

Abstract
Metabolic flux, the flow of metabolites through networks of enzymes, represents the dynamic productive output of cells. Improved understanding of intracellular metabolic fluxes will enable targeted manipulation of metabolic pathways of medical and industrial importance to a greater degree than is currently possible. Flux balance analysis (FBA) is a constraint-based approach to modeling metabolic fluxes, but its utility is limited by a lack of experimental measurements. Incorporation of experimentally measured fluxes as system constraints will significantly improve the overall accuracy of FBA. We applied a novel, two-tiered approach in the yeast Saccharomyces cerevisiae to measure nutrient consumption rates (extracellular fluxes) and a targeted intracellular flux using a 14C-labeled precursor with HPLC separation and flux quantitation by accelerator mass spectrometry (AMS). The use of AMS to trace the intracellular fate of 14C-glutamine allowed the calculation of intracellular metabolic flux through this pathway, with glutathione as the metabolic endpoint. Measured flux values provided global constraints for the yeast FBA model which reduced model uncertainty by more than 20%, proving the importance of additional constraints in improving the accuracy of model predictions and demonstrating the use of AMS to measure intracellular metabolic fluxes. Our results highlight the need to use intracellular fluxes to constrain the models. We show that inclusion of just one such measurement alone can reduce the average variability of model predicted fluxes by 10%.
Keywords: Metabolic flux analysis, Flux balance analysis, Accelerator mass spectrometry, glutathione, glutamate, glutamine, yeast
Metabolism consists of many interconnected networks of genes, enzymes, and metabolites, which interact to regulate cell function. While the genetic complement of the cell dictates what metabolic processes are possible, the mere knowledge of the metabolic reactions that can occur is insufficient for a thorough understanding of a cell’s metabolic state. It is important to understand both the structure and the output of these metabolic networks in order to more fully elucidate cell function and accurately predict cellular responses to various conditions. Two methods used for metabolic flux analysis (MFA) are the purely stoichiometric MFA that is commonly referred to as flux balance analysis (FBA) [1-5] and the 13C MFA which uses the pattern of distribution of labeled metabolites resulting from breakdown of a 13C labeled substrate to measure intracellular fluxes [6-11]. Thus, while FBA is a computational tool that models cellular metabolism based on the law of mass balance, 13C MFA adds to FBA an experimental component which is used to measure the time-dependent output of metabolic pathways.
Computational models have been developed to improve understanding of cellular metabolic activity for a wide variety of organisms [10, 12-17], but these models are limited by a lack of sufficient experimental data to make the models reflect nature more accurately. Because FBA is a constraint-based approach for assessing a genotype’s metabolic capabilities, implementation of any additional constraints (i.e. experimental results) will improve accuracy of FBA models’ predictions. Two of the most important classes of constraints that can be placed on FBA models are “internal flux constraints” and “external flux constraints”. The latter constraints ensure that import of nutrients and export of waste are set to experimentally measured values or capped to physiologically feasible levels. The former constraints ensure that the flux through each metabolic reaction has values that are biologically reasonable and in accord with experimental measurements. Unfortunately, the difficult task of measuring intracellular fluxes has meant that in the majority of developed FBA models the internal fluxes are not adequately constrained [18]. This shortcoming can increase the uncertainty of FBA model flux predictions for a variety of different situations such as those for parallel metabolic pathways or futile internal cycles which have no physiological relevance. The ideal solutions to overcome this problem would be to provide tighter constraints on the internal fluxes of the model as well as to include thermodynamic limitations on metabolic reactions [19, 20].
The primary methods currently in use for the measurement of metabolic fluxes are 13C-NMR, 13C-GC-MS and 13C-LC-MS [1, 5, 21-24]. Typical 13C tracer experiments do not measure fluxes directly, but make use of isotopic labeling patterns of metabolic endpoints, which are then incorporated into computational models to allow calculation of fluxes [5, 22]. While powerful in assessing the overall metabolic state of the cells under consideration, current techniques using 13C suffer from several limitations. Due to the relatively high natural abundance of 13C, it is necessary to heavily label the precursor compound to be traced [22]. In many cases it is difficult to follow low-abundance metabolites, including the pool of free amino acids and many other intermediary metabolites [18]. Most importantly, 13C tracer techniques generally require the use of a minimal medium with a single carbon source, as additional media nutrients can interfere with measurement techniques [22]. These growth conditions rarely mirror actual growth conditions of biological relevance. Attention has recently been drawn to the absence of experimental flux measurements in complex, nutrient-rich media [25]. Thus, lack of sensitivity, requirement of metabolic and isotopic steady states, and restriction of media components are important limitations of current metabolic flux measurement techniques [5, 22, 26-28].
The application of novel experimental techniques with improved sensitivity and the ability to use nutrient-rich growth media could improve measurement of intracellular metabolic fluxes. Although 13C flux measurement experiments are primarily performed using cells in metabolic and isotopic steady-states, a recent trend toward measuring isotopic and metabolic non-steady state cultures is emerging [4, 29, 30]. Whereas the steady-state experiments provide only isotope incorporation ratios for relatively high-abundance metabolic endpoints, it is possible to monitor metabolite fluxes using isotopic-dynamic cultures if a sufficiently sensitive isotope measurement technique is used. It has been suggested that more informative data could be obtained by measuring intracellular intermediate metabolites rather than biomass endpoints [30]. Although 13C kinetic metabolic profiling techniques have shown great promise for tracing intracellular metabolites [31], the usefulness of this approach is limited by the requirement of a heavily-labeled sole carbon source rather than the more biologically-relevant nutrient-rich media containing multiple carbon sources. Yanagimachi et al. reported the use of a 14C-labeled precursor to measure metabolic fluxes directly using HPLC coupled with radiometric detection [32], and thereby demonstrated the utility of using 14C for measuring intracellular metabolic fluxes. However, radiometric detection does not provide the optimal sensitivity necessary for measuring some of the important metabolic fluxes and still suffers from the need for the use of heavily-labeled substrates at unnaturally high concentrations. This measurement problem can potentially be solved by using accelerator mass spectrometry (AMS), which offers a 106-fold gain in 14C quantitation relative to decay counting [33]. Experiments have previously been performed using AMS to quantitatively trace 14C-labeled drugs and nutrients in organisms and tissues [33-40]. The sensitivity and inherent quantitative nature of AMS measurements suggest that this technique could be a useful adjunct to more traditional metabolic flux measurement methods. We therefore sought to extend the utility of AMS to the measurement of intracellular metabolic fluxes based on the rationale that intracellular fluxes can be measured directly in cells grown in nutrient-rich media using a targeted labeling approach with a sufficiently sensitive detection method. Experiments were performed using 14C-labeled glutamine to directly measure metabolic fluxes in Saccharomyces cerevisiae grown in synthetic complete media (SCM), with glutathione as the metabolic endpoint. Glutathione is a ubiquitous, thiol-containing antioxidant with a well-characterized biosynthetic pathway, and is maintained at nearly constant intracellular levels during non-stressed conditions, making it an appropriate test case for proof-of-principle experiments.
EXPERIMENTAL SECTION
Chemicals and Materials
Unless otherwise specified, all chemicals were obtained from Sigma Chemical Company (St. Louis, MO) and were of the highest purity available. Yeast Saccharomyces cerevisiae strain S288C was obtained from the American Type Culture Collection (ATCC, Manassas, VA). Uniformly-labeled 14C-Glutamine was purchased from Moravek Biochemicals (Brea, CA).
Cell growth conditions and experimental sampling
Saccharomyces cerevisiae strain S288C was grown in synthetic complete medium (SCM) supplemented with all 20 proteinogenic amino acids (Sigma yeast synthetic media supplement without uracil, with amino acids at 76 mg/L, leucine at 380 mg/L) and uracil (80 mg/L). Cells were grown and maintained in log-growth phase at 30° C with shaking at 230 rpm in a Gyrotory Water Bath Shaker (New Brunswick Scientific) for at least 24 hours before labeling experiments were started. Yeast cells were grown in the presence of 0.1 nCi/mL 14C glutamine, which corresponds to a ratio of 1 molecule labeled glutamine per 250,000 unlabeled glutamine molecules. Cell density was measured using a SpectraMax Plus 384 microplate spectrophotometer at 600 nm (Molecular Devices, Sunnyvale, CA). Cell numbers were calculated using a standard curve generated to correlate OD600 with cell number obtained by counting serially diluted yeast cells on a hemocytometer. All experiments were performed in triplicate. Cells were grown in log-phase, with aliquots collected every 30 minutes. Two- 2 mL aliquots were collected, cells were pelleted by centrifugation for 3 minutes at 10,000 rpm at −9° C, and the media was collected and removed. Media was further purified by centrifugation at 14,000 rpm for 10 minutes to remove remaining intact cells. Cell pellets were washed twice with 1 mL ice-cold PBS, after which polar metabolites were extracted as described below, or cells were frozen at −20° C for later analysis. Duplicate cell and media aliquots were collected for each time point. Polar metabolites were extracted as reported by Villas-Boas et al. [41]. Briefly, cell pellets were combined with 200 μL ice-cold chloroform, 100 μL methanol, and 100 μL of 3 mM PIPES-3 mM EDTA, pH 7.4, and vortexed for 45 minutes at −20° C. The upper, aqueous phase was collected in a fresh tube and stored at −20° C, and the organic phase was re-extracted with 100 μL methanol and 100 μL PIPES-EDTA. Extracts were spun at 14,000 rpm at −9° C for 10 minutes. Experiments showed greater than 90% recovery for glutamate, glutamine, and glutathione using this technique (data not shown). For cell analysis by AMS, cell pellets were re-suspended in 200 μL of sterile water.
HPLC measurement of amino acids and glutathione
Amino acids and glutathione in the polar metabolite extract were derivatized with ortho-phthalaldehyde and separated by reversed-phase HPLC essentially as described previously [42, 43], but using a 1:1 ratio of sample:ortho-phthalaldehyde-mercaptopropionic acid solution (Agilent Technologies, Santa Clara, CA). HPLC fractions were collected for AMS analysis using a Gilson fraction collector (Gilson, Middletown, WI). HPLC separations were performed on an Agilent 1100 instrument (Agilent Technologies) using an Agilent Eclipse Plus C18 5 μm 4.6 × 150 mm column at 30° C. The internal standard L-2-aminobutyric acid was added to each polar metabolite or media sample prior to HPLC analysis to a final concentration of 7.6 μg/mL. HPLC conditions were as follows: Flow rate, 1.5 mL/ min. Mobile phase A: 10 mM Potassium tetraborate tetrahydrate, 10 mM potassium phosphate, pH 8.15. Mobile phase B: 45% methanol, 45% acetonitrile, 10% water. Column: Agilent C18, flow rate: 1.5 mL/ minute. Gradient: Time 0, 2% B; 4 minutes, 6% B; 4.20 minutes, 8% B; 9.50 minutes, 23% B; 12.00 minutes, 27.5% B; 12.50 minutes, 30% B; 20.00 minutes; 50% B; 20.10 minutes, 100% B; 22.00 minutes, 100% B; 22.1 minutes, 2% B; stop time: 25 minutes. Detection was accomplished using an Agilent 1100 fluorescence detector, with excitation at 340 nm and emission at 450 nm [43]. Analyte concentrations were calculated using peak area under the curves (AUC) and standard curves generated using authentic standards for each metabolite of interest. Amino acids measured for external flux calculations were: glutamate, glutamine, arginine, histidine, aspartic acid, asparagines, alanine, serine, cysteine, and tyrosine.
Sample graphitization and analysis by accelerator mass spectrometry
Aliquots of cells, media, and polar metabolite extracts were converted to graphite and analyzed by AMS as previously described [34]. For AMS, the following sample volumes were used: HPLC fractions, 300 μL + 1 μL tributyrin carrier; Polar metabolite pool, 10 μL + 1 μL tributyrin; Media, 2 μL + 1 μL tributyrin; Cells, 2 μL + 1 μL tributyrin. Accelerator mass spectrometry results in fraction Modern 14C/C were converted to amol 14C based on the total C measured in each sample, consisting of analyte and tributyrin carrier. Because in all cases the quantity of carbon originating from the tributyrin carrier was much greater than the carbon due to the analyte, the following equation was used to determine amol 14C in the analyte:
where Rmeasured = total Fraction Modern measured, RCarrier = Fraction Modern of the tributyrin carrier, and CCarrier = the carbon present in the carrier (mg). All experiments were performed in triplicate. Error bars indicate standard error of the mean. GraphPad Prism 5 software was used to generate graphs and perform linear regression analysis to provide slopes used to calculate internal metabolic fluxes (GraphPad Software, La Jolla, CA).
Calculation of external flux values using HPLC measurements and internal flux values using 14C tracer measurements
The rates of uptake of ten amino acids (see Table 1) were calculated by measuring the amount of each amino acid consumed per unit volume (mM) for the duration of the experiment (3.5 hours) and dividing it by the number of cells that consumed the nutrient. To calculate the number of cells we calculated the area under the growth curve. The equation, f(t) (where t is time), for the curve was derived using linear regression. Glutamine uptake rates using AMS were calculated in the same manner, but using uptake of 14C label from media into cells, and converting label uptake to total glutamine uptake based on the knowledge that initially 1 in 250,000 glutamine molecules in the growth media contained the 14C label.
Table 1.
Extracellular and Intracellular Metabolite Flux Values
| Metabolic Process | Flux value (mmol/gDW·hr) |
RSD% |
|---|---|---|
| aspartate uptake | 0.72 | 5.16 |
| glutamine uptake (AMS) (F1) | 0.44 | 1.67 |
| glutamine uptake (HPLC) | 0.23 | 4.01 |
| glutamate uptake | 0.6 | 4.61 |
| asparagine uptake | 0.36 | 4.14 |
| serine uptake | 0.47 | 3.45 |
| histidine uptake | 0.26 | 3.68 |
| cysteine uptake | 0.78 | 11.29 |
| arginine uptake | 0.31 | 4.10 |
| alanine uptake | 0.1 | 3.28 |
| tyrosine uptake | 0.13 | 4.41 |
| glutamine → glutamate (F2) | 0.4 | 5.49 |
| glutamate → glutathione (F3) | 3.6×10−3 | 9.31 |
| glutamine → protein (BMGln) | 0.04 | 3.58 |
| glutamate → protein (BMGlu) | 0.11 | 6.47 |
| glutamate → other metabolites (CMGlu) | 0.89 | 6.47 |
Internal flux from glutamate to glutathione was calculated by tracing 14C label flowing to glutathione at each time point using the general formula:
Where Fraction 14C-Gluss indicates the fraction of glutamate in the free amino acid pool that contains the 14C label at isotopic steady state. The amount of glutamate converted to glutathione at each time point was calculated according to this equation, and plotted against time. Figure 1 displays a simplified diagram of glutamine metabolism in yeast in a nutrient rich environment that is based on a number of experimental observations [44, 45, 46] The slope of this line was taken as the flux of glutamate to glutathione, represented as F4 in Figure 1. Repression of glutamine synthetase when S. cerevisiae is in grown in the presence of glutamate has been reported previously [44, 45]. This observation indicates that conversion of glutamate to glutamine, an ATP-consuming process, should occur at a negligible rate in our experimental growth conditions.
Figure 1. Biosynthetic pathway for production of glutathione from glutamine.
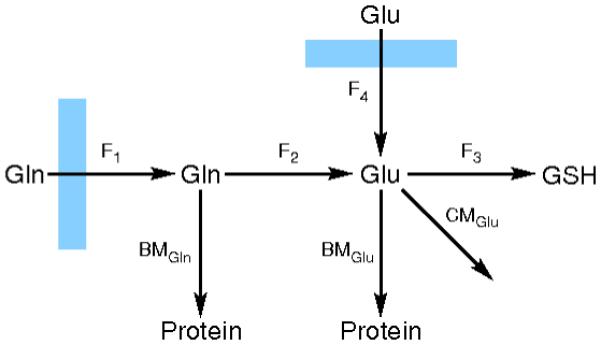
In nutrient-rich growth medium containing glutmate, glutamine is imported and converted to glutamate or protein (BM = rate of biomass production). Glutamate, a metabolic hub, can be used to produce protein and other cellular metabolites (CM) as well as glutathione. Metabolite fluxes are represented as F1, F2, F3, and F4.
Constraining the yeast FBA model with experimental measurements
The iND750 model of metabolism in yeast [17], was augmented to allow for the import of all constituent amino acids in the SCM medium up to a value of 1 mM/gDW/hr. Flux variability analysis [6] was conducted on the unconstrained model and the variability for each reaction was recorded. Using experimental measurements, the amino acid uptake rates were set and a new series of flux variability analyses were conducted.
For incorporation of intracellular flux values in the model, we determined that in nutrient (i.e. amino acid) rich media, the only pathways which require conversion of glutamine to glutamate are four reactions (catalyzed by enzymes EC. 2.4.2.14, EC. 6.3.5.2, EC. 6.3.5.3 and EC. 6.3.5.5) that are involved in the production of carbamoyl phosphate and purines. In order to use our experimental measurement of intracellular flux as a constraint, we fixed the sum of fluxes through these four reactions to the measured value and conducted a new set of flux variability analyses.
The improvements in global flux variability of the system were quantified using the equation:
Where VT is the normalized measure of global flux variability, N is the number of reactions in the model that exhibit flux variability, and represent the lower boundary and upper boundaries for flux of reaction i, and o denotes the values for the unconstrained model.
RESULTS AND DISCUSSION
Extracellular flux measurements by HPLC
Yeast cells were maintained in log-growth phase prior to labeling experiments and throughout the duration of the labeling experiments. Therefore, metabolic steady-state conditions were maintained for these experiments, as required for flux balance analysis. During labeling experiments, a doubling time of 1.84 hours was calculated. Media amino acids were taken up by yeast at different rates (see Table 1). A slight depletion of media 14C label was observed from the beginning of the labeling experiment to 3.5 hours.
Intracellular metabolite fluxes by AMS
The flow of 14C label through the metabolic pathway beginning at extracellular glutamine and proceeding to intracellular glutathione is shown in Figure 1. Specific metabolites in which the 14C label was traced include glutamine, glutamate, and glutathione. While glutathione is a relatively abundant, biologically important biomolecule in yeast, in log-phase growth, the vast majority of the free glutamate pool is used as an amine donor and for protein synthesis. Because the flux from glutamate to glutathione is small, and glutamine is readily taken up by yeast, this pathway was an appropriate target for the measurement of metabolic fluxes using AMS. Figure 2A shows uptake of glutamine, assessed as depletion of glutamine from the media (measured by HPLC) and by uptake of 14C label into cells (measured by AMS). Over time, the 14C-glutamine enters the free amino acid pool (polar metabolite pool) as shown in Figure 2B, and is metabolized as well as converted to biomass, especially proteins. Depletion of 14C label from the media correlated inversely with uptake of label into cells (data not shown). Although the total amount of 14C label continues to increase over time in both the polar metabolite pool and the total cell mass, free intracellular glutamate and glutamine reach isotopic steady state rapidly. Figure 2C shows 14C label normalized against the intracellular concentration of glutamine, glutamate, and glutathione respectively. Glutathione failed to reach isotopic steady state within the 3.5 hour timeframe of this study. In other words, the precursor pool for glutathione biosynthesis (glutamine) had reached isotopic steady state and could be used as a reference point, while the glutathione pool in cells was still accumulating label at a constant rate. The flux from glutamate to glutathione could therefore be determined by calculating the increase in 14C-glutathione over time. The ability to trace the flow of 14C through a relatively low-flux pathway in the presence of a large molar excess of glucose (approximately 100-fold compared to total glutamine) and other carbon sources at concentrations similar to that of glutamine, together with the low degree of labeling of the glutamine precursor (1 in 250,000 glutamine molecules, or approximately 4 × 10−7% of total media carbon) highlights the utility of AMS for the measurement of low-level intracellular metabolic fluxes.
Figure 2. Yeast 14C incorporation and label tracing.


A, Cellular uptake of media glutamine label measured by HPLC and AMS. B, Total cell 14C incorportation and 14C in the polar (free) metabolite pool. C, 14C labeling of free glutamate (Glu), glutamine (Gln), and glutathione (GSH). D, Metabolic flux from intracellular free glutamate to glutathione (GSH) calculated as the increase of 14C label in GSH over time (slope of the plot). Mean values for three independent replicates are plotted with error bars showing standard error of the means.
Model constraint using external and internal fluxes
Incorporation of the measured amino acid uptake rates (extracellular and intracellular metabolic fluxes) into the yeast FBA model provided global metabolic constraints, thus reducing the uncertainty present in the model (see Figure 3). In the absence of experimental constraints, it is mathematically likely that some of the predicted metabolic fluxes from the published yeast FBA model [17] may or may not be realizable in a biological system. The unconstrained model could provide solutions that include futile cycling or unrealistic flux values. In Figure 3, this uncertainty inherent in the unconstrained model is arbitrarily set to “1” (y-axis). Incorporation of most (excluding amino acids such as tyrosine and histidine with minimal metabolic activity) of the measured amino acid consumption rates into the FBA model provides biological constraints that individually or cumulatively reduce the uncertainty in the model by as much as 15%. Thus, measurement of extracellular metabolic fluxes and incorporation of these experimental results into the FBA model constrains the model at a global level.
Figure 3. Constraint of iND750 flux model with metabolic fluxes.

Quantification of reduction in average reaction flux variability inYeast following incorporation of amino acid consumption rates and intracellular flux values into the FBA model [17]. Bars represent the normalized value of total model uncertainty achieved by adding experimentally-determined flux values to the model as system constraints. Y-axis, normalized average system flux variability, where 1 represents maximum variability for the unconstrained model in a nutritionally rich medium and 0 represents no variability. X-axis, measured metabolite fluxes. DERN = system constraint using consumption rates of aspartic acid, glutamate, arginine, and asparagine.
Furthermore, our data show that by incorporating targeted intracellular flux values into FBA models, we may be able to drastically lower the level of uncertainty associated with predicted flux values and expand the utility of these important tools. Our results show (see Figure 3) that by fixing only the intracellular fluxes for conversion of glutamine to glutamate we were able to reduce the level of variability in the FBA flux predictions by nearly 10%. This value is greater than the combined fixing of extracellular fluxes for 4 important amino acids (DERN in Figure 3).
The ability to reliably measure intracellular metabolite fluxes in cells using spectrometric techniques, particularly the accurate and sensitive AMS-based method described here, will enable computational modeling of metabolism with greater accuracy and improved biological relevance. While AMS is extremely powerful in monitoring the fate of a 14C label, it is currently most effective in tracing the metabolic fate and kinetics of relatively few, low-abundance cellular metabolites. Our novel application of AMS has overcome some critical limitations of traditional flux measurement methods, and is able to directly measure intracellular metabolite fluxes using a low-level 14C label in cells grown in nutrient-rich complete media, beginning at isotopic non-steady-state and proceeding to approximate steady-state with respect to glutamine and glutamate. Further experiments using AMS to examine fluxes of critical intracellular intermediates, coupled with global flux measurement techniques, will significantly enhance our understanding of metabolism, leading to a greatly improved ability to manipulate metabolic pathways of interest. It is necessary to continue to improve techniques for measuring metabolic fluxes and increase the number of extracellular and intracellular metabolites that can be traced simultaneously. Because tracing metabolites using AMS requires the measurement of large numbers of samples, throughput of this method must be improved in order to broaden the utility of AMS for metabolic flux measurements. Efforts to couple HPLC directly to AMS are currently in progress at Lawrence Livermore National Laboratory (unpublished results), and are expected to dramatically increase sample throughput and reduce analysis time. Improvements in instrument throughput capabilities will enable routine, rapid tracing of large numbers of metabolites through cellular metabolic pathways.
CONCLUSIONS
We have demonstrated the utility of measurement of intracellular metabolic fluxes by AMS as an adjunct to existing techniques for measuring metabolic fluxes in this proof-of-principle study. This novel, two-tiered (extracellular and intracellular) approach provided global constraints for the Saccharomyces cerevisiae FBA model. The application of extracellular flux data to the FBA model only reduced model uncertainty by approximately 15%. However, by constraining the model with only one intracellular flux we were able to reduce the average variability of model predicted fluxes by nearly 10%. While the model remains underdetermined, this result clearly shows the critical need for targeted measurement of intracellular fluxes in order to improve the predictive power of FBA models. Our results verify that use of flux measurements provides global constraints for the yeast FBA model, and that the use of a targeted labeling approach allows the calculation of metabolite fluxes through a specific pathway using nCi (pmol) levels of 14C-labeled tracer in cells grown in the presence of multiple carbon sources. Thus, AMS can be an effective complement to traditional metabolic flux measurement methods by measuring otherwise intractable intracellular fluxes using complex growth media.
Acknowledgements
The authors would like to thank Professor Eivind Almaas and Dr. Jennifer Links for their help and advice on this project. This work was performed under the auspices of the US Department of Energy by Lawrence Livermore National Laboratory under Contract No. DE-AC52-07NA27344 and was supported by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program (P41RR013461) and LLNL LDRD 09-ERI-002. LLNL release number: LLNL-JRNL-446472.
References
- 1.Blank LM, Kuepfer L, Sauer U. Large-scale 13C-flux analysis reveals mechanistic principles of metabolic network robustness to null mutations in yeast. Genome Biol. 2005;6(6):R49. doi: 10.1186/gb-2005-6-6-r49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wiechert W. 13C metabolic flux analysis. Metab Eng. 2001;3(3):195–206. doi: 10.1006/mben.2001.0187. [DOI] [PubMed] [Google Scholar]
- 3.Fiaux J, et al. Metabolic-flux profiling of the yeasts Saccharomyces cerevisiae and Pichia stipitis. Eukaryot Cell. 2003;2(1):170–80. doi: 10.1128/EC.2.1.170-180.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iwatani S, Yamada Y, Usuda Y. Metabolic flux analysis in biotechnology processes. Biotechnol Lett. 2008;30(5):791–9. doi: 10.1007/s10529-008-9633-5. [DOI] [PubMed] [Google Scholar]
- 5.Sauer U. Metabolic networks in motion: 13C-based flux analysis. Mol Syst Biol. 2006;2:62. doi: 10.1038/msb4100109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mahadevan R, Schilling CH. The effects of alternate optimal solutions in constraint-based genome-scale metabolic models. Metab Eng. 2003;5(4):264–76. doi: 10.1016/j.ymben.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 7.Reed JL, Palsson BO. Thirteen years of building constraint-based in silico models of Escherichia coli. J Bacteriol. 2003;185(9):2692–9. doi: 10.1128/JB.185.9.2692-2699.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Price ND, et al. Genome-scale microbial in silico models: the constraints-based approach. Trends Biotechnol. 2003;21(4):162–9. doi: 10.1016/S0167-7799(03)00030-1. [DOI] [PubMed] [Google Scholar]
- 9.Orth JD, Thiele I, Palsson BO. What is flux balance analysis? Nat Biotechnol. 2010;28(3):245–8. doi: 10.1038/nbt.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cakir T, et al. Flux balance analysis of a genome-scale yeast model constrained by exometabolomic data allows metabolic system identification of genetically different strains. Biotechnol Prog. 2007;23(2):320–6. doi: 10.1021/bp060272r. [DOI] [PubMed] [Google Scholar]
- 11.Murabito E, et al. Capturing the essence of a metabolic network: a flux balance analysis approach. J Theor Biol. 2009;260(3):445–52. doi: 10.1016/j.jtbi.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 12.Raghunathan A, et al. Constraint-based analysis of metabolic capacity of Salmonella typhimurium during host-pathogen interaction. BMC Syst Biol. 2009;3:38. doi: 10.1186/1752-0509-3-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Navid A, Almaas E. Genome-scale reconstruction of the metabolic network in Yersinia pestis, strain 91001. Mol Biosyst. 2009;5(4):368–75. doi: 10.1039/b818710j. [DOI] [PubMed] [Google Scholar]
- 14.Feist AM, et al. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol Syst Biol. 2007;3:121. doi: 10.1038/msb4100155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nookaew I, et al. The genome-scale metabolic model iIN800 of Saccharomyces cerevisiae and its validation: a scaffold to query lipid metabolism. BMC Syst Biol. 2008;2:71. doi: 10.1186/1752-0509-2-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chavali AK, et al. Systems analysis of metabolism in the pathogenic trypanosomatid Leishmania major. Mol Syst Biol. 2008;4:177. doi: 10.1038/msb.2008.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duarte NC, Herrgard MJ, Palsson BO. Reconstruction and validation of Saccharomyces cerevisiae iND750, a fully compartmentalized genome-scale metabolic model. Genome Res. 2004;14(7):1298–309. doi: 10.1101/gr.2250904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee JM, Gianchandani EP, Papin JA. Flux balance analysis in the era of metabolomics. Brief Bioinform. 2006;7(2):140–50. doi: 10.1093/bib/bbl007. [DOI] [PubMed] [Google Scholar]
- 19.Beard DA, Liang SD, Qian H. Energy balance for analysis of complex metabolic networks. Biophys J. 2002;83(1):79–86. doi: 10.1016/S0006-3495(02)75150-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Henry CS, Broadbelt LJ, Hatzimanikatis V. Thermodynamics-based metabolic flux analysis. Biophys J. 2007;92(5):1792–805. doi: 10.1529/biophysj.106.093138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yuan Y, Hoon Yang T, Heinzle E. (13)C metabolic flux analysis for larger scale cultivation using gas chromatography-combustion-isotope ratio mass spectrometry. Metab Eng. 2010 doi: 10.1016/j.ymben.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 22.Zamboni N, et al. (13)C-based metabolic flux analysis. Nat Protoc. 2009;4(6):878–92. doi: 10.1038/nprot.2009.58. [DOI] [PubMed] [Google Scholar]
- 23.Costenoble R, et al. 13C-Labeled metabolic flux analysis of a fed-batch culture of elutriated Saccharomyces cerevisiae. FEMS Yeast Res. 2007;7(4):511–26. doi: 10.1111/j.1567-1364.2006.00199.x. [DOI] [PubMed] [Google Scholar]
- 24.Metallo CM, Walther JL, Stephanopoulos G. Evaluation of 13C isotopic tracers for metabolic flux analysis in mammalian cells. J Biotechnol. 2009;144(3):167–74. doi: 10.1016/j.jbiotec.2009.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zamboni N. (13)C metabolic flux analysis in complex systems. Curr Opin Biotechnol. 2010 doi: 10.1016/j.copbio.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 26.Schilling CH, et al. Combining pathway analysis with flux balance analysis for the comprehensive study of metabolic systems. Biotechnol Bioeng. 2000;71(4):286–306. [PubMed] [Google Scholar]
- 27.Wiechert W, Noh K. From stationary to instationary metabolic flux analysis. Adv Biochem Eng Biotechnol. 2005;92:145–72. doi: 10.1007/b98921. [DOI] [PubMed] [Google Scholar]
- 28.Mo ML, Palsson BO, Herrgard MJ. Connecting extracellular metabolomic measurements to intracellular flux states in yeast. BMC Syst Biol. 2009;3:37. doi: 10.1186/1752-0509-3-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Noh K, Wahl A, Wiechert W. Computational tools for isotopically instationary 13C labeling experiments under metabolic steady state conditions. Metab Eng. 2006;8(6):554–77. doi: 10.1016/j.ymben.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 30.Noh K, Wiechert W. Experimental design principles for isotopically instationary 13C labeling experiments. Biotechnol Bioeng. 2006;94(2):234–51. doi: 10.1002/bit.20803. [DOI] [PubMed] [Google Scholar]
- 31.Yuan J, Bennett BD, Rabinowitz JD. Kinetic flux profiling for quantitation of cellular metabolic fluxes. Nat Protoc. 2008;3(8):1328–40. doi: 10.1038/nprot.2008.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yanagimachi KS, et al. Application of radiolabeled tracers to biocatalytic flux analysis. Eur J Biochem. 2001;268(18):4950–60. doi: 10.1046/j.0014-2956.2001.02426.x. [DOI] [PubMed] [Google Scholar]
- 33.Turteltaub KW, Vogel JS. Bioanalytical applications of accelerator mass spectrometry for pharmaceutical research. Curr Pharm Des. 2000;6(10):991–1007. doi: 10.2174/1381612003400047. [DOI] [PubMed] [Google Scholar]
- 34.Brown K, Dingley KH, Turteltaub KW. Accelerator mass spectrometry for biomedical research. Methods Enzymol. 2005;402:423–43. doi: 10.1016/S0076-6879(05)02014-8. [DOI] [PubMed] [Google Scholar]
- 35.Brown K, Tompkins EM, White IN. Applications of accelerator mass spectrometry for pharmacological and toxicological research. Mass Spectrom Rev. 2006;25(1):127–45. doi: 10.1002/mas.20059. [DOI] [PubMed] [Google Scholar]
- 36.Lappin G, Garner RC. The use of accelerator mass spectrometry to obtain early human ADME/PK data. Expert Opin Drug Metab Toxicol. 2005;1(1):23–31. doi: 10.1517/17425255.1.1.23. [DOI] [PubMed] [Google Scholar]
- 37.Links J, et al. Quantitative metabolism using AMS: Choosing a labeled precursor. Nucl Instrum Methods Phys Res B. 2010;268(7-8):1309–1312. doi: 10.1016/j.nimb.2009.10.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vogel JS. Accelerator mass spectrometry for quantitative in vivo tracing. Biotechniques. 2005;(Suppl):25–9. doi: 10.2144/05386su04. [DOI] [PubMed] [Google Scholar]
- 39.Vogel JS, Love AH. Quantitating isotopic molecular labels with accelerator mass spectrometry. Methods Enzymol. 2005;402:402–22. doi: 10.1016/S0076-6879(05)02013-6. [DOI] [PubMed] [Google Scholar]
- 40.White IN, Brown K. Techniques: the application of accelerator mass spectrometry to pharmacology and toxicology. Trends Pharmacol Sci. 2004;25(8):442–7. doi: 10.1016/j.tips.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 41.Villas-Boas SG, et al. Global metabolite analysis of yeast: evaluation of sample preparation methods. Yeast. 2005;22(14):1155–69. doi: 10.1002/yea.1308. [DOI] [PubMed] [Google Scholar]
- 42.Hans MA, Heinzle E, Wittmann C. Quantification of intracellular amino acids in batch cultures of Saccharomyces cerevisiae. Appl Microbiol Biotechnol. 2001;56(5-6):776–9. doi: 10.1007/s002530100708. [DOI] [PubMed] [Google Scholar]
- 43.Kand’ar R, et al. Determination of reduced and oxidized glutathione in biological samples using liquid chromatography with fluorimetric detection. J Pharm Biomed Anal. 2007;43(4):1382–7. doi: 10.1016/j.jpba.2006.11.028. [DOI] [PubMed] [Google Scholar]
- 44.Legrain C, et al. Regulation of glutamine synthetase from Saccharomyces cerevisiae by repression, inactivation and proteolysis. Eur J Biochem. 1982;123(3):611–6. doi: 10.1111/j.1432-1033.1982.tb06576.x. [DOI] [PubMed] [Google Scholar]
- 45.Hofman-Bang J. Nitrogen catabolite repression in Saccharomyces cerevisiae. Mol Biotechnol. 1999;12(1):35–73. doi: 10.1385/MB:12:1:35. [DOI] [PubMed] [Google Scholar]
- 46.Magasanik B. Ammonia assimilation by Saccharomyces cerevisiae. Eukaryot Cell. 2003;2(5):827–9. doi: 10.1128/EC.2.5.827-829.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]