

Abstract
Peroxisomes are ubiquitous cellular organelles that perform vital functions including fatty acid beta-oxidation, plasmalogen synthesis, and detoxification of harmful oxidative species. In rodents numerous compounds that increase peroxisome biogenesis also alleviate metabolic syndrome (MetS)/type 2 diabetes (T2D) symptoms. However, compounds that increase peroxisome biogenesis in rodents largely do not increase peroxisome biogenesis in humans. We designed a novel genetically encoded high throughput screening (HTS) high content assay to identify small molecule compounds that function as peroxisome proliferators in human cells. From this assay we have confirmed that 4-phenylbutyrate (PBA), a PPAR independent peroxisome proliferator and chemical chaperone, increases peroxisome proliferation in human cells and serves as a positive control for our screen. We performed a small pilot and larger 15,000 compound production screen with an overall Z′ factor of 0.74 for 384-well plate format, providing a valuable screening tool for identifying peroxisome modulator compounds. From this screen we have identified 4 existing drugs and 10 novel compounds, some with common scaffolds 1000X more potent than PBA. It is hoped that these novel compounds may serve as scaffolds for testing for efficacy in alleviating MetS/T2D symptoms both in mouse models and ultimately human disease.
Keywords: high-content screening, high-content analysis, peroxisome biogenesis, diabetes, metabolic syndrome, 4-phenylbutyrate
Introduction
Peroxisomes are ubiquitous eukaryotic organelles that carry out metabolic reactions and responses to cellular oxidative stress. Peroxisomes represent a major site of fatty acid β-oxidation and the only site of very long-chain fatty acid (VLCFA) β-oxidation in the cell. In addition to oxidizing fatty acids, peroxisomes help synthesize vital plasmalogens,1 special ether phospholipids that include sphingolipids, a major component of both myelin and lipid rafts. Sphingolipids are highly enriched in the nervous system while other plasmalogens (eg, choline plasmalogen) are uniquely enriched in the heart. The vital function of peroxisomes was made clear with the molecular identification of mutations in a peroxisomal biogenesis disorder (PBD) first identified in 1992 in humans.2 There are four fatal clinical disease syndromes associated with PBDs – Zellweger syndrome (ZS), neonatal adrenoleukodystrophy (NALD), infantile Refsum disease (IRD) and rhizomelic chondrodysplasia punctata (RCDP).
Therapeutically targeting peroxisome biogenesis follows from the hypothesis that increasing fatty acid oxidation capacity could be beneficial for many aspects of metabolic syndrome by decreasing lipotoxicity particularly in key metabolic tissues like the liver. The lipotoxicity hypothesis posits that elevated plasma fatty acids may lead to ectopic lipid accumulation in non-adipose tissue, particularly the liver (“fatty liver”), that harms organ and cellular function.3 In addition, consumers of Western high-fat diets may benefit from increased fatty acid oxidation capacity as elevated plasma free fatty acids can contribute to insulin resistance and hyperglycemia, which are risk factors for both metabolic syndrome and T2D (type 2 diabetes).4 Indeed, it has long been known that many structurally unrelated compounds that increase peroxisome biogenesis in rodents are also successful in treating symptoms of dyslipidemia and metabolic syndrome (MetS) in rodents.5
The fibrates (Clofibrate, Bezafibrate, Ciprofibrate, Gemfibrozil, and Fenofibrate) represent one class of plasma lipid lowering drugs commonly prescribed in the 1970s. Although the mechanism of fibrate action was not known at the time, researchers screening for a fibrate-like drug in a diabetic mouse model identified Ciglitazone,6 a thiazolidinedione (“glitazone”) compound. Through Structure Activity Relationship (SAR) programs Ciglitazone gave rise to rosaglitazone (Avandia™) and pioglitazone (Actos™). Currently, fibrates are preferentially thought to activate peroxisome proliferator activated receptor α (PPARα) in the liver, while thiazolidinediones preferentially activate PPARγ in adipose tissue for their therapeutic effects.7 Despite the original PPAR name, current marketed PPAR agonists, particularly the glitazones, do not increase peroxisome biogenesis in humans. However, multiple compounds structurally unrelated to fibrates display both hypolipidemic effects and peroxisome proliferation in rodents5 suggesting that peroxisome proliferation itself may produce beneficial effects, perhaps through increased fatty acid oxidation.
Experimental evidence further supports peroxisome biogenesis as potentially therapeutic since rodents treated with fibrates display increased peroxisome proliferation and both enhanced insulin sensitivity and reduced fat deposition in the obese Zucker rat model.8 Interestingly, fibrates also enhance fatty acid oxidation in both lean and obese Zucker rats,9 Although both fibrates and rosiglitazone lower serum triglycerides in fatty Zucker rats, only fibrates lower body weight, while rosiglitazone leads to large undesirable weight gains in adipose tissue,10 an effect also seen in humans. This suggests that novel approaches to increase peroxisome biogenesis in humans could be superior to current PPAR treatments for diabetes and metabolic syndrome. Indeed, it has recently been documented that the FDA-approved drug 4-phenylbutyrate (PBA), a PPAR independent peroxisome proliferator and chemical chaperone, treats symptoms of MetS and T2D in ob/ob (obese diabetic) and db/db (diabetic) mice11,12 and in B6/HFD (high fat diet) mice,13 including restoration of glucose homeostasis, enhanced insulin sensitivity and decreased fatty liver disease. PBA serves as a positive control compound in our assays as a potent peroxisome proliferator in our human-liver cell line high throughput assay (see Results) and is a non-classical PPAR-independent peroxisome proliferator.14 Although PBA has been shown to be effective, it must be used at extremely high doses (1 g/Kg in rodents) and results in off-target side effects. PPAR-dependent compounds also display deleterious side effects (eg, FDA black box warning on rosiglitazone (Avandia™) and weight gain). We postulate that compounds that promote peroxisome biogenesis with high potency via PPAR-independent mechanisms may represent attractive therapeutic chemical entities.
During the study of fibrates and subsequent optimization of Ciglitazone through SAR programs in the 1970s and 1980s, the methodology for assaying peroxisome biogenesis involved laborious dissections of rodent livers and careful biochemical or anatomical electron micrograph analysis – not amenable to large-scale screening. Today, however, the technology exists to develop high content genetically encoded cell-based assays that directly monitor peroxisome biogenesis in vivo (see Results). Although peroxisome proliferation itself appears therapeutic in rodents, compounds that caused phenotypic peroxisome proliferation were largely abandoned in favor of pursuing molecular targets, including PPARs, despite their lack of efficacy in peroxisome proliferation in humans. Importantly, no previous assay that we are aware of has identified chemical compounds that can increase peroxisome biogenesis in human cells in an HTS format. This study describes the development and implementation of a new HTS assay to monitor peroxisome biogenesis in human cells.
Materials and methods
Cells and reagents
The human hepatocellular carcinoma (HepG2) cell line was genetically encoded with an enhanced peroxisome targeting fluorescent reporter (EPTFR) that expresses a GFP variant labeling peroxisomes in live cells. The reporter is based on a well-characterized carboxy terminal three amino acid peptide Peroxisome Targeting Sequence 1 (PTS1)15 motif sufficient to target polypeptides to the peroxisomal membrane. We have found through optimization/experimentation (data not shown) that adding a particular fourth amino acid (RSKL) increases targeting efficiency in human cells. We have analyzed both the stably GFP-transfected and wild type HepG2 cell lines with flow cytometric analysis and have concluded that the stable (G418 selected) cell line is monophasic, has remarkable GFP intensity allowing for short microscopic exposure times, and is ideal for screening purposes.
The cells were cultured according to the ATCC guidelines with EMEM growth medium (ATCC# 30-2003), supplemented with 10% FBS, 1% P/S and 400 μg/ml G418. The HepG2/GFP stable cell line was expanded to generate a master cell bank for high-content screening. Cryogenically preserved stocks were made at the same passage number with approximately 1 × 106-cells per 1 mL tube in cryo-preservation media consisting of growth media supplemented with 5% DMSO.
4-phenylbutyrate was used as the positive control in both single-point and dose-response studies and a 450 mM aqueous stock solution was prepared by dissolving 4.11 g 4-phenylbutyric acid (PBA, 99%, Sigma# P21005-25G, FW164.2) in 35 mL of autoclaved ddH2O. This solution was titrated with an equimolar amount of 1 N NaOH (approximately 20.4 mL) to pH 7.0 to form the sodium salt.
Compound libraries and management
The BRITE diversity library screened in this HCS effort consists of 33,600 compounds selected for chemical diversity from the 350,000 compound BRITE library, which was chosen for maximal diversity in chemical space using clustering tools provided with MOE (Molecular Operating Environment, Chemical Computing Group Inc.) using multi-objective optimization. This library was generated by combinatorial chemistry synthetic routes and was a gift from Biogen-Idec in 2005. All of the compound plates were stored in polypropylene deep-well blocks at 4°C without humidity controls. The BRITE compound library is plated in 384-well Greiner V-bottom polypropylene plates (Greiner# 781280) in 100% DMSO at 10 mM for dose/response studies and 1 mM for single-concentration screening.
Using the Biomek NX workstation (Beckman-Coulter, Brea, CA) and P20 pipette tips, 0.5 μl of 1 mM compound in DMSO was spotted into columns 3–22 of a Greiner 384-well Polypropylene V bottom square well plate for single-concentration screening. Columns 1 and 2 and 23 and 24 were spotted with 0.5 μl of DMSO and were used as negative and positive controls, respectively.
EC50 dose-response determination was performed in 384-well plates by cherry-picking 1 μl of compound from the corresponding 10 mM source plate, diluting to 5 μL total volume with DMSO for a final 2 mM compound concentration using a Biomek 3000 and P20 tips. Axygen conical 384-well PCR plates were used during the serial dilution scheme to facilitate mixing and small volume transfers. A 10-point 2-fold serial dilution was then performed across the plate with the Biomek® 3000. The serially diluted compounds were then spotted to columns 3–22 of a Greiner 384-well Polypropylene V bottom square well plate with the Biomek 3000 and P20 tips. Columns 1, 2, 23, and 24 were spotted with 0.5 μl of DMSO with the Biomek NX and P20 tips and used as positive and negative controls. We included a control dose response for PBA in wells A3–A12 for quality control purposes and measured 31 dose/responses per plate. Compounds that were confirmed with dose response were tested in intra-plate triplicate and cellular observations were averaged for each concentration and a single EC50 value was determined for each compound.
Peroxisome biogenesis assay
The peroxisome biogenesis assay was developed in 384-well format with a final assay volume of 50 μL. 384-well Corning black/clear thin-bottom tissue-culture-treated imaging plates (Corning #3712) were coated with a solution of 50 μg/mL Poly-D-lysine (Sigma-Aldrich) to aid in cell adherence overnight at 4°C, then washed twice with ddH2O and air-dried.
Cell plating
Cells were thawed from freezer-stocks, counted using a Vi-Cell XR (Beckman-Coulter, Brea, CA) and diluted to a final concentration of 0.16 × 106 viable-cells/mL. 20 μL of prewarmed growth medium were added to each well using a Mutidrop-384 (Thermo/Fisher) with a sterile head before transferring 20 μL of cell suspension (3200 viable cells/well) into each well. The pre-addition of 20 μL of media prior to dispensing the cell solution dramatically improved the distribution of cells within the well, yielding a homogenous cellular distribution across the center of the well. The cell plates were then allowed to attach overnight in the incubator.
Compound addition
An intermediate dilution step was used to limit the overall DMSO exposure to below 1%. Intermediate dilution plates were made by adding 40 μL of growth media to the spotted compound plates as described above (see: Compound Libraries and Management) and transferring 10 μL from this plate to two identical cell-plates in inter-plate duplicate format using the Biomek-NX workstation yielding a final single-point screening concentration of 2.5 μM and dose/response top concentration of 19.5 μM in 50 μL total assay volume. The plates were then incubated with compounds for three days. 4-phenylbutyrate was used as the positive control and 4.5 μL of a 450 mM stock solution was added to columns 23 and 24 of the intermediate dilution plates manually with an 8-channel P20 pipette. A final concentration of 9.1 mM 4-phenylbutyrate was achieved with the 10 μL transfer from intermediate dilution plate to cell plate.
Staining and fixation
Approximately 40 μL of media with compound was removed from each well using the Biomek NX workstation leaving enough liquid behind to wet cells. 30 μL of 10 μg/mL Hoechst-33342 and 2 μM CellTracker-Red (Invitrogen, Carlsbad, CA) were dissolved in media (omitting serum and antibiotics) and was added to each well and then incubated for 45 min at 37°C. 30 μL of media was aspirated to remove excess dye and was replaced with 30 μL of fresh 4% formaldehyde. The cell plates were incubated in fixative for 45 min at 37°C. The cells were then washed once with PBS using the Biomek-NX at the slowest dispense speed and were sealed with Thermo-ABgene plate sealer and were imaged on BD Pathway 855 bioimager (Becton Dickenson, San Jose CA) within 2 hours of processing.
Immunohistochemical staining
Colocalization studies
HepG2 cells were cultured on cover slips, fixed with 4% formaldehyde and made permeable with 0.5% Triton X-100 in PBS for 10 min. Non-specific binding was reduced by blocked in 3% BSA in PBS containing 0.5% Triton X-100 for 30 min at room temperature. Cells were incubated with the primary rabbit anti-PMP 70 antibody (Sigma, 1:200 dilution) for 2 hours at room temperature. The secondary Goat anti-rabbit IgG conjugated with the Cy3 probe (1:200 dilution) was used for fluorescent labeling. Cover slips were mounted on glass slides using VECTASHIELD Mounting Medium with DAPI. The slides were imaged and analyzed using a BD Pathway-855 and Nikon A1 confocal microscope for co-localization studies.
Lipid droplet staining
Oil-Red-O staining was performed on cover slip mounted HepG2 cells. Cells were allowed to attach on cover slips overnight, the media was removed and cells were fixed with 10% cold formalin (4% formaldehyde) for 10 minutes at room temperature. Cells were then washed in PBS twice and air-dried for 20 minutes. Slides were placed in 100% propylene glycol for 2 minutes, and were stained with a 0.5% w/v Oil-Red-O solution in propylene glycol for 10 minutes at 60°C. Slides were then differentiated in 85% propylene glycol in ddH2O for 2 minutes, then rinsed twice in ddH2O and stained with Mayer’s hematoxylin (diluted 1:2 in water) for 30 seconds. Cover slips were washed in running tap water followed by a ddH2O rinse and then mounted with aqueous mounting media (10% glycerol in water). Cells were imaged with an Olympus bright-field microscope at 20× magnification with a 4 megapixel color CCD camera and lipid droplets appear red in color and the nuclei appear pale blue.
Fluorescent imaging
The BD Pathway 855 was used to acquire multiplexed epifluorescent images in 384-well format with a 20×/0.7 NA Olympus UPlanApo objective lens using the BD Attovision software. Green fluorescent protein labeled peroxisomes were imaged with a 488/10 nm band-pass excitation filter, Fura/FITC epifluorescence dichroic, and a 515 nm long-pass emission filter. Hoechst dye labeled nucleic acids were imaged with a 380/10 nm band-pass excitation filter, a 400 nm dichroic long-pass, and a 435 nm long-pass emission filter. Laser-based autofocus was performed in each well prior to the collection of GFP and Hoechst images. Batches of six plates were processed and imaged at a time. Exposure times for both the GFP and Hoechst channels were adjusted prior to each batch to ensure optimal signal-to-noise ratio.
Cell detection, segmentation and quantitation of cellular morphology
Image processing
BD Attovision was used for cell detection, segmentation and quantification of peroxisomal and nuclear parameters. The first step for detection of individual cells using the nuclear/Hoechst channel used a shading algorithm to flatten the image background due to non-homogeneous illumination. Automatic thresholding was performed, followed by an optimized watershed algorithm to aid in separation of closely packed cells. A binary nuclear mask was generated by fitting a polygon to the perimeter of the remaining regions of interest (ROIs) for tabulation of nuclear fluorescent attributes for individual cells. The cytoplasmic mask was delineated by geometrically expanding the nuclear mask by 25 pixels (1 pixel = X μm) or until adjacent cell boundaries meet. The cytoplasmic binary mask was used to tabulate peroxisomal parameters on the GFP channel. Several GFP intensity and distribution moments were tabulated per cell to achieve a thorough phenotypic peroxisomal description. Spot-counting (sub-object counting) was also performed on the GFP channel inside the cytoplasmic mask to tabulate the number, size and density of peroxisomes per cell.
BD Image Data Explorer was used to generate a database table which contained cell-level results for an individual plate, that was directly imported into JMP8 (Statistical Analysis Software, Cary NC) for initial data exploration and visualization in assay development, for principal component analysis transformation of cell-level data, and for calculation of well-averaged results.
Well-averaged biological results were merged with chemical structure information using ActivityBase (IDBS Software) and were stored in the IDBS Oracle database. Nonlinear regressions were performed to fit dose response curves using a four-parameter one-site dose-response with the XLfit software (IDBS software).
Principal component regression during PBA dose-response to improve scoring system
Initial studies on the EPTFR-HepG2 cell line involved tabulating the total GFP intensity per cell for positive and negative control treatments. It was observed there was significant overlap between positive and negative control GFP expression leading to poor assay statistics (Z-prime close to zero). The difference between positive and negative samples was visually apparent but simple fluorescence intensity integration within a cell was inadequate to score the complex visual phenotype in this assay.
We have developed an image-processing pipeline to tabulate as many intensity/shape/size/distribution properties for each cell as necessary (currently <50 parameters are calculated per cell). Principal component analysis (PCA) is then used to reduce the dimensionality of the data while regressing against the variation in the compound concentration. In the conventional PCA approach, one makes linear combinations of the independent variables and mines for the combination that maximizes the variance in the data set. While performing PCA for analyzing peroxisomal biogenesis, a significant amount of the variance observed from cell to cell was due to natural variation and not due to the influence of the positive control PBA. In this case, the natural variability and the drug response were convoluted and a simple analysis of GFP intensity was quite misleading. We performed principal component regression, where we take the correlated independent variables (cell size, number of peroxisomes, size of peroxisomes, shape of peroxisome, etc.) and un-correlate them with standard PCA. Components of the PC1 used for hit-identification are shown in Table 1. We then take the un-correlated principal components describing the total variability in the data set and regress against the compound concentration in the dose-response study. This has the effect of eliminating a majority of the natural variability by focusing on the effects of the drug alone and un-correlating independent variables (such as cell size and peroxisome number). This data reduction procedure is critical during assay development to accurately characterize the effects of compounds during screening.
Table 1.
| Parameter | Eigenvalue |
|---|---|
| GFP Intesity | 0.52 |
| GFP Granularity | 0.39 |
| GFP Sub Object Count P1 | −0.01 |
| Hoechst Intensity | 0.34 |
| Cell Tracker Red Intensity | 0.44 |
| GFP Densitomentry Mean | 0.52 |
Quantitative western blot analysis
Peroxisome content was assessed by normalizing the PMP-70 signal to the alpha-tubulin signal to control for variation in sample loading. HepG2 cells were seeded at a density of 5 × 105 cells in 6-well dishes in complete growth medium and were allowed to attach overnight. Compounds were delivered at the appropriate concentration in DMSO (while maintaining less than 1% total DMSO concentration), and were allowed to incubate for 3 days. Cells were harvested, washed with cold PBS and total protein was extracted in lysis buffer (100 mM Tris-Cl pH 6.8, 2% SDS, 1 mM PMSF, Sigma protease inhibitor cocktail). Proteins were separated on 4%–12% NuPAGE BisTris gradient gels (Invitrogen) and then transferred to PVDF membrane. Blots were probed with a 1:3000 dilution of anti-PMP-70 and 1:16000 anti-alpha-tubulin (Sigma rabbit anti-PMP-70 and anti-alpha-tubulin clone B-5-1-2), followed with a 1:2000 dilution of secondary fluorescent antibodies (Li-Cor IRDye 680 Goat anti-rabbit and IRDye 800CW Goat anti-mouse). Western blot scanning and band quantification were performed with the Odyssey infrared imaging system (Li-Cor Biosciences, Lincoln NE) according to the manufacturer’s recommendations.
Results
Several small molecules in the literature are reported to induce peroxisomal biogenesis, including both PPAR and non-PPAR related compounds. We reasoned that the discovery of small molecules that act in a PPAR independent manner while increasing peroxisomal functional capacity could have a therapeutic benefit for the type-II diabetes mellitus and metabolic syndrome. Literature reports of PBA and peroxisomal biogenesis indicate that this fatty acid mimetic can induce hepatic peroxisomal proliferation and shows therapeutic benefits in animal models for T2D, but with potential significant side-effects relating to the high-dose required for therapeutic benefit (1 g/kg/day in mice). We searched for an assay to identify novel chemical probes that modulate peroxisomal biology similarly to PBA but at higher potencies.
Assay development
The initial phase of assay development to probe peroxisome biogenesis was done using a genetically encoded fluorescent reporter system to determine whether the fluorescent reporter would co-localize with peroxisomes and if the reporter would respond to stimulus. We chose a liver-derived cell line, HepG2, due to its past demonstration of robust peroxisomal plasticity.16
EPTFR-HepG2 cell line
We developed a novel stable cell line for this high-content assay utilizing an enhanced peroxisome targeting fluorescent reporter (EPTFR) that expresses a green fluorescent protein (GFP) variant that labels peroxisomes. The EPTFR can be assayed in live or fixed cells by simple fluorescent methods in real time. The reporter is based on a characterized carboxy terminal three amino acid peptide Peroxisome Targeting Sequence 1 (PTS1) motif sufficient to target polypeptides to the peroxisomal membrane. Generally, an SKL (serine-lysine-leucine) peptide at the extreme carboxy terminus is sufficient; however, we have found through optimization/experimentation (data not shown) that adding a particular fourth amino acid (arginine – RSKL) increases targeting efficiency in human cells. We have made a human liver cell-derived (HepG2) stable cell line with the genetically encoded EPTFR (Figure 1). Importantly, the EPTFR reporter in our cell line exactly co-localizes with antibody stains against endogenous peroxisomal antigens (Figure 1B), and both HepG2 cells expressing or not expressing our peroxisomal reporter show the same morphologies and numbers of peroxisomes (Figure 1C). We have selected a spectrally resolved fluorescent probe set (Figure 2A) and have analyzed both the stable and wild type cell lines with flow cytometric analysis (Figure 2B), concluding that the stable (G418 selected) cell line is monophasic, has remarkable GFP intensity allowing for short microscopic exposure times, and is ideal for screening purposes. The cells analyzed in Figure 2B were sampled from the low-passage EPTFR-HepG2 master cell bank that was used for this screening effort to reduce the day-to-day biological variability when screening from actively growing culture.
Figure 1.
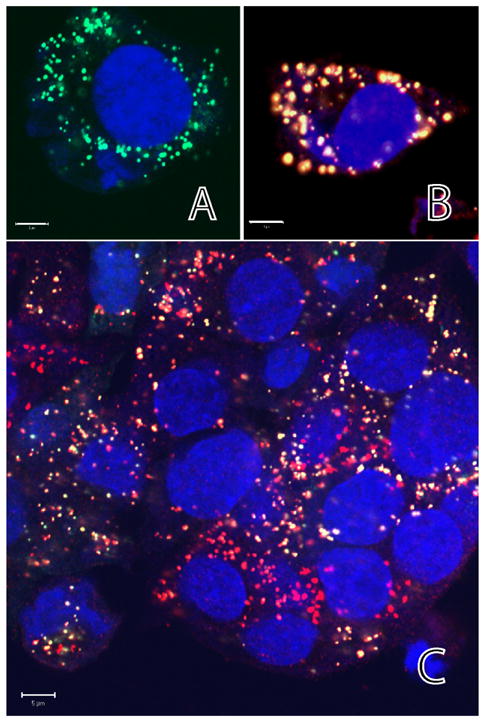
A) Enhanced peroxisome targeting fluorescent reporter (EPTFR) in HepG2 human cells. B) Double labeling of peroxisomal antigen PMP-70 (red) and EPTFR (Green) demonstrating co-localization. Note the general number, distribution and shape of peroxisomes is unchanged in EPTFR expressing cells (C, yellow). (Blue = Hoescht (nuclei)) (scale bar = 5 μm).
Figure 2.
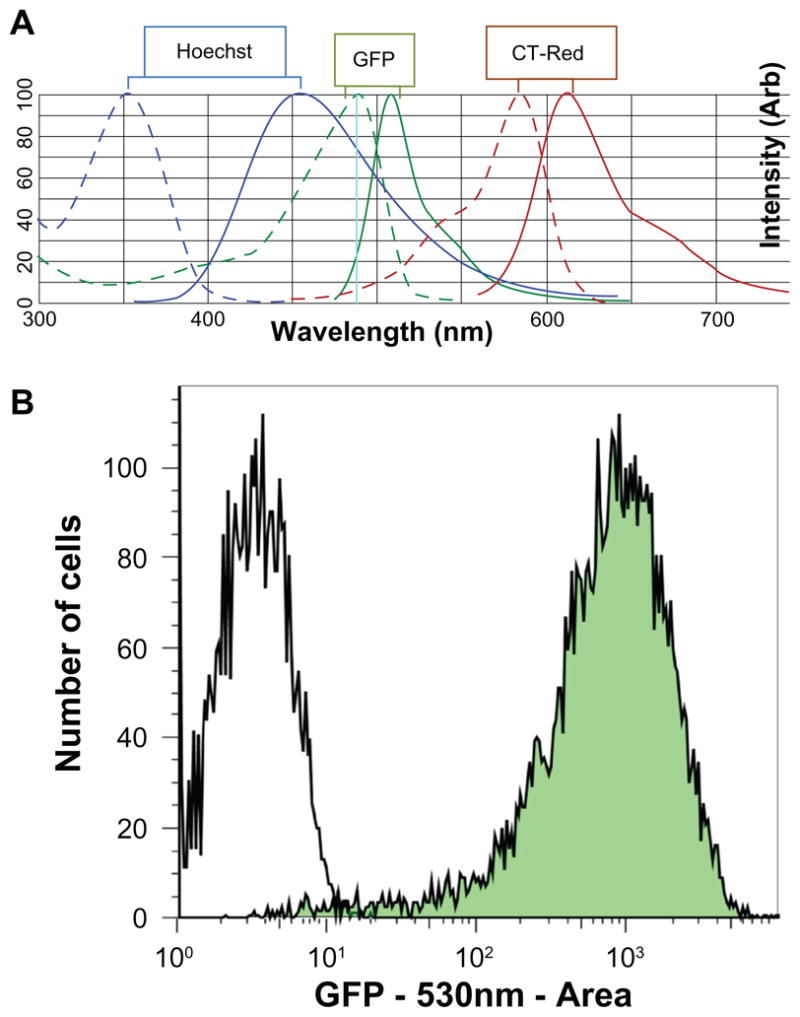
A) Spectral multiplexing of Hoechst (blue), GFP, and CellTracker-Red fluorescent probes. B) Flow cytometry analysis of a stable EPTFR-HepG2 line (green) and a wild-type HepG2 line showing suitability of the cell line for high-content screening purposes. The filled histogram shows a monophasic population of cells with substantial GFP expression (approximately 300× greater than the negative population). This cell population is sampled from the low-passage master cell-bank that was used in this screening effort.
4-Phenylbutyrate as a positive control for HCS assay development
Sodium 4-phenylbutyrate (PBA) is a fatty acid mimetic (Figure 3 – inset) that is FDA approved (marketed as Buphenyl ®) for the treatment of urea cycle disorders. PBA has recently been implicated as a therapeutic molecule for T2D/ MetS in mice11 and is a substantial non-PPAR peroxisome proliferator in both human cells in vitro and rodent liver in vivo.14 Figures 3B and 3C show confocal fluorescence images acquired on the Pathway 855 bioimager of normal peroxisomal distribution (negative control treated with DMSO vehicle) and under 9 mM PBA treatment, showing massive increase in total peroxisomal mass per cell. This dosage is consistent with the previous study in human primary hepatocytes that showed large peroxisomal antigen increases with 1–5 mM PBA.14 We have also demonstrated that PBA provides dose/response activity for peroxisome biogenesis in the GFP reporter system (Figure 3A) consistent with previously demonstrated microscopy studies and western blots.14 This result provides proof-of-principle for our hypothesis that the phenotypic end-point of non-classical peroxisome proliferation in human cell lines can be harnessed for high content imaging analysis and converted into HTS assays that might provide new tools for discovery of chemical probes and potential therapeutics. A validated reporter cell-line that co-localizes and responds to stimulus is requisite for high-content assay development.
Figure 3.

PBA validation as a positive control for high content assay development and screening. (A) Dose-response curve for GFP fluorescence (inset structure of PBA) exhibiting a 2.4 mM EC50. PBA causes significant increases in peroxisomes visualized with EPTFR-HepG2 cells (blue – nuclei, green – peroxisomes) showing normal peroxisome distribution (B) (DMSO vehicle) and (C) increased peroxisomal content (with PBA).
Primary high-content assay development probing peroxisome biogenesis
Assay development was conducted with the EPTFR-HepG2 cell line in 384 well plates to identify optimal assay timing, staining conditions, imaging conditions and data analysis procedures to yield the most sensitive assay for the detection of compounds than can influence peroxisomal biology. We optimized protocols for plating, drug incubation, staining and imaging the EPTFR-HepG2 cell line in the Pathway 855 high-content bioimager (BD Biosciences, Rockville, MD) with multiplexing to spectrally resolve the fluorophores; 1) green fluorescent protein-fusion labeling peroxisomes; 2) Hoechst 33342 for labeling the nuclei (blue); and 3) CellTracker Red for labeling the cytoplasm. Excellent spectral resolution was obtained in the peroxisome, nuclear, and cytoplasmic images for accurate binary masking and subsequent quantification of cellular responses (Figure 2A).
Cell detection, segmentation and quantitation of cellular morphology
Accurate feature extraction for individual cells enables the detection of meaningful phenotypic change. Our general strategy for image analysis is to calculate as many cellular features as possible and use multivariate data reduction techniques including principal component analysis and partial least squares regression, coupled with biological insight to identify meaningful phenotypic change. Bioimage data generated from this assay consists of a multiplexed set of three images (blue-nucleus, red-cytoplasm and green-peroxisomes) of a field of cells acquired in the well-center using a 20× microscope objective. Refinement of automated image pre-processing and segmentation parameters for quantization of cellular response have been performed to ensure fidelity over a wide range of cell densities, GFP expression ranges, and minor fluctuations in staining. This process involves careful acquisition of reference images for background subtraction, image pre-processing, cell detection/segmentation, and feature extraction (Figure 4).
Figure 4.
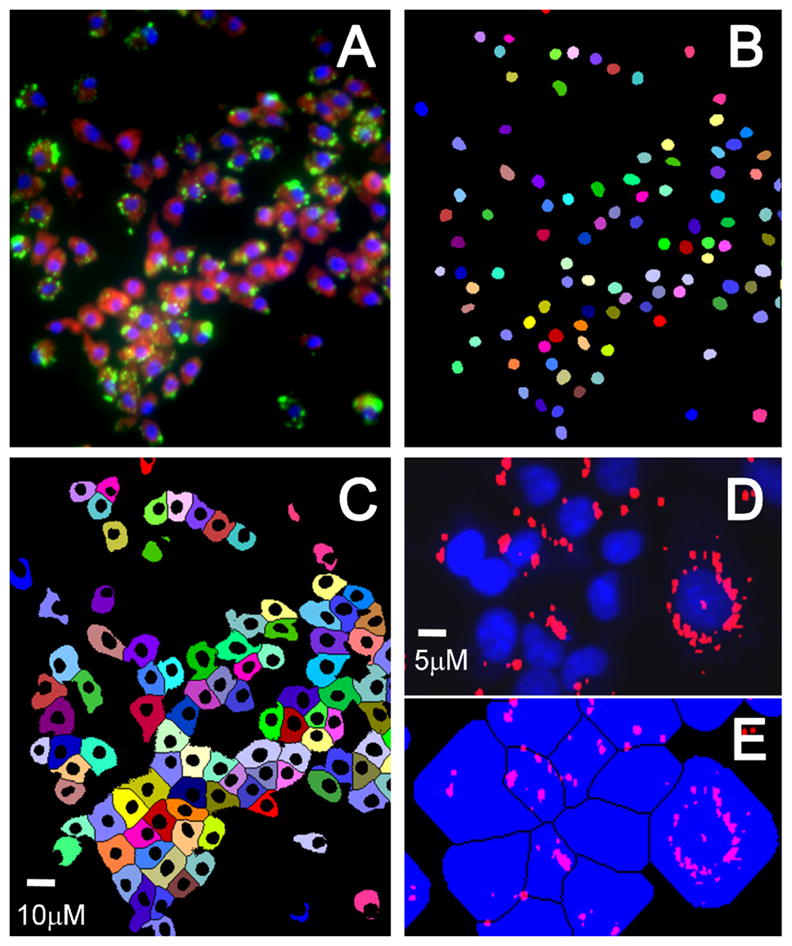
Image segmentation showing (A) merged color image with blue nuclei, red cytoplasm and green peroxisomes, (B) nuclear binary mask, and (C) cytoplasmic mask. Each colored object in the masks indicates a unique cell, showing effective segmentation. (D) Peroxisomes masked (red) with nuclei (blue), and (E) assignment of peroxisomes to individual cells inside the respective cell mask.
Image pre-processing was performed in the following two steps: 1) Correction of background illumination variation using a shading algorithm for all channels to ensure a flat field for segmentation; and 2) 10-pixel rolling-ball background subtraction in the GFP-peroxisome channel to aid in the separation and quantitation of individual peroxisomes.
Segmentation starts with identifying the well-separated individual nuclei in the Hoechst image by first thresholding the background and then applying the watershed algorithm for edge detection based on interpreting a grayscale image as a topographical surface. This topographical surface is then flooded from its minima using upper boundary conditions to detect the edges and effectively segment closely spaced nuclei. A binary nuclear mask is generated as shown in Figure 4B. The nuclear mask is then used as the basis for generating the cytoplasmic mask. The edge-detection algorithm is then used to mask the boundary of each cell’s cytoplasm area using the CellTracker-Red image. All quantification of peroxisomal number/attributes is done inside of the individual cell cytoplasmic mask as shown in Figure 4E. Peroxisomal parameters for each cell were calculated inside the cytoplasmic mask and include the total integrated GFP intensity (representing total peroxisomal mass), peroxisomal number through sub-object counting, and various shape/size attributes. These per-cell statistics were then compiled into a SQL database for visualization, statistical analysis and tabulation of screening results.
Assay validation
The assay was evaluated for screening robustness by performing a Min-Max experiment to determine the variance in the negative and positive controls. Ideally, the standard deviation in the cellular response will be small for all wells treated with a specific concentration of our positive control PBA, indicating a good signal-to-noise ratio (SNR) and a desirable Z-prime statistic (above 0.5).
Data from this Min-Max study show effective use of 9 mM PBA concentration as a positive control in two 384-well plates with 0 mM and 9 mM (appropriate carrier solvent volume added to negative control) as shown in Figure 5. Each data point plotted in Figure 5 represents a well-averaged response demonstrating a suitable assay window (max-minus-min). The response (normalized peroxisomal response) is a linear combination of all measured peroxisomal attributes derived from partial least squares regression analysis and through rejection of a sub-population of cells that have a zero detectable peroxisomes (less than 5% of the population). Assay conditions have been optimized to ensure that at least 100 cells per CCD field in a well with a 20× objective are captured to ensure adequate statistics even in the presence of anti-proliferative or cytotoxic compounds. Cells with less than one identifiable peroxisome were omitted from the analysis due to low potential for peroxisomal plasticity, further reducing variability in the assay.
Figure 5.

Min-Max assay validation in two 384 well plates showing incubation with positive control (Max – 9.0 mM), and assay negative control (Min – DMSO carrier). Inset table shows assay statistics, resulting in a calculated Z of 0.72.
We have also analyzed the plate-to-plate variation in typical triplicate assay plates and have found an average coefficient of variation (CV) of 4.1%, and the day-to-day variation of less than 5%. Assessment of the initial quality of the assay from this Min-Max experiment results in a calculated Z-prime for individual plates at approximately 0.72 (Figure 5). Our Z-prime value indicates the robustness of the high-content cell-based assay for high-throughput screening. We are ultimately interested in identifying small molecule potentiators of peroxisome biogenesis that also increase peroxisomal function, which can be validated by multiple secondary (orthogonal) assays such as fatty-acid uptake and oxidation in the future.
The primary screening assay was single-point (single concentration) in an inter-plate duplicate format. The calculated peroxisomal response per cell, and per well was averaged for duplicate data and was analyzed for agreement between assay plates. Hits are identified by using a standard deviation based hit-threshold (signal mean plus 3× or 6× the standard deviation in the compound data). Duplicate wells that are identified as active that have a substantial disagreement (>15%) are manually inspected and a decision is made depending on the visual inspection whether the compound effect is significant. Typically upon disagreement, one of the wells contains a foreign object or is out of focus and can be easily triaged out of the hit list. Compounds that significantly reduce the number of cells per well, as compared to controls, are flagged as toxic/anti-proliferative for future categorization in the cell database. Compounds identified as single-point hits were confirmed using dose response/ EC50 determination. Figure 6B shows the dose response curve for niclosamide (a positive compound identified in the Prestwick pilot screen) exhibiting a 2.4 μM EC50 and a correlating western blot (inset) showing that, upon stimulation with niclosamide at the EC50 dose, there is a 33% increase in the peroxisomal specific antigen PMP-70 (peroxisomal membrane protein-70) indicating increased peroxisomal content. Importantly, niclosamide has no known PPAR activity in multiple PPAR assays available in public databases and in the scientific literature. Thus, corroboration between the GFP reporter system and the western blot showing substantial increase in the peroxisomal antigen PMP-70 provides proof-of-principle that this assay is capable of identifying non-PPAR modulators of peroxisome biology.
Figure 6.

Pilot screening results from the Prestwick FDA-approved drug library. (A) Pilot screen scatter for the 1120 compound Prestwick library. Response in this assay is calculated as a percent-effect as compared with the positive control. A cursory hit threshold is drawn at <32% effect, representing the μ + 3σ (B) Dose response confirmation for the active compound niclosamide (inset) showing a 2.4 μM EC50 and correlating western blot (left lane = DMSO, right lane = positive control) normalized to tubulin. Increased GFP expression correlates with increased peroxisomal specific antigen PMP-70 (↑33%).
Abbrevation: U, arbitrary fluorescent units.
Screening results
Pilot screen – FDA approved drug library
For concentration range-finding and to validate assay development parameters, we screened the commercially available Prestwick library, consisting of 1120 highly-diverse FDA-approved drugs that have established biological activities. Seven compounds were active from single-point screening (blue open diamonds in Figure 6A) and four were confirmed with dose/response in this assay, including niclosamide as shown in Figure 6B (inset), yielding an overall hit-rate of 0.4% and a confirmation rate of 66%.
Production screen – BRITE diversity set
15,360 compounds were screened for their ability to increase cellular peroxisome content and/or to significantly changing peroxisomal morphology. The normalized peroxisomal response (a linear combination of the most important peroxisomal parameters chosen from PCA analysis of the effects of PBA in the response including; GFP intensity, peroxisomal number per cell, cell size, nuclear content and peroxisomal shape statistics) is plotted for each compound as shown in Figure 7A. The screening data is transformed using the first principal component eigenvector, which reflects increases in the number and size of peroxisomes per-cell. The compound distribution data scatter as shown in the histogram and box plot in Figures 7B and 7C shows a normal (un-skewed) distribution with a mean set to zero and a standard deviation in compound data of 7.63, leading to a Z-factor = 0.63 and an overall Z-prime of 0.74 for this 15 k compound screen.
Figure 7.

Production HCA screening data for 15 k compounds for peroxisomal biogenesis. (A) Scatterplot for 15,000 compounds screened, (B) histogram for the screen showing a normal (Gaussian) statistical distribution and (C) box plot for the screen showing distribution statistics and outliers (points).
Dose response confirmation – EC50 determination
Of the 15 k compounds screened, 21 compounds were above a μ + 6σ threshold of 45 percent effect and were carried forward into dose-response studies yielding a primary hit rate of 0.14%. Of the 21 compounds tested in dose-response, 10 compounds exhibited dose-response behavior for a 48% confirmation rate. Of the compounds that did not confirm, several were trending upwards at 19.5 μM in the last or last-two data points but were not considered active due to low potency. Of the surviving 10 compounds, four were of the same scaffold with the most potent at 2 μM EC50. Two compounds from another promising scaffold were discovered and had potency between 10–20 μM. The remaining four compounds were structurally unrelated and have potencies ranging from 7–25 μM.
Decreased lipid droplet accumulation in HepG2 under 4-phenylbutyrate treatment
Although ultimately we hope that increased peroxisome biogenesis will correlate with increased fatty acid oxidation, the primary screening assay does not directly measure peroxisome function. Numerous commercially available quantitative assays exist to accurately measure ascribed peroxisomal functions including for instance fatty acid uptake. However, there are also simple cost effective assays to measure lipid content in cells including Oil-Red O staining to visualize intracellular lipid droplets. PBA treatment of diabetic mice resolves fatty liver and lowers liver triglyceride content.11 Treating HepG2 cells with PBA consistently decreased both the size and staining intensity of lipid droplets (Figure 8).
Figure 8.

20× color micrographs of Oil Red O staining of lipid droplets in HepG2 cells for untreated and PBA treated conditions showing a significant decrease in the size and intensity of staining with PBA. The respective histograms (lower panels) show the distribution of lipid droplet size per cell. Untreated mean lipid droplet area of ∼300 pixels2 (0.02 μm2) compared 50 pixels2 (0.003 μm2) for PBA treated.
There is a left skew in the histogram of the untreated cells (Figure 8a lower panel). This non-Gaussian distribution is likely an artifact of the limited resolution of the chromogenic dye causing multiple distinct lipid droplets to appear superimposed. However, the peak of the distribution does accurately reflect the increased isolated lipid-droplet size. Decreases in numbers and sizes of lipid droplets generally reflect increased lipid catabolism as lipid droplets are the major triglyceride stores in eukaryotic cells.17 Although, more direct measures of compound-induced peroxisomal function will be pursued in the future, Oil-Red O staining provides a cheap, simple secondary assay to measure whether a compound crudely affects cellular lipid content.
Discussion
Liver cells represent ideal targets for peroxisome biogenesis regulation. In rodents, liver cells in vivo show the most peroxisome plasticity (the ability to increase peroxisome biogenesis). Also, the liver may be the most relevant physiological site for targeting lipid and glycemic disorders as the liver is a central effector of energy metabolism. Liver steatosis for instance occurs in many metabolic diseases and obesity and also contributes to insulin resistance.3 In this assay we use a liver-derived cell line, HepG2 cells, which have previously demonstrated plasticity for peroxisome biogenesis.18
Stratification of compounds for future study
There are several logical experiments that should be performed once new compounds have been identified in the primary HTS screen. Initially, secondary screens should be conducted using primary liver cells. Secondary assays should also be performed to establish that novel peroxisome modulators function in the absence of the EPTFR reporter: for instance by simple use of anti-peroxisomal immunocytochemistry or Western blot. Exclusion of compounds that directly activate PPARs should also be performed using commercial PPAR-activation assays. In addition, primary liver cells or liver cell lines from other species (including rodents) should also be utilized to test peroxisome biogenesis compounds for conserved function as an important step before testing these compounds in rodent models in vivo.
Of the 10 compounds identified, we have identified two promising scaffolds that were multiply represented and appear to have good medicinal chemistry properties and potencies between 2 μM and 25 μM. The confirmation rate was relatively low at 48%, however many of the compounds that did not confirm had an upward trend at higher concentrations. Had the dose-response concentration been extended to 100–200 μM, we most likely would have observed greater than 50% effect. We chose to exclude these compounds from future study due to low potency. Several of the unconfirmed compounds were sufficiently cytotoxic (measured by the reduction in cell count per well as compared with the negative control wells) but were included in the event that they were very potent and cytotoxicity at the screening concentration might prove to be irrelevant. It is possible that compounds that interfere with processes that lead to peroxisomal turnover (eg, pexophagy or autophagy) can increase peroxisomal content. These compounds would likely cause cellular stress/cytotoxicity and would be observed through a reduction in total cell count per well as compared with negative control wells. Alternatively, these compounds could be eliminated from future study if they lead to decreased autophagic flux in secondary assays.
Peroxisome biogenesis disorders
It is possible that compounds identified herein could eventually lead to therapeutics either for treatment of PBDs or other metabolic disorders. Independent genetically defined mutations affecting peroxisomes result in a range of phenotypes in humans from specific peroxisomal enzyme deficiencies to the complete absence of peroxisomes depending on the molecular lesion. In the most extreme cases, complete absence of peroxisomes results in death within the first year of life (severe Zellwegers), while many other PBDs result in neurodegenerative-like conditions later in life. For investigators modeling human PBDs in the mouse, compounds that inhibit peroxisome biogenesis could be used as chemical tools to provide an alternative to laborious, time-consuming mouse genetic knockouts to model disease. Mouse gene knockout models have been made for selected PEX genes, however, mouse knockouts without functioning peroxisomes die near birth16,19–22 making them a useful model only for the most severe PBDs. Compounds that inhibit peroxisome biogenesis that could be given at any time point – acutely or chronically – in the adult animal could be useful tools to model less severe forms of peroxisomal diseases and to elucidate normal contributions of peroxisomes for maintaining health and metabolism.
Other benefits of increased peroxisome biogenesis
In addition to fatty acid and VLCFA metabolism, peroxisomes are a major site of reactive oxygen species (ROS) inactivation and thus provide beneficial protection against free radicals. Peroxisomal catalase decomposes hydrogen peroxide while other peroxisomal enzymes metabolize harmful reactive oxygen species in the cell.23 Further, another function of the peroxisome is retroconversion of VLCFAs to docosahexaenoic acid (DHA) (found in fish oil), which may mediate numerous beneficial effects of very long chain n-3 (ω-3) fatty acids for treating cardiovascular disease including reduced mortality and myocardial infarction.24 Indeed, DHA synthesis is impaired in ZS fibroblasts lacking peroxisomes25 and DHA is also believed to have a required role in brain development and nervous system function.26 These other beneficial capacities of peroxisomes-increased free radical/reactive oxygen species inactivation and production of beneficial fatty acids (distinguish the fatty acid oxidation in peroxisomes from that of mitochondria). Although mitochondria are major sites of fatty acid oxidation, mitochondria also produce toxic entities through oxidative phosphorylation and are known to lead to the induction of apoptosis through the release of cytochrome c., which can lead to activation of capases and apoptosis. In this regard increasing fatty acid oxidation through peroxisomal rather than mitochondria biogenesis could in theory produced larger net benefits.
Although we hypothesize that novel compounds promoting peroxisomal activity independent of PPARs may provide treatments for metabolic disorders by preventing lipid accumulation in non-adipose tissue and decreasing fatty acids in plasma, these compounds need to be tested both in primary liver cells as well as in vivo in mice, including the high fat diet (HFD) diet induced obesity (DIO) model in mice. 4-phenylbutryate (PBA) is a PPAR-independent compound that functions as a positive control for peroxisome biogenesis in this high content assay and has been demonstrated to be therapeutic in diabetic/ obese (ob/ob and db/db) mice.11,12 Thus the roadmap for testing these compounds is well established. The underlying peroxisome biogenesis machinery is largely conserved.
Although we hope that non-PPAR peroxisome proliferators may have therapeutic value for MetS and T2D, there are other applications for small molecules that increase peroxisome biogenesis. These compounds could be used as chemical probes to identify their targets, perhaps elucidating new machinery regulating peroxisome formation not previously known. Yeast undergo massive peroxisome proliferation from single to hundreds of peroxisomes per cell when fatty acids are the sole nutrient source instead of sugars and thus have even greater proliferation than mammals. Forward genetic screens in yeast have indeed identified a series of mutations in conserved “PEX” (PEroXin) genes required for peroxisome biogenesis. The importance of this conserved machinery is evidenced by human mutations in 13 different conserved PEX genes that cause ZS, NALD, IRD or RCDP.27 Although most of the peroxisomal biogenesis genes are conserved from yeast to man (20 genes) the upstream transcriptional regulation of these genes cannot be conserved as yeast have no PPAR-like genes to regulate transcription. Thus, exploring peroxisome biogenesis in human cells should increase our understanding of peroxisome regulation in higher organisms, which for a subset of genes should not always be identical to yeast.
Conclusion
In summary, we have developed a genetically encoded peroxisomal reporter cell-line and a novel 3-channel high-content primary assay to identify compounds that increase cellular peroxisomal mass. We validated 4-phenylbutarate as a positive control for this assay and also demonstrated that this screen is capable of identifying potent novel chemical probes by the identification and validation of niclosamide, which is 1000 times more potent that PBA. From the production screen we observed a Z′ factor of 0.74 and have confirmed 10 compounds, representing four scaffolds that are three orders of magnitude higher potency than PBA. We intend to explore these structurally distinct novel scaffolds in mouse models of type II diabetes in further studies.
Acknowledgments
Support to J S and J B comes from NINDS grant # 1R21NS063858. We would also like to thank Sam Witherspoon for assistance with flow cytometry.
Footnotes
Disclosures
The authors report no conflict of interest in this work.
This is an Open Access article which permits unrestricted noncommercial use, provided the original work is properly cited.
References
- 1.Heymans HS, Schutgens RB, Tan R, et al. Severe plasmalogen deficiency in tissues of infants without peroxisomes (Zellweger syndrome) Nature. 1983;306(5938):69–70. doi: 10.1038/306069a0. [DOI] [PubMed] [Google Scholar]
- 2.Shimozawa N, Tsukamoto T, Suzuki Y, et al. A human gene responsible for Zellweger syndrome that affects peroxisome assembly. Science. 1992;255(5048):1132–1134. doi: 10.1126/science.1546315. [DOI] [PubMed] [Google Scholar]
- 3.Song S. The role of increased liver triglyceride content: a culprit of diabetic hyperglycaemia? Diabetes Metab Res Rev. 2002;18(1):5–12. doi: 10.1002/dmrr.260. [DOI] [PubMed] [Google Scholar]
- 4.Boden G. Fatty acid-induced inflammation and insulin resistance in skeletal muscle and liver. Curr Diab Rep. 2006;6(3):177–181. doi: 10.1007/s11892-006-0031-x. [DOI] [PubMed] [Google Scholar]
- 5.Reddy JK, Krishnakantha TP. Hepatic peroxisome proliferation: induction by two novel compounds structurally unrelated to clofibrate. Science. 1975;190(4216):787–789. doi: 10.1126/science.1198095. [DOI] [PubMed] [Google Scholar]
- 6.Fujita T, Sugiyama Y, Taketomi S, et al. Reduction of insulin resistance in obese and/or diabetic animals by 5-[4-(1-methylcyclohexylmethoxy) benzyl]-thiazolidine-2,4-dione (ADD-3878, U-63,287, ciglitazone), a new antidiabetic agent. Diabetes. 1983;32(9):804–810. doi: 10.2337/diab.32.9.804. [DOI] [PubMed] [Google Scholar]
- 7.Willson TM, Brown PJ, Sternbach DD, et al. The PPARs: from orphan receptors to drug discovery. J Med Chem. 2000;43(4):527–550. doi: 10.1021/jm990554g. [DOI] [PubMed] [Google Scholar]
- 8.Guerre-Millo M, Gervois P, Raspe E, et al. Peroxisome proliferator-activated receptor alpha activators improve insulin sensitivity and reduce adiposity. J Biol Chem. 2000;275(22):16638–16642. doi: 10.1074/jbc.275.22.16638. [DOI] [PubMed] [Google Scholar]
- 9.Clouet P, Henninger C, Niot I, et al. Short term treatment by fenofibrate enhances oxidative activities towards long-chain fatty acids in the liver of lean Zucker rats. Biochem Pharmacol. 1990;40(9):2137–2143. doi: 10.1016/0006-2952(90)90246-h. [DOI] [PubMed] [Google Scholar]
- 10.Chaput E, Saladin R, Silvestre M, et al. Fenofibrate and rosiglitazone lower serum triglycerides with opposing effects on body weight. Biochem Biophys Res Commun. 2000;271(2):445–450. doi: 10.1006/bbrc.2000.2647. [DOI] [PubMed] [Google Scholar]
- 11.Ozcan U, Yilmaz E, Ozcan L, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313(5790):1137–1140. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ozcan L, Ergin AS, Lu A, et al. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 2009;9(1):35–51. doi: 10.1016/j.cmet.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 13.Basseri S, Lhotak S, Sharma A, et al. The chemical chaperone 4-phenylbutyrate inhibits adipogenesis by modulating the unfolded protein response. The Journal of Lipid Research. 2009;50(12):2486–2501. doi: 10.1194/jlr.M900216-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gondcaille C, Depreter M, Fourcade S, et al. Phenylbutyrate up-regulates the adrenoleukodystrophy-related gene as a nonclassical peroxisome proliferator. J Cell Biol. 2005;169(1):93–104. doi: 10.1083/jcb.200501036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neuberger G, Maurer-Stroh S, Eisenhaber B, et al. Prediction of peroxisomal targeting signal 1 containing proteins from amino acid sequence. J Mol Biol. 2003;328(3):581–592. doi: 10.1016/s0022-2836(03)00319-x. [DOI] [PubMed] [Google Scholar]
- 16.Grabenbauer M, Sätzler K, Baumgart E, et al. Three-dimensional ultra-structural analysis of peroxisomes in HepG2 cells. Cell Biochemistry and Biophysics. 2000;32(1):37–49. doi: 10.1385/cbb:32:1-3:37. [DOI] [PubMed] [Google Scholar]
- 17.Kobayashi K, Inoguchi T, Maeda Y, et al. The lack of the C-terminal domain of adipose triglyceride lipase causes neutral lipid storage disease through impaired interactions with lipid droplets. J Clin Endocrinol Metab. 2008;93(7):2877–2884. doi: 10.1210/jc.2007-2247. [DOI] [PubMed] [Google Scholar]
- 18.Schrader M, Krieglstein K, Fahimi HD. Tubular peroxisomes in HepG2 cells: selective induction by growth factors and arachidonic acid. Eur J Cell Biol. 1998;75(2):87–96. doi: 10.1016/s0171-9335(98)80051-4. [DOI] [PubMed] [Google Scholar]
- 19.Baes M, Gressens P, Baumgart E, et al. A mouse model for Zellweger syndrome. Nature Genetics. 1997;17(1):49–57. doi: 10.1038/ng0997-49. [DOI] [PubMed] [Google Scholar]
- 20.Gray RH, de la Iglesia FA. Quantitative microscopy comparison of peroxisome proliferation by the lipid-regulating agent gemfibrozil in several species. Hepatology. 1984;4(3):520–530. doi: 10.1002/hep.1840040328. [DOI] [PubMed] [Google Scholar]
- 21.Hoivik DJ, Qualls CW, Jr, Mirabile RC, et al. Fibrates induce hepatic peroxisome and mitochondrial proliferation without overt evidence of cellular proliferation and oxidative stress in cynomolgus monkeys. Carcinogenesis. 2004;25(9):1757–1769. doi: 10.1093/carcin/bgh182. [DOI] [PubMed] [Google Scholar]
- 22.Srinivasan K, Ramarao P. Animal models in type 2 diabetes research: an overview. Indian J Med Res. 2007;125(3):451–472. [PubMed] [Google Scholar]
- 23.Schrader M, Fahimi HD. Mammalian peroxisomes and reactive oxygen species. Histochem Cell Biol. 2004;122(4):383–393. doi: 10.1007/s00418-004-0673-1. [DOI] [PubMed] [Google Scholar]
- 24.Wang C, Harris WS, Chung M, et al. n-3 Fatty acids from fish or fish-oil supplements, but not alpha-linolenic acid, benefit cardiovascular disease outcomes in primary- and secondary-prevention studies: a systematic review. Am J Clin Nutr. 2006;84(1):5–17. doi: 10.1093/ajcn/84.1.5. [DOI] [PubMed] [Google Scholar]
- 25.Ferdinandusse S, Denis S, Mooijer PA, et al. Identification of the peroxisomal beta-oxidation enzymes involved in the biosynthesis of docosahexaenoic acid. J Lipid Res. 2001;42(12):1987–1995. [PubMed] [Google Scholar]
- 26.Moore SA, Hurt E, Yoder E, et al. Docosahexaenoic acid synthesis in human skin fibroblasts involves peroxisomal retroconversion of tetracosahexaenoic acid. J Lipid Res. 1995;36(11):2433–2443. [PubMed] [Google Scholar]
- 27.Steinberg SJ, Dodt G, Raymond GV, et al. Peroxisome biogenesis disorders. Biochim Biophys Acta. 2006;1763(12):1733–1748. doi: 10.1016/j.bbamcr.2006.09.010. [DOI] [PubMed] [Google Scholar]