

Abstract
Importance of the field
Chemokine receptors are G protein-coupled receptors (GPCRs) most noted for their role in cell migration. However, inappropriate utilization or regulation of these receptors is implicated in many inflammatory diseases, cancer and HIV, making them important drug targets.
Areas covered in this review
Allostery, oligomerization, and ligand bias are presented as they pertain to chemokine receptors and their associated pathologies. Specific examples of each are described from the recent literature and their implications are discussed in terms of drug discovery efforts targeting chemokine receptors.
What the reader will gain
Insight into the expanding view of the multitude of pharmacological variables that need to be considered or that may be exploited in chemokine receptor drug discovery.
Take home message
Since 2007, two drugs targeting chemokine receptors have been approved by the FDA, Maraviroc for preventing HIV infection and Mozobil™ for hematopoietic stem cell mobilization. While these successes permit optimism for chemokine receptors as drug targets, only recently has the complexity of this system begun to be appreciated. The concepts of allosteric inhibitors, biased ligands and functional selectivity raise the possibility that drugs with precisely-defined properties can be developed. Other complexities such as receptor oligomerization and tissue-specific functional states of receptors also offer opportunities for increased target and response specificity, although it will be more challenging to translate these ideas into approved therapeutics compared to traditional approaches.
Keywords: G protein-coupled receptors, chemokine receptors, ligand bias, allosteric inhibitors, functional selectivity, oligomerization, drug discovery
Article Highlights.
Two drugs targeting the chemokine system have been approved and many more are currently in clinical trials.
In addition to orthosteric sites, allosteric binding sites have been identified in chemokine receptors, which increases their potential druggability, and encourages drug discovery efforts to go beyond traditional ligand displacement assays. Such drugs may be more effective in inhibiting the binding of large proteins, like chemokines, or selectively blocking specific signaling pathways while preserving others.
Ligand bias complicates drug discovery but also illustrates the potential of developing functionally selective drugs that minimize off-target effects.
Receptor homo- and hetero-oligomerization may affect drug responses in cells expressing multiple chemokine receptors and needs to be considered for better predictability of in vivo efficacy.
Exploiting the complex pharmacology of chemokine receptors, it should be possible to taylor therapeutics with unique properties such as the ability to inhibit ligand binding and specific signaling pathways coupled with the ability to internalize receptor and prevent recycling.
1. Introduction
Chemokine receptors are an important subfamily of class A G Protein-Coupled Receptors (GPCRs) that function as critical mediators of cell migration during development, immune surveillance, and inflammation associated with protection from pathogens [1–5]. In this context, the ligands act as traffic signals to guide the migration of receptor bearing cells, and function by inducing conformational changes in the receptors that trigger intracellular signaling pathways involved in cell movement and activation [1]. Four major families have been classified (CC, CXC, CX3C and XC) according to the pattern of cysteine residues in the ligands, which are small 70–120 amino acid proteins [6,7]. To date, approximately 44 ligands and 22 receptors have been identified in humans alone [8], and in the past decade, over half of these receptors have been targeted in clinical trials [9–12].
The most validated disease indication involving chemokine receptors is AIDS where HIV enters leukocytes by exploiting two primary receptors, CXCR4 and CCR5, along with the HIV surface glycoprotein gp120 and the host receptor CD4. The observation that chemokines block viral entry suggested the concept of developing entry inhibitors for the treatment of AIDS, which has now been validated with an FDA approved drug [13]. Interestingly, Plasmodium Vivax the most widespread of the four human malaria species, enters red blood cells via the chemokine receptor DARC (Duffy Antigen Receptor for Chemokines) and can also be blocked by chemokines [14]. Thus similar entry inhibitors could provide viable treatments for malaria where new drugs are desperately needed [15]. Many inflammatory diseases, including rheumatoid arthritis, multiple sclerosis, Crohn’s disease, transplant rejection and atherosclerosis are more related to the deregulation of the chemokine system where uncontrolled cell migration and activation can lead to collateral cell damage. Thus inhibitors of many inflammatory chemokine receptors have been extensively pursued. Additionally the migratory functions of chemokines/receptors have also been shown to be exploited by tumor cells to promote metastasis as well as angiogenesis of endothelial precursors to supply the tumor cells with nutrients, oxygen and other growth promoting factors [16–18]. More recently identified functions of chemokines/receptors in cancer include their ability to promote growth and survival signaling of solid tumors and hematological malignancies [19]. Overall, given their extraordinary association with disease, the chemokine system seems to be a fertile territory for drug discovery.
Recent reviews have extensively described the successes, failures and current status of drug discovery efforts targeting chemokine receptors primarily for HIV and inflammatory diseases. The approaches have predominantly involved small molecule inhibitors although monoclonal antibodies and antagonist peptide analogs are also in clinical trials [9–12,20]. These reviews have discussed the motivation and validation for targeting specific chemokine receptors, challenges such as cross-reactivity with other GPCRs and the lack of cross-species reactivity which is problematic for pre-clinical animal models, and the failure of some compounds due to differences in biology between rodents and humans [9–12]. By contrast, with less consideration of specific receptors and pathologies, the purpose of this review is to describe some emerging concepts that may improve traditional approaches of directly targeting receptors; these concepts include allosteric inhibitors and biased ligands, as well as complexities in receptor pharmacology that may need to be taken into account or that could be exploited such as receptor oligomerization and internalization, respectively.
2. Strategies for Small Molecule Inhibition of Chemokine Receptors: Orthosteric vs Allosteric Inhibitors
Finding small molecules that target GPCRs has been the bread and butter of the pharmaceutical industry. Indeed, roughly 30% of FDA-approved small molecule drugs are thought to target GPCRs [21]. The first small molecule chemokine receptor antagonist to enter clinical trials was Berlex Bioscience’s CCR1 inhibitor, BX471, in 2000 [22]. Unfortunately, BX471 failed to meet minimum efficacy set-points in a phase II trial for multiple sclerosis and was discontinued [23]. Currently, over 30 compounds targeting chemokine receptors are in various stages of clinical trial and two have been approved by the FDA to date [12]. The first, approved in 2007 to fight HIV infection, was the CCR5-specific antagonist Selzentry® (Maraviroc, Pfizer), which has the ability to block R5-tropic HIV infection [13]. The second, Mozobil™ (AMD3100, Genzyme, Cambridge, USA), targets CXCR4 and was approved in 2008 for hematopoietic stem cell mobilization after previously failing as an HIV inhibitor due to low oral bioavailability and cardiovascular effects [24,25]. Currently, one of the more promising candidates is Chemocentryx/GlaxoSmithKline’s GSK-1605786 (CCX-282; Traficet-EN™), a CCR9 antagonist for the potential treatment of inflammatory bowel disease, including Crohn’s and celiac disease. A recently completed phase II/III trial of GSK-1605786 demonstrated evidence of clinical efficacy in patients with moderate to severe Crohn’s disease [26].
Initially, most lead compounds targeting chemokine receptors were thought to be orthosteric ligands. Orthosteric compounds by definition bind to the same site as the natural ligand, thus competitively and sterically blocking the interaction (Figure 1A, right). Certainly, the use of competitive inhibition of radiolabeled natural ligand as the initial screen for compound binding would likely bias the discovery of orthosteric small molecule competitors, over noncompetitive allosteric ligands. Allosteric ligands have non-overlapping binding sites with the natural ligand and because they may affect signaling via direct modulation of receptor conformation, do not necessarily need to displace the natural ligand (Figure 1B, middle and right). Interestingly, in more recent assessments many compounds have been reclassified as allosteric based on the fact that they possess “probe dependence”, a feature of allosteric behavior observed as differing effects on the binding kinetics, affinity, saturability, or signaling efficacy of different ligands that target the same receptor. Such compounds may have advantages, both in terms of inhibiting the binding of large protein ligands, like chemokines, as well as providing more selectivity in signaling inhibition, as discussed below.
Figure 1.
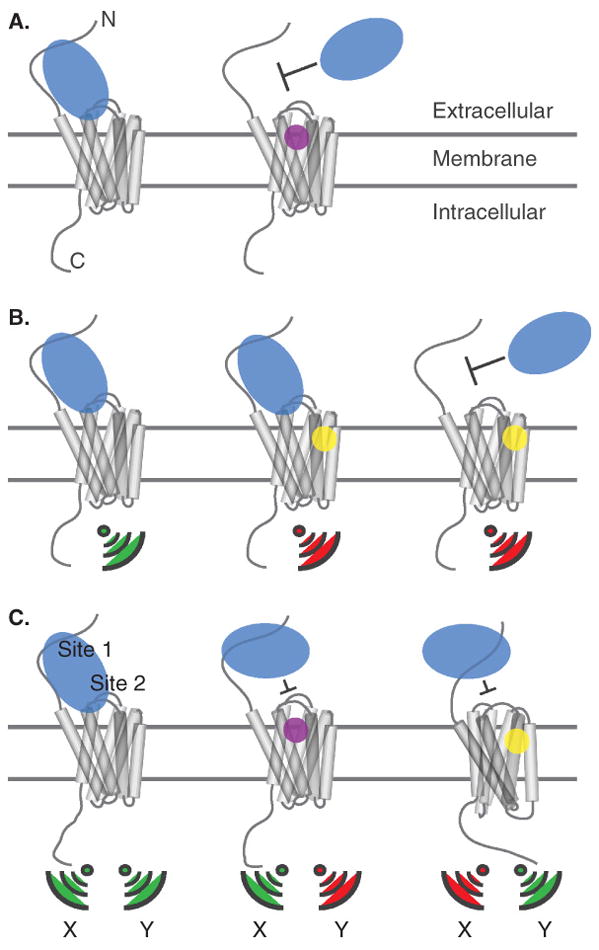
Cartoon depicting various scenarios for the inhibition of chemokine binding and signaling by small molecules. The N- and C-termini of the receptor are labeled. Chemokine ligands are shown in blue, receptors in grey, orthosteric antagonists in purple, and allosteric antagonists in yellow. Green signal icons below receptor indicate activated signaling, red signal icons indicate blocked signaling. Panel A depicts orthosteric inhibition of chemokine binding. Panel B shows allosteric inhibition of signaling with and without concomitant blocking of chemokine binding. Panel C depicts partial blockage of the chemokine:receptor interaction by orthosteric and allosteric compounds. Sites 1 and 2 in the two-site model are labeled. Different generic signaling profiles highlight the idea that functional selectivity may be observed with both orthosteric and allosteric molecules.
Unlike most class A GPCRs whose cognate ligands are small molecules or short peptides, chemokine receptors are regulated by globular protein ligands, and as such may present difficulties for blocking with small molecules. Additionally, the interaction between the HIV protein gp120 and the chemokine coreceptors CCR5 and CXCR4 involves a relatively large protein:protein interface. As a way of circumventing this issue, it has been suggested that small molecules acting as allosteric modulators could inhibit chemokine or gp120 binding, by producing sufficiently large conformational changes in the receptor, such that the ligand sees a completely different and incompatible binding surface [27].
In contrast to the situation with AIDS where complete blockade of HIV gp120 binding to CCR5/CXCR4 is the goal, for many if not most cases of targeting chemokine receptors for treatment of inflammatory or other diseases, modulation of signaling is an acceptable and sometimes preferable outcome, and inhibition of chemokine-induced functions by complete displacement may not be necessary (Figure 1B, middle). In this vein, Kenakin and coworkers have shown that the compound aplaviroc (873140) is an allosteric noncompetitive CCR5 inhibitor of CCL5/RANTES that modulates signaling but not binding [28]. These researchers also conducted co-administration binding assays, which suggested that four other previously characterized CCR5 antagonists bind to a common allosteric site (see section 3). Moreover, the CXCR1 antagonist repertaxin was described as an allosteric ligand that does not block CXCL8 binding but potently inhibits human neutrophil migration [29], and the CCR1 antagonist BX471, mentioned above, was later labeled as allosteric based on the fact that an iodinated analogue could not be displaced by CCL3, CCL5, or CCL7 [30]. In light of these and other studies [28], it seems possible that allosteric antagonism of chemokine receptors by small molecule compounds may be the norm rather than the exception. This observation underscores the importance of the use of appropriate functional assays as opposed to binding displacement assays in primary screens for discovering inhibitors.
However, the distinction between allostery and orthostery may not be so straightforward. Mutational as well as NMR studies indicate that the chemokine:receptor binding event involves at least two discrete interaction sites on the chemokine and the receptor [31–33]. Accordingly, a two-site binding model has been proposed involving a “site one” interaction between the receptor N-terminus (and possibly the extracellular loops) and the core globular domain of the chemokine, as well as a “site two” interaction between the chemokine N-terminal signaling domain and the transmembrane domains and extracellular loops of the receptor (Figure 1C, left) [31,33]. One implication of this model is that orthosteric small molecule interference at one site may not preclude binding to the other site, but could still modulate signaling. As an example where this behavior is observed, Shimada and coworkers showed by NMR that Mozobil could displace the N-terminus of CXCL12/SDF-1 from binding CXCR4, presumably within the site two helical bundle, without displacing the core domain binding interaction at site one [34]. We note that this scenario provides an alternate mechanism to explain the incomplete displacement of the natural ligand by small molecules; although a behavior normally ascribed to allosteric inhibitors, incomplete displacement can in principle occur with orthosteric ligands (Figure 1C, middle). In general, the presence of parallel orthosteric sites as well as nearby or partially overlapping allosteric sites may blur the line between purely orthosteric and allosteric mechanisms of action in some cases. Future studies will certainly add resolution to these binding events and provide insight into how chemokines activate various receptor functions, information that will be useful for therapeutic strategies.
In another interesting new twist, allosteric inhibition of chemokine receptors by small molecules acting at a novel intracellular site has recently been reported for CCR4/CCR5 [35] and CXCR1/CXCR2 [36]. Both of the publications describe chimeric constructs of the receptor pair studied, which led to changes in the small molecule inhibition of chemokine-induced calcium flux compared to the WT receptors. These data suggested a new, uncharacterized binding site which was determined to be much deeper in the transmembrane domain and bounded on one side by the C-terminal tail of the receptor. As further evidence to support this claim, both papers note that intracellular access was required for full effect, a phenomenon not seen for ligands binding at the better known extracellular site. In a more recent follow up study, Salchow et al. used more extensive mutational analysis to better delineate the location of this site and characterize the interaction of three CXCR2 antagonists, which are shown to likely bind to this cytoplasmic pocket [37]. As far as we know these are the first publications to delineate a small molecule binding pocket within the intracellular region of a chemokine receptor. While this site also clearly displays allosteric behavior, being far removed from the endogenous chemokine binding site and blocking signaling without affecting chemokine binding, the question remains whether the blockade occurs through conformational constraint of the receptor, or by blockage of G protein coupling or other interactions with proteins at the intracellular interface. Further discussion of intracellular modulation is presented below.
3. Biased Agonism/Antagonism for Signaling Selectivity; Evidence from Chemokine-Mediated Receptor Responses
One aspect of GPCR signaling that has recently become an area of intense interest, particularly for drug discovery, is that of biased agonism/antagonism, also known as functional selectivity. Here, the “bias” or “selectivity” refers to the ability of different ligands that bind the same receptor to elicit differential signaling responses. And in fact, chemokine receptors and their ligands display a high degree of redundancy in that many receptors bind multiple ligands and vice versa. Critics argue that this redundancy may be therapeutically problematic in that blocking a single receptor may never produce clinical efficacy due to the presence of overlapping functions of multiple receptors. However, there is mounting evidence that in at least some cases, different ligands for the same receptor elicit different signaling response to fine-tune the biological readout (Figure 2). The idea then with respect to drug discovery is that it may be possible to selectively inhibit certain signaling pathways while sparing others, thereby avoiding unwanted effects. While most examples illustrating differential signaling of receptors are in response to endogenous or modified chemokine ligands, in principle, similar control of receptor signaling could be commandeered by small molecules as has been demonstrated for other GPCRs [38].
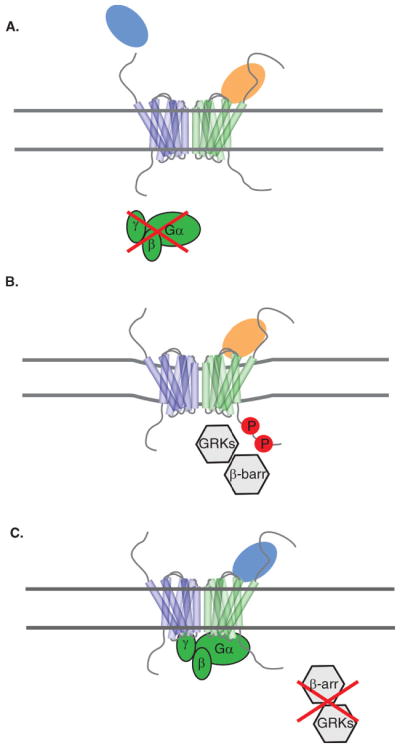
Figure 2.

Potential scenarios of functional selectivity of chemokine receptor signaling. Panel A depicts a scenario for ligand bias in which one ligand may induce typical G-protein activation and β-arrestin recruitment leading to receptor desensitization and internalization while another ligand for the same receptor activates G-proteins but does not recruit β-arrestins or lead to receptor internalization. Alternatively, another ligand for the receptor could activate β-arrestins, GRKs and receptor internalization but not G-protein signaling as shown in Panel B. It is also possible that two different ligands may similarly activate G-protein and β-arrestin pathways, but then the trafficking of the receptor could differ. For example, as depicted in Panel C, the receptor could be targeted to the lysosome for degradation or recycled to the cell surface through recycling endosomes. Internalization is depicted as a slight indentation of the membrane, and the three panels are meant to be independent scenarios.
Chemokine receptors transduce many different types of signals including those for calcium mobilization, inhibition of adenylyl cyclase and activation of kinase cascades (e.g. Raf/MEK/ERK1/2) that mediate migration, proliferation and cell survival responses. These signaling responses and their regulation through receptor desensitization, internalization, and recycling or degradation are modulated by interactions of specific proteins with the intracellular loops and C-termini of the receptors. The key modulators include G proteins, G protein-coupled receptor kinases (GRKs), and β-arrestins [39]. Thus despite the redundancy within the chemokine network with respect to ligand-receptor binding, there are numerous mechanisms in place for unique signaling properties, or ligand bias. While the structural basis for such ligand bias is largely unknown, it is likely that slight differences in receptor conformation accommodate or block interactions with certain intracellular modulators [40–44] (Figure 2).
Some well established evidence for ligand bias within the chemokine network involves the CCR7 ligands, CCL19 and CCL21. Both CCL19 and CCL21 bind CCR7 with similar affinities, induce signaling through Gi, and have similar efficacies in terms of inducing calcium flux and cell migration [45]. However, CCL19 but not CCL21 effectively internalizes CCR7 and induces strong phosphorylation of the C-terminus of the receptor (similar to Figure 2A) [45–47]. Recently, it was shown that CCL19 was unique in engaging GRK3, which could possibly account for some of the differences in the receptor responses to these two ligands [47]. The role of β-arrestin2 (arrestin 3) in regulating CCL19 but not CCL21 endocytosis and migration has also been proposed, suggesting the possibility for distinct interactions with intracellular modulators depending on the ligand [48]. As CCL19 and CCL21 are endogenous ligands that can compete with one another for CCR7 binding [49], these results suggest that functional selectivity can be achieved through orthosteric modulation of receptor conformation, not just allosteric modulation (Figure 1C, middle and right panel).
Another example of functional selectivity within the chemokine network involves the decoy receptor D6, which is believed to be important for dampening the immune response by scavenging a variety of inflammatory chemokines. D6 binds multiple chemokines, one of which is CCL14. CCL14 can be proteolyzed to forms that are either active (9–74) or inactive (1–74 and 11–74) for CCR1/CCR3/CCR5 signaling. Interestingly, D6 has the ability to bind all of forms of CCL14, but selectively trafficks only the 9–74 active form into intracellular degradation pathways [50]. Additionally, extreme examples of orthosteric chemokines with biased signaling are single point mutants of MCP-1/CCL2 which have variable effects on inhibition of adenylyl cyclase, calcium mobilization and chemotaxis [51] and the CCL5/RANTES analogues discussed below.
The ability to bias signaling responses is clearly attractive for the development of therapeutic targets, whereby one could selectively target signaling events that may be contributing to disease while preserving other functions, and thereby reduce some of the negative off-target effects of therapeutics. One of the early discoveries in the chemokine field demonstrating this potential is aminooxypentane (AOP)-RANTES, a chemically modified form of CCL5/RANTES that functions as a CCR5 biased agonist/HIV inhibitor [52,53]. Unlike classic receptor antagonists that compete for endogenous chemokine binding without promoting receptor activation, AOP-RANTES was found to maintain many agonist functions including calcium mobilization, chemotaxis and receptor internalization. However, upon internalization, it inhibited recycling of the receptor to the cell surface in contrast to wild type (WT) RANTES. Thus the potent anti-HIV activity of AOP-RANTES (for R5 tropic HIV strains) has been largely attributed to its internalization and recycling properties which promote overall low CCR5 surface expression [53]. This effect is consistent with the observations that high levels of endogenous CCR5 chemokines can suppress HIV infection and that elevated gene copy number of CCL3L1 correlates with lower susceptibility to HIV [54,55]. Interestingly, like CCL5/RANTES which binds CCR1, CCR3 and CCR5, AOP-RANTES can bind and elicit chemotaxis and calcium flux responses through these other receptors as well [56]. However, unlike the effects of AOP-RANTES on inhibiting CCR5 recycling in eosinophils, it actually promotes CCR1 recycling and does not alter CCR3 down-modulation or recycling, suggesting that functional selectivity for different receptors can also be achieved [57].
Taking the AOP-RANTES concept one step further, Hartley and coworkers attempted to engineer a RANTES analogue that had antagonist activity rather than agonist activity, while retaining high affinity for CCR5 and the ability to internalize the receptor [58]. The underlying premise was that for entry inhibitors, it may be preferable to have a chemokine analogue that doesn’t promote cell migration and other Gi-linked activation processes in order to avoid local inflammation. Using phage display, they successfully produced the recombinant variant, 5P14-RANTES, which showed no detectable G protein activation upon binding, but had a strong propensity to internalize, indicating that it is indeed possible to identify molecules with this fairly unique combination of biased properties.
In addition to chemokine ligands, numerous small molecule antagonists of CCR5 that are allosteric modulators exhibit functional selectivity [59]. Allosteric antagonists can exhibit functional selectivity by directly inducing conformational changes in the receptor that may block interactions with or functions of select intracellular modulators [60–63]. Alternatively, functional selectivity may also refer to the ability of different small molecules that bind the same receptor to display differential inhibitory potencies in blocking the functions induced by natural ligands [60]. As an example of the latter, a compound may block function X with nanomolar potency and function Y with micromolar potency, thus defining a window of selectivity in which this compound may be used to selectively block function X. In a follow up to the aplaviroc:CCR5 binding study mentioned in section 2, Kenakin and coworkers showed that a series of small molecule CCR5 inhibitors including aplaviroc display functional selectivity, as evidenced by the differential inhibitory potencies of the compounds with respect to gp120 mediated HIV infection and CCL3L1-induced CCR5 internalization [64]. This finding led to the idea (most strikingly demonstrated by the compound TAK652) that small molecules are capable of blocking HIV infection through CCR5, while allowing the receptor to remain sensitive to native chemokine induced functions. One can imagine a number of ways in which this situation may be beneficial. For instance, allowing the receptor to function normally during treatment may be important for maintaining strong immune system function, another important requirement for combating HIV. Yet another possibility is that compounds that specifically allow chemokine-induced internalization of CCR5 could actually produce enhanced clinical efficacy, as receptor internalization has been shown to be an effective means of protecting against HIV entry (see AOP-RANTES discussion above). These ideas are fairly new, and as mentioned in the Kenakin article, it remains to be seen if the ability to inhibit specific functions proves to be therapeutically useful, but the idea of sparing normal receptor function while inhibiting HIV infection is intriguing to say the least.
The prevalence of biased agonism emphasizes the limitations of identifying antagonists simply based on binding properties. To gain a more complete understanding of agonist and antagonist functions, it is important to perform a range of assays to examine the effects on G protein and β-arrestin dependent pathways as well as potential alterations in receptor trafficking and processing (recycling/degradation) (Figure 2). As mentioned above with the AOP-RANTES HIV inhibitor, antagonists that cross react with multiple receptors may still provide selectivity for the receptor of interest in terms of function, again underscoring the importance of functional assays to complement binding studies. Additionally, as chemokines can often bind to multiple receptors, the concept of biased receptors, in addition to biased ligands, should be mentioned. Biased receptors refer to a situation in which the same ligand binding to different receptors elicits different responses [65]. This idea of receptor bias may be an important consideration, particularly in the context of cross regulation and receptor heterodimerization, as discussed below.
4. Important Considerations for Drug Efficacy: Homo- and Hetero-Oligomerization of Chemokine Receptors
The above sections describe some emerging advances in understanding the pharmacological nuances of drug action involving a single receptor. When one adds in the ability of receptors to affect one another though oligomerization, it becomes clear that one must consider the in vivo state of the receptor and its interaction network when interpreting the pharmacological effects of compounds. It is now widely accepted that chemokine receptors can constitutively form functional hetero- and homo- oligomers, and that such complexes can profoundly affect the pharmacological properties of the constituent receptors. Recently, a major focus of study has involved the identification of the functional repercussions of chemokine receptor oligomer formation, with an emphasis on agonist/antagonist ligand binding, signaling and receptor trafficking. Multiple mechanisms for cross regulation of chemokine receptors have been reported, including ligand mediated-transinhibition or transactivation, ligand sequestration, as well as modulation of receptor desensitization and internalization [66–69]. Here we cite several examples of chemokine receptor homo- and hetero-oligomerization to highlight the effects that oligomerization can have on modulating chemokine receptor activity (Figure 3), as there are corresponding implications for responses to drugs.
Figure 3.
Potential effects of hetero-oligomerization on chemokine receptor signaling responses. As depicted in Panel A, binding of a ligand specific for one receptor in a heterodimer could prevent the binding of ligands to the other receptor and/or alter its ability to interact with/signal through intracellular modulators, such as G-proteins. Another potential effect of hetero-oligomerization or receptor cross-desensitization is the internalization of one receptor (purple) through ligand activation of another receptor (green) as represented in Panel B. Here, internalization is depicted as a slight indentation of the membrane. Alternatively, hetero-oligomerization of a ligand activated receptor (green) with another receptor (purple) could prevent desensitization of the activated receptor (green) in the complex (Panel C). This is effectively the scenario described in WHIM syndrome where heterodimers occur between WT and C-terminally truncated forms of CXCR4.
The identification of the CCR5Δ32 allelic truncation variant present in a small population of HIV-1 resistant individuals [52,70,71] first shed light on the biological impact that chemokine receptor oligomerization can have on disease phenotype. In homozygous CCR5Δ32 individuals, resistance is conferred by the inability of the mutant receptor to reach the cell surface and facilitate HIV entry as a result of its retention in the endoplasmic reticulum (ER). Furthermore, heterozygous CCR5Δ32 individuals are partially protected and progress more slowly to AIDS when infected [72,73], despite the fact that they produce WT CCR5, an observation which is thought to be due to ER retention of WT CCR5 through dimerization with CCR5Δ32 [74]. In the case of WHIM (Warts, Hypogammaglobulinemia, Infections, and Myelokathexis) syndrome, oligomerization is also thought to result in aberrant function of the WT CXCR4 receptor. WHIM syndrome is a rare genetic immunodeficiency disorder that results in the expression of C-terminal truncation mutants of CXCR4. These receptors show enhanced signaling responses to CXCL12/SDF-1 as a consequence of their reduced ability to undergo ligand-mediated receptor desensitization and internalization [66,75]. As WHIM syndrome is primarily a heterozygous disease, constitutive heterodimerization of WT and mutant CXCR4 has been proposed as a mechanism by which WHIM CXCR4 can retain WT CXCR4 on the cell surface, leading to similarly enhanced CXCL12-induced responses [66,76] (Figure 3C).
In addition to effects on receptor trafficking as the above examples illustrate, heterodimerization can influence ligand-mediated signaling responses of the involved receptors by affecting ligand binding, G-protein coupling or association with other signaling modulators. Since the discovery of the novel chemokine receptor, CXCR7 in 2005 [77], and the finding that CXCL12 is a high affinity ligand for both CXCR4 and CXCR7, several groups have demonstrated that these receptors can form constitutive heterodimers [78–80]. The functional role of the CXCR4:CXCR7 heterodimer is particularly interesting as both receptors are linked to a number of cancers, and little is known regarding how heterodimerization modulates the ligand mediated activity of each receptor in the hetero complex compared to their function alone. In addition to CXCL12, CXCR7 also has a second ligand, CXCL11, though neither triggers Gi mediated signaling of CXCR7, which has been attributed to its lack of the classic “DRYLAIV” motif important for G protein coupling [79,81]. However, Zabel et al. and others [67,82] have shown that although CXCR7 does not couple to Gi, stimulation by CXCL11 or CXCL12 readily induces association with β-arrestin2, suggesting unique signaling properties of this receptor. Zabel et al. recently probed the role of CXCR7 in tumor cell transendothelial migration (TEM) using the CXCR7 specific small molecule antagonist, CCX771, as well as its natural protein ligand, CXCL11 [83]. Interestingly, CXCR7 was critical for CXCL12-mediated CXCR4 TEM of CXCR4+/CXCR7+ bearing NC-37 tumor cells, though it did not impact in vitro “bare filter” cell migration. Furthermore, CXCR4-dependent TEM of these cells was potently antagonized by the CXCR7 ligands CCX771 and CXCL11. Notably, CCX771 does not alter the binding affinity of CXCL12 for CXCR4. Nevertheless, it is approximately 20-fold more potent at inhibiting CXCL12-mediated TEM compared to Mozobil, a direct and specific CXCR4 small molecule antagonist. Surprisingly, the antagonist, CCX771, also triggers β-arrestin2 association and with greater potency than CXCL12 [83]. Together these results show several mechanisms by which ligand engagement of CXCR7 is transmitted into the functional regulation of CXCR4, a finding which has important implications for the effectiveness of drugs targeted against the individual receptors.
In yet another level of complexity, there is emerging evidence that chemokine receptors can exist not only as homo- and hetero- dimers, but also as higher order oligomeric complexes [68]. Building on earlier findings that CCR2, CCR5 and CXCR4 form functional (homodimers) and heterodimers [84–88], Sohy et al. recently demonstrated the presence of CCR2, CCR5, and CXCR4 hetero-oligomeric complexes recombinantly expressed in HEK293 cells, through a combination of luciferase complementation and bioluminescence resonance energy transfer (BRET) assays. Importantly, they also found these hetero-complexes endogenously expressed in primary leukocytes through radioligand binding competition assays using receptor-specific chemokines, blocking antibodies, and small molecule antagonists [68]. Furthermore, addition of CCR2, CCR5 or CXCR4 receptor specific antagonists (e.g. TAK-779 and Mozobil) in cells expressing all three receptors resulted in transinhibition of chemokine binding to the other receptors in the oligomeric complex demonstrating their functional interaction in leukocytes. As for CXCR4:CXCR7 oligomers, this example further illustrates that it is important to consider that an antagonist directed at one receptor can potentially inhibit the function of other receptors in the same cell. Depending on the biological context, transinhibition could prove to be beneficial (e.g. by inhibiting the function of other receptors expressed in an inflammatory setting) or alternatively, transinhibition could lead to unfavorable effects with unwanted inhibition of other receptors in the complex that are needed to support normal tissue function. While the focus here has been exclusively on chemokine receptor hetero- and homo- oligomerization, there are also examples of chemokine receptors oligomerizing with non-chemokine receptors such as the T cell receptor, opioid receptors (delta and kappa) and glycoproteins (e.g. CD26, CD4), or chemokine receptor crosstalk with other signaling molecules (reviewed in [89]) that should also be considered.
Obviously without the ability to study specific receptors in the oligomerization states as they naturally exist in target tissues, it will be difficult to predict how the efficacy of inhibitors developed against individual receptors in vitro will be translated in vivo. On the otherhand, the existence of tissue-specific organizations of receptors alone or in complex with other receptors might enable the development of drugs that target unique receptor complexes.
5. Expert Opinion
Previous reviews have highlighted some of the major challenges in targeting the chemokine receptor network for therapeutic applications citing examples of specific compounds, their receptor targets and the relevant disease indications [9,12]. These challenges include (i) lack of efficacy due to poor pharmacokinetics such as binding to serum components, (ii) lack of efficacy due to the inappropriateness of the disease models, and of course (iii) off target effects and toxicity. While these problems are likely relevant to all GPCRs, more unique to the chemokine network is the aforementioned problem of redundancy where most receptors have multiple ligands and many ligands interact with multiple receptors. This issue has often been invoked as one of the reasons that chemokine receptors may prove to be more difficult targets than other GPCRs. For example, CCR1 has at least 8 ligands, many of which also bind other chemokine receptors with similar affinities. As a result, redundancy in receptor-ligand pairing presents a general challenge in identifying appropriate targets for specific disease indications based on elevated ligand or receptor expression levels [9,12]; given an up-regulated ligand, what receptor is relevant? Functional redundancy could also provide an explanation for poor efficacy results where blocking one ligand/receptor is not sufficient to achieve the expected endpoint, or alternatively where it produces undesired side effects. “Polypharmacology” where one develops compounds that inhibit multiple receptors has been suggested as a solution to this problem [9–12], and in fact several compounds with dual specificity have been reported [90–93]. Independent of the redundancy issue in the chemokine network, this approach may be important in complex diseases where more than one receptor is likely relevant such as multiple sclerosis and rheumatoid arthritis [9,12].
On the otherhand, as described in this review, there is growing evidence for ligand and receptor bias (instead of redundancy) which could be exploited therapeutically to obtain better selectivity; for example, if only one of multiple receptor interactions with a ligand were blocked, side effects might be avoided. Along these lines, we have been able to use phage display to select antagonists based on the CXCL12 scaffold, that are specific for CXCR4 but have poor affinity for its other receptor, CXCR7, as well as antagonists with dual specificity (polypharmacology) by appropriate panning schedules against both receptors (Handel et al, unpublished results). Conversely, inhibiting only one ligand of a receptor could have therapeutic advantages, and here the example of compounds that block HIV with preservation of chemokine function was discussed in Section 3. This scenario could, in principle, allow for direct HIV blockade coupled with the added benefit of allowing chemokine-induced receptor internalization (also favorable for HIV inhibition, while preserving the desired immunity associated effects of the ligand [64]. In reality, it is likely that there will be situations where redundancy is problematic and polypharmacology is needed, and other cases where it can be exploited. To this end, a better understanding of the underlying biology of different diseases will be required. Additionally the recent successes since 2007 in determining the crystal structures of GPCRs, including CXCR4 in complex with a small molecule and peptide inhibitor of HIV entry (Figure 4) [94], will certainly facilitate the structure-based design and fine tuning of molecules with promiscuous or functionally selective effects.
Figure 4.

Structure of CXCR4 bound to a small molecule isothiourea compound [94]. Panel A illustrates a side view of the receptor (grey) with the isothiourea compound shown as a cpk model. Panel B shows the binding pocket which is formed primarily by polar residues from transmembrane (TM) helices 1, 2, 3, and 7 and extracellular loop 2 (ECL2), with the ligand being stabilized by a network of hydrogen bonds, including the critical hydrogen bonding interactions with E288 (7.39 in Ballesteros notation) and D97 (2.63).
Another well-appreciated challenge with the chemokine network is the lack of cross-species reactivity (binding) with receptors which complicates testing of drugs screened against human receptors in rodent models due to the differences in the respective immune systems [9,12]. Whether this is more challenging with chemokine receptors relative to other GPCRs is currently unclear. However binding cross reactivity with human and rodent receptors is likely just the tip of the iceberg with respect to cross species challenges, as their inherent biology may also differ significantly. Examples include species differences in chemokine/receptor expression patterns making the relevance of the receptor in the rodent model possibly irrelevant to the human disease indication. Other differences, discussed below, could include how specific receptors are coupled into downstream signaling pathways, and functional effects of the downstream mediators. Obviously, the only solution to this problem is better understanding of the disease and improvements in disease models, a challenging endeavor and a topic outside the scope of this review.
Nevertheless, better in vitro pre-clinical strategies that take into account the complexities of the biology, even just within humans, may increase the chance of a drug having the desired outcome and reaching the clinic. Receptor oligomerization is probably the most well-studied complexity with regard to the development of drugs targeting chemokine receptors and other GPCRs. Transinhibition as a consequence of hetero-oligomerization, previously discussed in Section 4, could be detrimental for the desired responses to drugs, or it could be advantageous with respect to polypharmacology. However, one can imagine many other aspects of receptor pharmacology that also deserve consideration. The environment, and in particular the cell type, may have a profound influence on the effectiveness of drugs. Although considerable drug screening and information regarding chemokine receptor function has been obtained from HEK293 cells and other non-native over-expression systems, receptors in these cells may behave quite differently from receptors in their natural in vivo environment. For one, it is established that different cells express varying levels of intracellular modulators of receptor function including β-arrestins, G-proteins, and GRKs [95,96]. Thus some signaling “biases” may be inherent to the cell type. Theoretically, if the cell type being targeted expresses high levels of β-arrestins but low levels of G-proteins, selective inhibition of G-protein function might be much more readily achieved, and vice versa, but the same may not true of the receptor in the natural cellular environment. Also, the over-expression of receptors in non-native cell types may influence the interaction of the receptor under study with intracellular modulators. For example, G proteins that would not normally couple to receptors in their native environment, might be recruited in the presence of excess receptor, altering down stream signaling and possibly even ligand affinity. Even the lipid environment is known to affect signaling [97,98]. Thus it is not surprising that receptors can show different responses to ligands in different cell types, especially in the context of normal versus disease states. Along these lines, we recently showed that chronic lymphocytic leukemia cells (CLL) B cells migrate poorly to CXCL12 compared to normal B cells despite the fact that CXCR4 levels are higher in the CLL cells. Instead, CXCL12/CXCR4 signaling may be more directed towards cell survival in CLL [99]. Similarly, a P2G variant of CXCL12 antagonizes lymphocyte migration, ligand binding, and calcium mobilization of lymphocytes in vitro, and inhibits T cell-dependent immune responses in vivo, but it has no effect on CXCL12-induced calcium mobilization or migration of MDA-MB-231 or MDA-MB-361 cancer cells (Shaun McColl, University of Adelaide, personal communications). Again, the bad news is that all of these issues can complicate the drug discovery process, but the good news is that in some cases there may be specificity built into the cells one wishes to target, like cancer cells, by virtue of the receptor microenvironment. Thus compounds developed from pre-clinical in vitro studies that target receptors in their natural cellular environment may show the most efficacious and specific effects as they progress through the various stages of drug discovery.
There are other complexities of chemokine biology that are also noteworthy, and can either present challenges or can be exploited. In addition to their G protein-coupled receptors, many chemokines require interactions with glycosaminoglycans (GAGs) for function in vivo [100]. For one, GAG-binding appears to allow immobilization, accumulation and retention of chemokines on cell surfaces near their sites of production in order to provide directional signals to migrating cells. Additionally, GAG interactions are involved in the transport of chemokines across cell surfaces, and they may even have roles in signaling [101]. The importance of these interactions has been demonstrated with GAG-binding deficient mutants that are fully capable of promoting cell migration across bare filters in vitro, indicating an intact ability to activate their receptors; however when tested in vivo such mutants are not capable of promoting migration [100,102–107]. Furthermore, these mutants have anti-inflammatory effects in vivo. While the complete mechanisms underlying their anti-inflammatory effects is not fully understood, a major component appears to be the systemic distribution of the chemokine mutants due to their inability to bind GAGs, coupled with their intact agonist properties allowing desensitization and internalization of receptors prior to their normal engagement with endothelium-bound chemokines. Moreover, these types of mutants can show “polypharmacology” by desensitizing and/or internalizing multiple chemokine receptors; examples include a GAG-binding deficient variants of MCP-3/CCL7, CXCL12 and a modified variant of CCL14 [106]. In some cases like CCL14, these effects are direct because CCL14 acts on multiple receptors [106]. However, indirect effects through heterologous desensitization of receptors have also been observed with GAG-binding deficient mutants of CXCL12 and CCL7 [103,107]. If small molecules could achieve the same ability to block chemokine binding to GAGs, keeping chemokines in circulation, they might provide another solution to the redundancy issue.
In summary, although the development of chemokine therapeutics has been slow and dominated by failed clinical trails, recent insights into these failures [12] and consideration of the full complexity of the receptor pharmacology as described here, may reinvigorate the field with many different and more refined strategies that have a better chance of success.
Acknowledgments
We thank Irina Kufareva (UCSD) for the preparation of Figure 4.
Footnotes
Declaration of Interest
M O’Hayre was supported by a California Breast Cancer Research Program Dissertation Award (14GB-0147) while CL Salanga was supported by a Ruth L. Kirschstein NIGMS MARC Predoctoral Fellowship (F31). DJ Hamel was supported by an NIH Postdoctoral Fellowship (F32 GM083463), while T Handel received awards from NIAID (RO1-AI37113), NIGMS (RO1-GM081763) and was supported by the Lymphoma Foundation
References
- 1.Baggiolini M. Chemokines and leukocyte traffic. Nature. 1998;392(6676):565–568. doi: 10.1038/33340. [DOI] [PubMed] [Google Scholar]
- 2.Murphy PM. The molecular biology of leukocyte chemoattractant receptors. Annu Rev Immunol. 1994;12:593–633. doi: 10.1146/annurev.iy.12.040194.003113. [DOI] [PubMed] [Google Scholar]
- 3.Baggiolini M. Chemokines in pathology and medicine. J Intern Med. 2001;250(2):91–104. doi: 10.1046/j.1365-2796.2001.00867.x. [DOI] [PubMed] [Google Scholar]
- 4.Rossi D, Zlotnik A. The biology of chemokines and their receptors. Annu Rev Immunol. 2000;18:217–242. doi: 10.1146/annurev.immunol.18.1.217. [DOI] [PubMed] [Google Scholar]
- 5.Wells TN, Proudfoot AE, Power CA. Chemokine receptors and their role in leukocyte activation. Immunol Lett. 1999;65(1–2):35–40. doi: 10.1016/s0165-2478(98)00121-7. [DOI] [PubMed] [Google Scholar]
- 6.Zlotnik A, Yoshie O. Chemokines: a new classification system and their role in immunity. Immunity. 2000;12(2):121–127. doi: 10.1016/s1074-7613(00)80165-x. [DOI] [PubMed] [Google Scholar]
- 7.Bacon K, Baggiolini M, Broxmeyer H, Horuk R, Lindley I, Mantovani A, Maysushima K, Murphy P, Nomiyama H, Oppenheim J, Rot A, et al. Chemokine/chemokine receptor nomenclature. J Interferon Cytokine Res. 2002;22(10):1067–1068. doi: 10.1089/107999002760624305. [DOI] [PubMed] [Google Scholar]
- 8.Nomiyama H, Osada N, Yoshie O. The evolution of mammalian chemokine genes. Cytokine Growth Factor Rev. doi: 10.1016/j.cytogfr.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 9.Horuk R. Chemokine receptor antagonists: overcoming developmental hurdles. Nat Rev Drug Discov. 2009;8(1):23–33. doi: 10.1038/nrd2734. [DOI] [PubMed] [Google Scholar]
- 10.Pease JE, Horuk R. Chemokine receptor antagonists: part 2. Expert Opin Ther Pat. 2009;19(2):199–221. doi: 10.1517/13543770802641353. [DOI] [PubMed] [Google Scholar]
- 11.Pease JE, Horuk R. Chemokine receptor antagonists: Part 1. Expert Opin Ther Pat. 2009;19(1):39–58. doi: 10.1517/13543770802641346. [DOI] [PubMed] [Google Scholar]
- 12.Proudfoot AEI, Power CA, Schwarz MK. Anti-chemokine small molecule drugs: a promising future? Expert Opinion on Investigational Drugs. 2010;19(3):345–355. doi: 10.1517/13543780903535867. [DOI] [PubMed] [Google Scholar]
- 13.Fatkenheuer G, Pozniak AL, Johnson MA, Plettenberg A, Staszewski S, Hoepelman AIM, Saag MS, Goebel FD, Rockstroh JK, Dezube BJ, Jenkins TM, et al. Efficacy of short-term monotherapy with maraviroc, a new CCR5 antagonist, in patients infected with HIV-1. Nat Med. 2005;11(11):1170–1172. doi: 10.1038/nm1319. [DOI] [PubMed] [Google Scholar]
- 14.Horuk R, Chitnis CE, Darbonne WC, Colby TJ, Rybicki A, Hadley TJ, Miller LH. A receptor for the malarial parasite Plasmodium vivax: the erythrocyte chemokine receptor. Science. 1993;261(5125):1182–1184. doi: 10.1126/science.7689250. [DOI] [PubMed] [Google Scholar]
- 15.Handel TM, Horuk R. Duffy antigen inhibitors: Useful therapeutics for malaria? Trends in Parasitology. 26(7):329–333. doi: 10.1016/j.pt.2010.03.010. [DOI] [PubMed] [Google Scholar]
- 16.Ben-Baruch A. Host microenvironment in breast cancer development: Inflammatory cells, cytokines and chemokines in breast cancer progression - reciprocal tumor-microenvironment interactions. Breast Cancer Res. 2003;5(1):31–36. doi: 10.1186/bcr554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410(6824):50–56. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 18.Murakami T, Cardones AR, Hwang ST. Chemokine receptors and melanoma metastasis. Journal of Dermatological Science. 2004;36(2):71–78. doi: 10.1016/j.jdermsci.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 19.Burger JA, Kipps TJ. CXCR4: a key receptor in the crosstalk between tumor cells and their microenvironment. Blood. 2006;107(5):1761–1767. doi: 10.1182/blood-2005-08-3182. [DOI] [PubMed] [Google Scholar]
- 20.Allegretti M, Bertini R, Bizzarri C, Beccari A, Mantovani A, Locati M. Allosteric inhibitors of chemoattractant receptors: opportunities and pitfalls. Trends in Pharmacological Sciences. 2008;29 (6):280–286. doi: 10.1016/j.tips.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 21.Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1(9):727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 22.Liang M, Mallari C, Rosser M, Ng HP, May K, Monahan S, Bauman JG, Islam I, Ghannam A, Buckman B, Shaw K, et al. Identification and Characterization of a Potent, Selective, and Orally Active Antagonist of the CC Chemokine Receptor-1. Journal of Biological Chemistry. 2000;275:19000–19008. doi: 10.1074/jbc.M001222200. [DOI] [PubMed] [Google Scholar]
- 23.Zipp F, Hartung HP, Hillert J, Schimrigk S, Trebst C, Stangel M, Infante-Duarte C, Jakobs P, Wolf C, Sandbrink R, Pohl C, et al. Blockade of chemokine signaling in patients with multiple sclerosis. Neurology. 2006;67(10):1880–1883. doi: 10.1212/01.wnl.0000244420.68037.86. [DOI] [PubMed] [Google Scholar]
- 24.Hendrix CWMD, Collier ACMD, Lederman MMMD, Schols DP, Pollard RBMD, Brown SMD, Jackson JBMD, Coombs RWMDP, Glesby MJMD, Flexner CWMD, Bridger GJP, et al. Safety, Pharmacokinetics, and Antiviral Activity of AMD3100, a Selective CXCR4 Receptor Inhibitor. HIV-1 Infection. :1525–4135. doi: 10.1097/01.qai.0000137371.80695.ef. [DOI] [PubMed] [Google Scholar]
- 25.Broxmeyer HE, Orschell CM, Clapp DW, Hangoc G, Cooper S, Plett PA, Liles WC, Li X, Graham-Evans B, Campbell TB, Calandra G, et al. Rapid mobilization of murine and human hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. The Journal of Experimental Medicine. 2005;201(8):1307–1318. doi: 10.1084/jem.20041385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eksteen B, Adams DH. GSK-1605786, a selective small-molecule antagonist of the CCR9 chemokine receptor for the treatment of Crohn’s disease. IDrugs. 2010;13(7):472–781. [PubMed] [Google Scholar]
- 27.Kazmierski W, Bifulco N, Yang H, Boone L, DeAnda F, Watson C, Kenakin T. Recent progress in discovery of small-molecule CCR5 chemokine receptor ligands as HIV-1 inhibitors. Bioorg Med Chem. 2003;11(13):2663–2676. doi: 10.1016/s0968-0896(03)00161-5. [DOI] [PubMed] [Google Scholar]
- 28.Watson C, Jenkinson S, Kazmierski W, Kenakin T. The CCR5 receptor-based mechanism of action of 873140, a potent allosteric noncompetitive HIV entry inhibitor. Mol Pharmacol. 2005;67 (4):1268–1282. doi: 10.1124/mol.104.008565. [DOI] [PubMed] [Google Scholar]
- 29.Casilli F, Bianchini A, Gloaguen I, Biordi L, Alesse E, Festuccia C, Cavalieri B, Strippoli R, Cervellera MN, Bitondo RD, Ferretti E, et al. Inhibition of interleukin-8 (CXCL8/IL-8) responses by repertaxin, a new inhibitor of the chemokine receptors CXCR1 and CXCR2. Biochemical Pharmacology. 2005;69(3):385–394. doi: 10.1016/j.bcp.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 30.Vaidehi N, Schlyer S, Trabanino RJ, Floriano WB, Abrol R, Sharma S, Kochanny M, Koovakat S, Dunning L, Liang M, Fox JM, et al. Predictions of CCR1 Chemokine Receptor Structure and BX 471 Antagonist Binding Followed by Experimental Validation. Journal of Biological Chemistry. 2006;281(37):27613–27620. doi: 10.1074/jbc.M601389200. [DOI] [PubMed] [Google Scholar]
- 31.Gupta SK, Pillarisetti K, Thomas RA, Aiyar N. Pharmacological evidence for complex and multiple site interaction of CXCR4 with SDF-1alpha: implications for development of selective CXCR4 antagonists. Immunol Lett. 2001;78(1):29–34. doi: 10.1016/s0165-2478(01)00228-0. [DOI] [PubMed] [Google Scholar]
- 32.Veldkamp CT, Seibert C, Peterson FC, De la Cruz NB, Haugner JC, 3rd, Basnet H, Sakmar TP, Volkman BF. Structural basis of CXCR4 sulfotyrosine recognition by the chemokine SDF-1/CXCL12. Sci Signal. 2008;1(37):ra4. doi: 10.1126/scisignal.1160755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crump MP, Gong JH, Loetscher P, Rajarathnam K, Amara A, Arenzana-Seisdedos F, Virelizier JL, Baggiolini M, Sykes BD, Clark-Lewis I. Solution structure and basis for functional activity of stromal cell-derived factor-1; dissociation of CXCR4 activation from binding and inhibition of HIV-1. EMBO J. 1997;16(23):6996–7007. doi: 10.1093/emboj/16.23.6996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kofuku Y, Yoshiura C, Ueda T, Terasawa H, Hirai T, Tominaga S, Hirose M, Maeda Y, Takahashi H, Terashima Y, Matsushima K, et al. Structural Basis of the Interaction between Chemokine Stromal Cell-derived Factor-1/CXCL12 and Its G-protein-coupled Receptor CXCR4. Journal of Biological Chemistry. 2009;284(50):35240–35250. doi: 10.1074/jbc.M109.024851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Andrews G, Jones C, Wreggett KA. An intracellular allosteric site for a specific class of antagonists of the CC chemokine G protein-coupled receptors CCR4 and CCR5. Mol Pharmacol. 2008;73 (3):855–867. doi: 10.1124/mol.107.039321. [DOI] [PubMed] [Google Scholar]
- 36.Nicholls DJ, Tomkinson NP, Wiley KE, Brammall A, Bowers L, Grahames C, Gaw A, Meghani P, Shelton P, Wright TJ, Mallinder PR. Identification of a putative intracellular allosteric antagonist binding-site in the CXC chemokine receptors 1 and 2. Mol Pharmacol. 2008;74(5):1193–1202. doi: 10.1124/mol.107.044610. [DOI] [PubMed] [Google Scholar]
- 37.Salchow K, Bond ME, Evans SC, Press NJ, Charlton SJ, Hunt PA, Bradley ME. A common intracellular allosteric binding site for antagonists of the CXCR2 receptor. Br J Pharmacol. 159(7):1429–1439. doi: 10.1111/j.1476-5381.2009.00623.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rajagopal S, Rajagopal K, Lefkowitz RJ. Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat Rev Drug Discov. 9(5):373–386. doi: 10.1038/nrd3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kohout TA, Lefkowitz RJ. Regulation of G protein-coupled receptor kinases and arrestins during receptor desensitization. Mol Pharmacol. 2003;63(1):9–18. doi: 10.1124/mol.63.1.9. [DOI] [PubMed] [Google Scholar]
- 40.Bhattacharya S, Hall SE, Li H, Vaidehi N. Ligand-stabilized conformational states of human beta(2) adrenergic receptor: insight into G-protein-coupled receptor activation. Biophys J. 2008;94 (6):2027–2042. doi: 10.1529/biophysj.107.117648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bhattacharya S, Hall SE, Vaidehi N. Agonist-induced conformational changes in bovine rhodopsin: insight into activation of G-protein-coupled receptors. J Mol Biol. 2008;382(2):539–555. doi: 10.1016/j.jmb.2008.06.084. [DOI] [PubMed] [Google Scholar]
- 42.Kobilka BK, Deupi X. Conformational complexity of G-protein-coupled receptors. Trends Pharmacol Sci. 2007;28(8):397–406. doi: 10.1016/j.tips.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 43.Yao X, Parnot C, Deupi X, Ratnala VR, Swaminath G, Farrens D, Kobilka B. Coupling ligand structure to specific conformational switches in the beta2-adrenoceptor. Nat Chem Biol. 2006;2 (8):417–422. doi: 10.1038/nchembio801. [DOI] [PubMed] [Google Scholar]
- 44.Kenakin T, Miller LJ. Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol Rev. 2010;62 (2):265–304. doi: 10.1124/pr.108.000992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kohout TA, Nicholas SL, Perry SJ, Reinhart G, Junger S, Struthers RS. Differential desensitization, receptor phosphorylation, beta-arrestin recruitment, and ERK1/2 activation by the two endogenous ligands for the CC chemokine receptor 7. J Biol Chem. 2004;279(22):23214–23222. doi: 10.1074/jbc.M402125200. [DOI] [PubMed] [Google Scholar]
- 46.Otero C, Groettrup M, Legler DF. Opposite fate of endocytosed CCR7 and its ligands: recycling versus degradation. J Immunol. 2006;177(4):2314–2323. doi: 10.4049/jimmunol.177.4.2314. [DOI] [PubMed] [Google Scholar]
- 47.Zidar DA, Violin JD, Whalen EJ, Lefkowitz RJ. Selective engagement of G protein coupled receptor kinases (GRKs) encodes distinct functions of biased ligands. Proc Natl Acad Sci U S A. 2009;106(24):9649–9654. doi: 10.1073/pnas.0904361106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Byers MA, Calloway PA, Shannon L, Cunningham HD, Smith S, Li F, Fassold BC, Vines CM. Arrestin 3 mediates endocytosis of CCR7 following ligation of CCL19 but not CCL21. J Immunol. 2008;181(7):4723–4732. doi: 10.4049/jimmunol.181.7.4723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yoshida R, Nagira M, Kitaura M, Imagawa N, Imai T, Yoshie O. Secondary lymphoid-tissue chemokine is a functional ligand for the CC chemokine receptor CCR7. J Biol Chem. 1998;273(12):7118–7122. doi: 10.1074/jbc.273.12.7118. [DOI] [PubMed] [Google Scholar]
- 50.Savino B, Borroni EM, Torres NM, Proost P, Struyf S, Mortier A, Mantovani A, Locati M, Bonecchi R. Recognition versus adaptive up-regulation and degradation of CC chemokines by the chemokine decoy receptor D6 are determined by their N-terminal sequence. J Biol Chem. 2009;284(38):26207–26215. doi: 10.1074/jbc.M109.029249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jarnagin K, Grunberger D, Mulkins M, Wong B, Hemmerich S, Paavola C, Bloom A, Bhakta S, Diehl F, Freedman R, McCarley D, et al. Identification of surface residues of the monocyte chemotactic protein 1 that affect signaling through the receptor CCR2. Biochemistry. 1999;38(49):16167–16177. doi: 10.1021/bi9912239. [DOI] [PubMed] [Google Scholar]
- 52.Simmons G, Clapham PR, Picard L, Offord RE, Rosenkilde MM, Schwartz TW, Buser R, Wells TN, Proudfoot AE. Potent inhibition of HIV-1 infectivity in macrophages and lymphocytes by a novel CCR5 antagonist. Science. 1997;276(5310):276–279. doi: 10.1126/science.276.5310.276. [DOI] [PubMed] [Google Scholar]
- 53.Mack M, Luckow B, Nelson PJ, Cihak J, Simmons G, Clapham PR, Signoret N, Marsh M, Stangassinger M, Borlat F, Wells TN, et al. Aminooxypentane-RANTES induces CCR5 internalization but inhibits recycling: a novel inhibitory mechanism of HIV infectivity. J Exp Med. 1998;187(8):1215–1224. doi: 10.1084/jem.187.8.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gonzalez E, Kulkarni H, Bolivar H, Mangano A, Sanchez R, Catano G, Nibbs RJ, Freedman BI, Quinones MP, Bamshad MJ, Murthy KK, et al. The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science. 2005;307(5714):1434–1440. doi: 10.1126/science.1101160. [DOI] [PubMed] [Google Scholar]
- 55.Cocchi F, DeVico AL, Garzino-Demo A, Arya SK, Gallo RC, Lusso P. Identification of RANTES, MIP-1 alpha, and MIP-1 beta as the major HIV-suppressive factors produced by CD8+ T cells. Science. 1995;270(5243):1811–1815. doi: 10.1126/science.270.5243.1811. [DOI] [PubMed] [Google Scholar]
- 56.Proudfoot AE, Buser R, Borlat F, Alouani S, Soler D, Offord RE, Schroder JM, Power CA, Wells TN. Amino-terminally modified RANTES analogues demonstrate differential effects on RANTES receptors. J Biol Chem. 1999;274(45):32478–32485. doi: 10.1074/jbc.274.45.32478. [DOI] [PubMed] [Google Scholar]
- 57.Elsner J, Mack M, Bruhl H, Dulkys Y, Kimmig D, Simmons G, Clapham PR, Schlondorff D, Kapp A, Wells TN, Proudfoot AE. Differential activation of CC chemokine receptors by AOP-RANTES. J Biol Chem. 2000;275(11):7787–7794. doi: 10.1074/jbc.275.11.7787. [DOI] [PubMed] [Google Scholar]
- 58.Gaertner H, Cerini F, Escola J-M, Kuenzi G, Melotti A, Offord R, Rossitto-Borlat In, Nedellec R, Salkowitz J, Gorochov G, Mosier D, et al. Highly potent, fully recombinant anti-HIV chemokines: Reengineering a low-cost microbicide. Proceedings of the National Academy of Sciences. 2008;105(46):17706–17711. doi: 10.1073/pnas.0805098105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Muniz-Medina VM, Jones S, Maglich JM, Galardi C, Hollingsworth RE, Kazmierski WM, Ferris RG, Edelstein MP, Chiswell KE, Kenakin TP. The relative activity of “function sparing” HIV-1 entry inhibitors on viral entry and CCR5 internalization: is allosteric functional selectivity a valuable therapeutic property? Mol Pharmacol. 2009;75(3):490–501. doi: 10.1124/mol.108.052555. [DOI] [PubMed] [Google Scholar]
- 60.Kenakin T. Functional selectivity through protean and biased agonism: who steers the ship? Mol Pharmacol. 2007;72(6):1393–1401. doi: 10.1124/mol.107.040352. [DOI] [PubMed] [Google Scholar]
- 61.Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, Javitch JA, Roth BL, Christopoulos A, Sexton PM, Miller KJ, et al. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther. 2007;320(1):1–13. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- 62.Conn PJ, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov. 2009;8(1):41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Leach K, Sexton PM, Christopoulos A. Allosteric GPCR modulators: taking advantage of permissive receptor pharmacology. Trends Pharmacol Sci. 2007;28(8):382–389. doi: 10.1016/j.tips.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 64.Muniz-Medina VM, Jones S, Maglich JM, Galardi C, Hollingsworth RE, Kazmierski WM, Ferris RG, Edelstein MP, Chiswell KE, Kenakin TP. The Relative Activity of Function Sparing HIV-1 Entry Inhibitors on Viral Entry and CCR5 Internalization: Is Allosteric Functional Selectivity a Valuable Therapeutic Property? Molecular Pharmacology. 2009;75(3):490–501. doi: 10.1124/mol.108.052555. [DOI] [PubMed] [Google Scholar]
- 65.Rajagopal S, Kim J, Ahn S, Craig S, Lam CM, Gerard NP, Gerard C, Lefkowitz RJ. Beta-arrestin- but not G protein-mediated signaling by the “decoy” receptor CXCR7. Proc Natl Acad Sci U S A. 2010;107(2):628–632. doi: 10.1073/pnas.0912852107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lagane B, Chow KY, Balabanian K, Levoye A, Harriague J, Planchenault T, Baleux F, Gunera-Saad N, Arenzana-Seisdedos F, Bachelerie F. CXCR4 dimerization and beta-arrestin-mediated signaling account for the enhanced chemotaxis to CXCL12 in WHIM syndrome. Blood. 2008;112(1):34–44. doi: 10.1182/blood-2007-07-102103. [DOI] [PubMed] [Google Scholar]
- 67.Luker KE, Steele JM, Mihalko LA, Ray P, Luker GD. Constitutive and chemokine-dependent internalization and recycling of CXCR7 in breast cancer cells to degrade chemokine ligands. Oncogene. 2010 doi: 10.1038/onc.2010.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sohy D, Yano H, de Nadai P, Urizar E, Guillabert A, Javitch JA, Parmentier M, Springael JY. Hetero-oligomerization of CCR2, CCR5, and CXCR4 and the protean effects of “selective” antagonists. J Biol Chem. 2009;284(45):31270–31279. doi: 10.1074/jbc.M109.054809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang J, Norcross M. Dimerization of chemokine receptors in living cells: key to receptor function and novel targets for therapy. Drug Discov Today. 2008;13(13–14):625–632. doi: 10.1016/j.drudis.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 70.Mellado M, Rodriguez-Frade JM, Vila-Coro AJ, de Ana AM, Martinez AC. Chemokine control of HIV-1 infection. Nature. 1999;400(6746):723–724. doi: 10.1038/23382. [DOI] [PubMed] [Google Scholar]
- 71.Agrawal L, Lu X, Qingwen J, VanHorn-Ali Z, Nicolescu IV, McDermott DH, Murphy PM, Alkhatib G. Role for CCR5Delta32 protein in resistance to R5, R5X4, and X4 human immunodeficiency virus type 1 in primary CD4+ cells. J Virol. 2004;78(5):2277–2287. doi: 10.1128/JVI.78.5.2277-2287.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dean M, Carrington M, Winkler C, Huttley GA, Smith MW, Allikmets R, Goedert JJ, Buchbinder SP, Vittinghoff E, Gomperts E, Donfield S, et al. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort, ALIVE Study. Science. 1996;273(5283):1856–1862. doi: 10.1126/science.273.5283.1856. [DOI] [PubMed] [Google Scholar]
- 73.Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, Farber CM, Saragosti S, Lapoumeroulie C, Cognaux J, Forceille C, Muyldermans G, et al. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature. 1996;382(6593):722–725. doi: 10.1038/382722a0. [DOI] [PubMed] [Google Scholar]
- 74.Benkirane M, Jin DY, Chun RF, Koup RA, Jeang KT. Mechanism of transdominant inhibition of CCR5-mediated HIV-1 infection by ccr5delta32. J Biol Chem. 1997;272(49):30603–30606. doi: 10.1074/jbc.272.49.30603. [DOI] [PubMed] [Google Scholar]
- 75.Hernandez PA, Gorlin RJ, Lukens JN, Taniuchi S, Bohinjec J, Francois F, Klotman ME, Diaz GA. Mutations in the chemokine receptor gene CXCR4 are associated with WHIM syndrome, a combined immunodeficiency disease. Nat Genet. 2003;34(1):70–74. doi: 10.1038/ng1149. [DOI] [PubMed] [Google Scholar]
- 76.Balabanian K, Lagane B, Pablos JL, Laurent L, Planchenault T, Verola O, Lebbe C, Kerob D, Dupuy A, Hermine O, Nicolas JF, et al. WHIM syndromes with different genetic anomalies are accounted for by impaired CXCR4 desensitization to CXCL12. Blood. 2005;105(6):2449–2457. doi: 10.1182/blood-2004-06-2289. [DOI] [PubMed] [Google Scholar]
- 77.Balabanian K, Lagane B, Infantino S, Chow KY, Harriague J, Moepps B, Arenzana-Seisdedos F, Thelen M, Bachelerie F. The chemokine SDF-1/CXCL12 binds to and signals through the orphan receptor RDC1 in T lymphocytes. J Biol Chem. 2005;280(42):35760–35766. doi: 10.1074/jbc.M508234200. [DOI] [PubMed] [Google Scholar]
- 78.Levoye A, Balabanian K, Baleux F, Bachelerie F, Lagane B. CXCR7 heterodimerizes with CXCR4 and regulates CXCL12-mediated G protein signaling. Blood. 2009;113(24):6085–6093. doi: 10.1182/blood-2008-12-196618. [DOI] [PubMed] [Google Scholar]
- 79.Sierro F, Biben C, Martinez-Munoz L, Mellado M, Ransohoff RM, Li M, Woehl B, Leung H, Groom J, Batten M, Harvey RP, et al. Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proc Natl Acad Sci U S A. 2007;104(37):14759–14764. doi: 10.1073/pnas.0702229104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Luker KE, Gupta M, Luker GD. Imaging chemokine receptor dimerization with firefly luciferase complementation. FASEB J. 2009;23(3):823–834. doi: 10.1096/fj.08-116749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Burns JM, Summers BC, Wang Y, Melikian A, Berahovich R, Miao Z, Penfold ME, Sunshine MJ, Littman DR, Kuo CJ, Wei K, et al. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J Exp Med. 2006;203(9):2201–2213. doi: 10.1084/jem.20052144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kalatskaya I, Berchiche YA, Gravel S, Limberg BJ, Rosenbaum JS, Heveker N. AMD3100 is a CXCR7 ligand with allosteric agonist properties. Mol Pharmacol. 2009;75(5):1240–1247. doi: 10.1124/mol.108.053389. [DOI] [PubMed] [Google Scholar]
- 83.Zabel BA, Wang Y, Lewen S, Berahovich RD, Penfold ME, Zhang P, Powers J, Summers BC, Miao Z, Zhao B, Jalili A, et al. Elucidation of CXCR7-mediated signaling events and inhibition of CXCR4-mediated tumor cell transendothelial migration by CXCR7 ligands. J Immunol. 2009;183(5):3204–3211. doi: 10.4049/jimmunol.0900269. [DOI] [PubMed] [Google Scholar]
- 84.Contento RL, Molon B, Boularan C, Pozzan T, Manes S, Marullo S, Viola A. CXCR4-CCR5: a couple modulating T cell functions. Proc Natl Acad Sci U S A. 2008;105(29):10101–10106. doi: 10.1073/pnas.0804286105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.El-Asmar L, Springael JY, Ballet S, Andrieu EU, Vassart G, Parmentier M. Evidence for negative binding cooperativity within CCR5-CCR2b heterodimers. Mol Pharmacol. 2005;67(2):460–469. doi: 10.1124/mol.104.003624. [DOI] [PubMed] [Google Scholar]
- 86.Hernanz-Falcon P, Rodriguez-Frade JM, Serrano A, Juan D, del Sol A, Soriano SF, Roncal F, Gomez L, Valencia A, Martinez AC, Mellado M. Identification of amino acid residues crucial for chemokine receptor dimerization. Nat Immunol. 2004;5(2):216–223. doi: 10.1038/ni1027. [DOI] [PubMed] [Google Scholar]
- 87.Issafras H, Angers S, Bulenger S, Blanpain C, Parmentier M, Labbe-Jullie C, Bouvier M, Marullo S. Constitutive agonist-independent CCR5 oligomerization and antibody-mediated clustering occurring at physiological levels of receptors. J Biol Chem. 2002;277(38):34666–34673. doi: 10.1074/jbc.M202386200. [DOI] [PubMed] [Google Scholar]
- 88.Mellado M, Rodriguez-Frade JM, Vila-Coro AJ, Fernandez S, Martin de Ana A, Jones DR, Toran JL, Martinez AC. Chemokine receptor homo- or heterodimerization activates distinct signaling pathways. EMBO J. 2001;20(10):2497–2507. doi: 10.1093/emboj/20.10.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Salanga CL, O’Hayre M, Handel T. Modulation of chemokine receptor activity through dimerization and crosstalk. Cell Mol Life Sci. 2009;66(8):1370–1386. doi: 10.1007/s00018-008-8666-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sabroe I, Peck MJ, Van Keulen BJ, Jorritsma A, Simmons G, Clapham PR, Williams TJ, Pease JE. A small molecule antagonist of chemokine receptors CCR1 and CCR3. Potent inhibition of eosinophil function and CCR3-mediated HIV-1 entry. J Biol Chem. 2000;275(34):25985–25992. doi: 10.1074/jbc.M908864199. [DOI] [PubMed] [Google Scholar]
- 91.Baba M, Nishimura O, Kanzaki N, Okamoto M, Sawada H, Iizawa Y, Shiraishi M, Aramaki Y, Okonogi K, Ogawa Y, Meguro K, et al. A small-molecule, nonpeptide CCR5 antagonist with highly potent and selective anti-HIV-1 activity. Proc Natl Acad Sci U S A. 1999;96(10):5698–5703. doi: 10.1073/pnas.96.10.5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Naya A, Kobayashi K, Ishikawa M, Ohwaki K, Saeki T, Noguchi K, Ohtake N. Structure-activity relationships of 2-(benzothiazolylthio)acetamide class of CCR3 selective antagonist. Chem Pharm Bull (Tokyo) 2003;51(6):697–701. doi: 10.1248/cpb.51.697. [DOI] [PubMed] [Google Scholar]
- 93.Walters I, Austin C, Austin R, Bonnert R, Cage P, Christie M, Ebden M, Gardiner S, Grahames C, Hill S, Hunt F, et al. Evaluation of a series of bicyclic CXCR2 antagonists. Bioorg Med Chem Lett. 2008;18 (2):798–803. doi: 10.1016/j.bmcl.2007.11.039. [DOI] [PubMed] [Google Scholar]
- 94.Wu B, Chien EYT, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi C, Hamel DJ, et al. Structures of the CXCR4 chemokine receptor in complex with small molecule and cyclic peptide antagonists. Science, accepted for publication. 2010 doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kinzer-Ursem TL, Linderman JJ. Both ligand- and cell-specific parameters control ligand agonism in a kinetic model of g protein-coupled receptor signaling. PLoS Comput Biol. 2007;3(1):e6. doi: 10.1371/journal.pcbi.0030006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Vroon A, Heijnen CJ, Kavelaars A. GRKs and arrestins: regulators of migration and inflammation. J Leukoc Biol. 2006;80(6):1214–1221. doi: 10.1189/jlb.0606373. [DOI] [PubMed] [Google Scholar]
- 97.Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1(1):31–39. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- 98.Cardaba CM, Kerr JS, Mueller A. CCR5 internalisation and signalling have different dependence on membrane lipid raft integrity. Cell Signal. 2008;20(9):1687–1694. doi: 10.1016/j.cellsig.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 99.O’Hayre M, Salanga C, Kipps T, Messmer D, Dorrestein P, Handel T. Elucidating the CXCL12/CXCR4 signaling network in chronic lymphocytic leukemia through phosphoproteomics analysis. PLoS One. 2010 doi: 10.1371/journal.pone.0011716. Accepted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Proudfoot AE, Handel TM, Johnson Z, Lau EK, LiWang P, Clark-Lewis I, Borlat F, Wells TN, Kosco-Vilbois MH. Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc Natl Acad Sci U S A. 2003;100(4):1885–1890. doi: 10.1073/pnas.0334864100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang L, Fuster M, Sriramarao P, Esko JD. Endothelial heparan sulfate deficiency impairs L-selectin- and chemokine-mediated neutrophil trafficking during inflammatory responses. Nat Immunol. 2005;6(9):902–910. doi: 10.1038/ni1233. [DOI] [PubMed] [Google Scholar]
- 102.Ali S, O’Boyle G, Hepplewhite P, Tyler JR, Robertson H, Kirby JA. Therapy with nonglycosaminoglycan-binding mutant CCL7: a novel strategy to limit allograft inflammation. Am J Transplant. 10(1):47–58. doi: 10.1111/j.1600-6143.2009.02868.x. [DOI] [PubMed] [Google Scholar]
- 103.Ali S, O’Boyle G, Mellor P, Kirby JA. An apparent paradox: chemokine receptor agonists can be used for anti-inflammatory therapy. Mol Immunol. 2007;44(7):1477–1482. doi: 10.1016/j.molimm.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 104.Ali S, Robertson H, Wain JH, Isaacs JD, Malik G, Kirby JA. A non-glycosaminoglycan-binding variant of CC chemokine ligand 7 (monocyte chemoattractant protein-3) antagonizes chemokine-mediated inflammation. J Immunol. 2005;175(2):1257–1266. doi: 10.4049/jimmunol.175.2.1257. [DOI] [PubMed] [Google Scholar]
- 105.Johnson Z, Kosco-Vilbois MH, Herren S, Cirillo R, Muzio V, Zaratin P, Carbonatto M, Mack M, Smailbegovic A, Rose M, Lever R, et al. Interference with heparin binding and oligomerization creates a novel anti-inflammatory strategy targeting the chemokine system. J Immunol. 2004;173(9):5776–5785. doi: 10.4049/jimmunol.173.9.5776. [DOI] [PubMed] [Google Scholar]
- 106.Gupta S, Rieder S, Richter R, Schulz-Maronde S, Manns J, Escher SE, Heitland A, Mack M, Forssmann WG, Elsner J, Forssmann U. CCR1- and CCR5-mediated inactivation of leukocytes by a nonglycosaminoglycan (non-GAG)-binding variant of n-nonanoyl-CCL14 (NNY-CCL14) J Leukoc Biol. 88(2):383–392. doi: 10.1189/jlb.0509366. [DOI] [PubMed] [Google Scholar]
- 107.O’Boyle G, Mellor P, Kirby JA, Ali S. Anti-inflammatory therapy by intravenous delivery of non-heparan sulfate-binding CXCL12. FASEB J. 2009;23(11):3906–3916. doi: 10.1096/fj.09-134643. [DOI] [PMC free article] [PubMed] [Google Scholar]