additional pathologic evidence that different pathogenic mechanisms are responsible for brain damage in NMO and MS. Understanding why the cortex is preferentially spared in NMO despite abundant cortical expression of the target antigen, AQP4, is a challenging question for understanding the disease pathogenesis and may lead to new therapeutic approaches aimed at relapse prevention.
ACKNOWLEDGMENT
The authors thank Patricia Ziemer, Mayo Clinic, for technical assistance. The Thomas Willis Oxford Brain Collection provided 3 of the cases described in this report.
DISCLOSURE
Dr. Popescu reports no disclosures. Dr. Parisi serves on scientific advisory boards for the US Government Defense Health Board and the Subcommittee for Laboratory Services and Pathology; serves as a Section Editor for Neurology; receives royalties from the publication of Principles & Practice of Neuropathology, 2nd ed. (Oxford University Press, 2003); and receives research support from the NIH (NS32352-13 [coinvestigator]). Dr. Cabrera-Gómez reports no disclosures. Dr. Newell receives/has received research support from the NIH (U01 AG031106 [coinvestigator] and R21 AG027419 [coinvestigator]). Dr. Mandler reports no disclosures. Dr. Pittock may accrue revenue for patents re: Aquaporin-4 associated antibodies for diagnosis of neuromyelitis optica and aquaporin-4 autoantibody as a cancer marker; and has received research support from Alexion Pharmaceuticals, Inc. and the Guthy Jackson Charitable Foundation. Dr. Lennon is a named investor on a patent application filed by the Mayo Foundation for Medical Education and Research that relates to the NMO antigen and its application to the diagnosis of NMO; may accrue revenue for a patent re: Aquaporin-4 associated antibodies for diagnosis of neuromyelitis optica; receives research support from the NIH (R01 DK71209-05 [PI]) and the Guthy Jackson Charitable Foundation. Dr. Weinshenker serves on data safety monitoring boards for Novartis and Biogen Idec; serves on the editorial boards of the Canadian Journal of Neurological Sciences and the Turkish Journal of Neurology; has received research support from Genzyme Corporation and the Guthy-Jackson Charitable Foundation; and receives license royalties from RSR Ltd. for a patent re: Aquaporin-4 associated antibodies for diagnosis of neuromyelitis optica. Dr. Lucchinetti may accrue revenue for a patent re: Aquaporin-4 associated antibodies for diagnosis of neuromyelitis optica; receives royalties from the publication of Blue Books of Neurology: Multiple Sclerosis 3 (Saunders Elsevier, 2010); and receives research support from the NIH (NS49577-R01 [PI]), the Guthy Jackson Charitable Foundation (PI), and the National MS Society (RG 3185-B-3 [PI]).
Address correspondence and reprint requests to Dr. Claudia F. Lucchinetti, Neurology, Mayo Clinic, College of Medicine, 200 First St. SW, Rochester, MN 55905 lucchinetti.claudia@mayo.edu
Study funding: Supported by the NIH (RO1-NS049577-01-A2 to C.F.L.), the National Multiple Sclerosis Society (RG 3185-B-3 to C.F.L.), and the Guthy Jackson Foundation (to C.F.L.).
Disclosure: Author disclosures are provided at the end of the article.
Received July 1, 2010. Accepted in final form September 1, 2010.
REFERENCES
- 1.Lucchinetti CF, Mandler RN, McGavern D, et al. A role for humoral mechanisms in the pathogenesis of Devic's neuromyelitis optica. Brain 2002;125:1450–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lennon VA, Wingerchuk DM, Kryzer TJ, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 2004;364:2106–2112. [DOI] [PubMed] [Google Scholar]
- 3.Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med 2005;202:473–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bradl M, Misu T, Takahashi T, et al. Neuromyelitis optica: pathogenicity of patient immunoglobulin in vivo. Ann Neurol 2009;66:630–643. [DOI] [PubMed] [Google Scholar]
- 5.Kinoshita M, Nakatsuji Y, Kimura T, et al. Anti-aquaporin-4 antibody induces astrocytic cytotoxicity in the absence of CNS antigen-specific T cells. Biochem Biophys Res Commun 2010;394:205–210. [DOI] [PubMed] [Google Scholar]
- 6.Hinson SR, McKeon A, Lennon VA. Neurological autoimmunity targeting aquaporin-4. Neuroscience 2010;168:1009–1018. [DOI] [PubMed] [Google Scholar]
- 7.Roemer SF, Parisi JE, Lennon VA, et al. Pattern-specific loss of aquaporin-4 immunoreactivity distinguishes neuromyelitis optica from multiple sclerosis. Brain 2007;130:1194–1205. [DOI] [PubMed] [Google Scholar]
- 8.Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology 2006;66:1485–1489. [DOI] [PubMed] [Google Scholar]
- 9.Blanc F, Zephir H, Lebrun C, et al. Cognitive functions in neuromyelitis optica. Arch Neurol 2008;65:84–88. [DOI] [PubMed] [Google Scholar]
- 10.Rocca MA, Agosta F, Mezzapesa DM, et al. Magnetization transfer and diffusion tensor MRI show gray matter damage in neuromyelitis optica. Neurology 2004;62:476–478. [DOI] [PubMed] [Google Scholar]
- 11.Badaut J, Lasbennes F, Magistretti PJ, Regli L. Aquaporins in brain: distribution, physiology, and pathophysiology. J Cereb Blood Flow Metab 2002;22:367–378. [DOI] [PubMed] [Google Scholar]
- 12.Peterson JW, Bo L, Mork S, Chang A, Trapp BD. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann Neurol 2001;50:389–400. [DOI] [PubMed] [Google Scholar]
- 13.Kutzelnigg A, Faber-Rod JC, Bauer J, et al. Widespread demyelination in the cerebellar cortex in multiple sclerosis. Brain Pathol 2007;17:38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vass K, Lassmann H, Wekerle H, Wisniewski HM. The distribution of Ia antigen in the lesions of rat acute experimental allergic encephalomyelitis. Acta Neuropathol 1986;70:149–160. [DOI] [PubMed] [Google Scholar]
- 15.Pittock SJ, Lennon VA, Krecke K, Wingerchuk DM, Lucchinetti CF, Weinshenker BG. Brain abnormalities in neuromyelitis optica. Arch Neurol 2006;63:390–396. [DOI] [PubMed] [Google Scholar]
- 16.Pittock SJ, Weinshenker BG, Lucchinetti CF, Wingerchuk DM, Corboy JR, Lennon VA. Neuromyelitis optica brain lesions localized at sites of high aquaporin 4 expression. Arch Neurol 2006;63:964–968. [DOI] [PubMed] [Google Scholar]
- 17.Suzuki M, Obara K, Sasaki Y, et al. Comparison of perivascular astrocytic structure between white matter and gray matter of rats. Brain Res 2003;992:294–297. [DOI] [PubMed] [Google Scholar]
- 18.Hirano A, Kawanami T, Llena JF. Electron microscopy of the blood-brain barrier in disease. Microsc Res Tech 1994;27:543–556. [DOI] [PubMed] [Google Scholar]
- 19.Dallasta LM, Pisarov LA, Esplen JE, et al. Blood-brain barrier tight junction disruption in human immunodeficiency virus-1 encephalitis. Am J Pathol 1999;155:1915–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ellison MD, Povlishock JT, Merchant RE. Blood-brain barrier dysfunction in cats following recombinant interleukin-2 infusion. Cancer Res 1987;47:5765–5770. [PubMed] [Google Scholar]
- 21.Fenton RA, Moeller HB, Zelenina M, Snaebjornsson MT, Holen T, MacAulay N. Differential water permeability and regulation of three aquaporin 4 isoforms. Cell Mol Life Sci 2010;67:829–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bo L, Vedeler CA, Nyland HI, Trapp BD, Mork SJ. Subpial demyelination in the cerebral cortex of multiple sclerosis patients. J Neuropathol Exp Neurol 2003;62:723–732. [DOI] [PubMed] [Google Scholar]
- 23.Kutzelnigg A, Lassmann H. Cortical demyelination in multiple sclerosis: a substrate for cognitive deficits? J Neurol Sci 2006;245:123–126. [DOI] [PubMed] [Google Scholar]
- 24.Kutzelnigg A, Lucchinetti CF, Stadelmann C, et al. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 2005;128:2705–2712. [DOI] [PubMed] [Google Scholar]
- 25.Wingerchuk DM, Pittock SJ, Lucchinetti CF, Lennon VA, Weinshenker BG. A secondary progressive clinical course is uncommon in neuromyelitis optica. Neurology 2007;68:603–605. [DOI] [PubMed] [Google Scholar]
- 26.Christodoulou C, Krupp LB, Liang Z, et al. Cognitive performance and MR markers of cerebral injury in cognitively impaired MS patients. Neurology 2003;60:1793–1798. [DOI] [PubMed] [Google Scholar]