

Abstract
Sepsis-induced muscle atrophy is produced in part by decreased protein synthesis mediated by inhibition of mTOR (mammalian target of rapamycin). The present study tests the hypothesis that alteration of specific protein-protein interactions within the mTORC1 (mTOR complex 1) contributes to the decreased mTOR activity observed after cecal ligation and puncture in rats. Sepsis decreased in vivo translational efficiency in gastrocnemius and reduced the phosphorylation of eukaryotic initiation factor (eIF) 4E-binding protein (BP) 1, S6 kinase (S6K) 1, and mTOR, compared with time-matched pair-fed controls. Sepsis decreased T246-phosphorylated PRAS40 (proline-rich Akt substrate 40) and reciprocally increased S792-phosphorylated raptor (regulatory associated protein of mTOR). Despite these phosphorylation changes, sepsis did not alter PRAS40 binding to raptor. The amount of the mTOR-raptor complex did not differ between groups. In contrast, the binding and retention of both 4E-BP1 and S6K1 to raptor were increased, and, conversely, the binding of raptor with eIF3 was decreased in sepsis. These changes in mTORC1 in the basal state were associated with enhanced 52032-AMP activated kinase activity. Acute in vivo leucine stimulation increased muscle protein synthesis in control, but not septic rats. This muscle leucine resistance was associated with coordinated changes in raptor-eIF3 binding and 4E-BP1 phosphorylation. Overall, our data suggest the sepsis-induced decrease in muscle protein synthesis may be mediated by the inability of 4E-BP1 and S6K1 to be phosphorylated and released from mTORC1 as well as the decreased recruitment of eIF3 necessary for a functional 48S complex. These data provide additional mechanistic insight into the molecular mechanisms by which sepsis impairs both basal protein synthesis and the anabolic response to the nutrient signal leucine in skeletal muscle.
Keywords: mTOR, PRAS40, raptor, eIF3, atrophy
INTRODUCTION
Negative nitrogen balance and the erosion of lean body mass are defining characteristics of bacterial infection and can adversely affect morbidity and mortality in sepsis (1). The observed atrophy is undoubtedly multifactorial, but it is in part mediated by decreased synthesis of both myofibrillar and sarcoplasmic proteins in skeletal muscle preferentially composed of fast-twitch (e.g., gastrocnemius) fibers (2). This sepsis-induced decrease in muscle protein synthesis results from reduced translational efficiency and is predominantly controlled at the level of peptide-chain initiation (3). In turn, translation initiation is regulated by a number of specialized proteins termed eukaryotic initiation factors (eIFs), many of which are sensitive to hormonal and nutritional signals functioning in a cooperative manner to adjust translation to match cellular requirements. A rate-controlling step in translation initiation is mediated by eIF4F and involves the binding of mRNA to the 43S preinitiation complex. This multimeric eIF4F complex is composed of several proteins including eIF4E, which directly binds the m7GTP-cap structure; eIF4A, which together with eIF4B unwinds secondary structure via its ATP-dependent RNA helicase activity; and eIF4G, which serves as an adaptor protein. eIF4G is the nucleus for the formation of the initiation complex, as evidenced by its binding sites not only for eIF4E but also for eIF4A and eIF3 (4). As a result, eIF4G recruits the 40S subunit to the 52032 end of mRNA and coordinates the circularization of mRNA via its interactions with eIF4E, poly(A)-binding protein (PABP), and eIF3.
Formation of the eIF4F complex controls cap-dependent initiation by regulating the availability of “free” eIF4E. Although sepsis does not alter the total amount of cellular eIF4E, it shifts the distribution of eIF4E from the active to the inactive complex (2, 5). The binding of eIF4E to eIF4E-BP-1 (4E-BP1) allows association with mRNA, but it prevents binding to eIF4G and consequently the formation of the active eIF4F complex (6) (Fig. 1). Nutrients and growth factors positively modulate the ordered phosphorylation of 4E-BP1, releasing 4E-BP1 from eIF4E, and thereby stimulating cap-dependent mRNA translation by enhancing formation of the active eIF4E 00B7 eIF4G complex. The phosphorylation of 4E-BP1 is mediated by the conserved serine (Ser)/threonine (Thr) protein kinase mammalian target of rapamycin (mTOR) (7). A consensus has emerged pertaining to the ability of sepsis to impair eIF4F formation in skeletal muscle. In general, septic insults—such as those imposed by cecal ligation and puncture (CLP) and endotoxin (LPS)—decrease 4E-BP1 phosphorylation (5, 8). This change increases the amount of the inactive eIF4E 00B7 4E-BP1 complex and reciprocally decreases the active eIF4E 00B7 eIF4G complex. The sepsis-induced reduction in eIF4E 00B7 eIF4G diminishes mRNA binding with the ribosome and thereby limits muscle protein synthesis.
Fig. 1.

Regulation of muscle protein synthesis. Simplified schematic of the integrating role of mTOR in controlling diverse cellular signals, such as hormones, nutrients, and stress, regulating protein synthesis. The mTORC1 consisting of mTOR, raptor (regulatory associated protein of mTOR), GβL (also known as mLST8), PRAS40, and DEPTOR stimulates translation initiation and protein synthesis by increasing phosphorylation of both 4E-BP1 (eIF4E BP-1) and S6K1 (ribosomal protein S6K1). In contrast, mTORC2, consisting of mTOR, rictor (rapamycin-insensitive companion of mTOR), GβL, DEPTOR, mSIN1 (mammalian stress-activated protein kinase-interacting protein), and PRR5 (proline-rich protein 5), seems to have minimal impact on muscle protein synthesis. The phosphorylation of 4E-BP1 decreases the inactive 4E-BP1 00B7 eIF4E complex and increases the eIF4E 00B7 eIF4G complex, thereby enhancing cap-dependent translation. Activation of S6K1 phosphorylates a host of proteins, which can differentially regulate protein synthesis. Growth factors, such as insulin and IGF-1 (insulinlike growth factor 1), signal through binding to their cognate receptors and regulate mTORC1 via a PI3K-Akt–dependent pathway. Akt destabilizes the TSC1/TSC2 protein-protein complex, thereby activating mTOR via the small GTPase Rheb (Ras homolog enriched in brain). In addition, nutrients, such as the branched-chain amino acid leucine, increase translation via a mechanism affecting mTOR probably distal to or at the level of Rheb and possibly mediated by the family of Rag G-proteins or Vps34 (vacuolar protein sorting 34). The cellular energy status (i.e., AMP/ATP ratio) is transduced by LKB1 modulation of AMPK (52032-AMP activated protein kinase) activity.
The metabolic consequences of various catabolic stresses and anabolic stimuli are integrated by mTOR (3) (Fig. 1). Sepsis and endotoxin decrease mTOR kinase activity as evidenced by the coordinate decrease in mTOR autophosphorylation at Ser2481 as well as the phosphorylation of the mTOR substrates 4E-BP1 and S6 kinase 1 (S6K1) (2, 5, 8, 9). However, despite its importance, there is a paucity of data pertaining to the mechanism mediating the sepsis-induced decrease in skeletal muscle mTOR activity. In general, mTOR is partitioned between two large multiprotein complexes having distinct functions. One of the mTOR complexes, mTORC2, is considered rapamycin-insensitive and, although an important regulator of the cytoskeleton organization, has little influence on mRNA translation (10). Conversely, the other mTOR complex 1 (mTORC1), consisting of mTOR, GβL (G-protein β-subunit–like protein/mLST8), PRAS40 (proline-rich Akt substrate 40), raptor (regulatory associated protein of mTOR), and DEPTOR (also known as DEP domain containing 6; DEPDC) (7, 11), regulates mTOR activity in a rapamycin-sensitive manner (7) and is altered by sepsis (2). Consistent with this observation, muscle-specific inactivation of raptor, but not rictor, in mice produces muscle atrophy (12). In this regard, raptor is a scaffold protein, which regulates mTOR kinase activity and the subsequent downstream phosphorylation of 4E-BP1 and S6K1. The importance of 4E-BP1 has been discussed above, and the ordered phosphorylation of S6K1 activates the protein resulting in the phosphorylation of more than a dozen substrates, many of which affect cap-dependent translation (13). However, whether sepsis modulates the mTOR 00B7 raptor interaction as well as other newly recognized protein-protein interactions regulating cap-dependent translation in skeletal muscle has not been investigated, and this gap is addressed by the data provided herein.
MATERIALS AND METHODS
Experimental protocols
Male Sprague-Dawley rats (200–225 g; Charles River Breeding Laboratories, Cambridge, Mass) were acclimated for 1 week in a controlled environment. Water and standard rat chow were provided ad libitum. All experiments were approved by the Institutional Animal Care and Use Committee of The Pennsylvania State University College of Medicine and adhered to the National Institutes of Health guidelines for the use of experimental animals. Sepsis was induced by CLP (5, 9). Rats were anesthetized with pentobarbital (50–60 mg/kg), and a midline laparotomy was performed. The cecum was ligated at its base and punctured twice using a 20-gauge needle. The cecum was returned to the peritoneal cavity, the muscle and skin layers were closed separately, and rats were resuscitated with 10 mL of 0.9% sterile saline (3700B0C) administered subcutaneously. The nonseptic control animals received a laparotomy with intestinal manipulation and fluid resuscitation. After surgery, all rats were fasted, but permitted free access to water. Hence, any observed change between septic and nonseptic rats cannot be attributed to differences in nutritional status. In a second study, septic and nonseptic rats were administered 1.35 g/kg body weight of leucine (54 g/L) by oral gavage 24 h after CLP, and muscle was excised 30 min thereafter. This leucine dose maximally stimulates muscle protein synthesis and indices of translation initiation in nonseptic rats at the time point examined (14). Although we have previously reported there is no difference in the plasma leucine concentrations between control and septic rats gavaged with leucine (5, 9), we quantified leucine concentrations in the current study by high-pressure liquid chromatography.
Muscle protein synthesis
In vivo protein synthesis in gastrocnemius was determined approximately 24 h after CLP or sham surgery using the flooding-dose technique (15). Briefly, rats were anesthetized with pentobarbital i.p. (100 mg/kg), and a catheter was placed in the carotid artery. A bolus injection of l-[2,3,4,5,6 –3H]phenylalanine (Phe; 150 mM, 30 μCi/mL; 1 mL/100 g body weight) was injected via the jugular vein, and serial blood samples were drawn after 2, 6, and 10 min for measurement of Phe concentration and radioactivity. Immediately after the final blood sample, the gastrocnemius from 1 leg was frozen in vivo between aluminum blocks precooled to the temperature of liquid nitrogen, and the other muscle was rapidly excised with a portion being homogenized directly, and the remainder freeze clamped. Blood was centrifuged, and plasma was collected. All tissue and plasma samples were stored at –8000B0C until analyzed. The frozen muscle was powdered under liquid nitrogen, and a portion used to estimate the rate of incorporation of [3H]Phe into protein (15). Total RNA was determined, and translational efficiency was calculated by dividing the rate of synthesis by the total RNA per tissue.
Immunoprecipitation and Western blot analysis
The tissue preparation was the same as previously described by our laboratory (2, 5, 8, 9). Muscle was homogenized in ice-cold buffer (pH 7.4) composed of the following (in millimolars): 20 HEPES, 2 EGTA, 50 NaF, 100 KCl, 0.2 EDTA, 50 β-glycerophosphate, 1 DTT, 0.1 PMSF, 1 benzamidine, 0.5 sodium vanadate, plus one protease inhibitor cocktail tablet from Roche. After clarification by centrifugation, the protein concentration was determined, and an equal amount of protein per sample was subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). The blots were incubated with primary antibodies to total and phosphorylated proteins (Cell Signaling, Beverly, Mass, unless otherwise noted) including total and Thr1462-phosphorylated TSC2 (tuberous sclerosis complex 2), total and Thr37/46-phosphorylated 4E-BP1 (Bethyl Laboratories, Montgomery, Tex), total and phosphorylated (both Ser2448 and Ser2481) mTOR, total and Ser1108-phosphorylated eIF4G, total and Ser240/244-phosphorylated-S6, total and Ser428-phosphorylated LKB1, total and Thr172-phosphorylated 52032-AMP–activated protein kinase (AMPK), total and Ser79-phosphorylated acetyl-CoA carboxylase (ACC), total and Thr246-phosphorylated PRAS40 (Biosource, Camarillo, Calif), total and phosphorylated Akt (both Thr308 and Ser473), total and Ser21/9-glycogen synthase kinase (GSK)-α/β, total and Ser422-phosphorylated eIF4B, and total GβL, TSC1, REDD1 (REgulated in Development and DNA damage responses; Peprotech, Inc, Chicago, Ill), eIF4A, PABP, DEPTOR (Millipore, Billerica, Mass), and eIF3f or eIF3b (Abcam, Cambridge, Mass). The blots were developed with enhanced chemiluminescence Western blotting reagents (Amersham, Piscataway, NJ), and the blots were exposed to x-ray film to achieve a signal within the linear range. After development, the film was scanned (Microtek ScanMaker IV) and quantified with Scion Image 3b software (Scion Corp, Frederick, Md).
The eIF4E 00B7 4E-BP1 and eIF4E 00B7 eIF4G complexes were quantified as described (2, 5, 8, 9). Briefly, eIF4E was immunoprecipitated from aliquots of supernatants using an anti-eIF4E monoclonal antibody (kindly provided by Drs. Jefferson and Kimball, Hershey, Pa). Antibody-antigen complexes were collected using magnetic beads, subjected to SDS-PAGE, and finally transferred to a polyvinylidene fluoride membrane. Blots were incubated with a mouse anti–human eIF4E antibody, rabbit anti–rat 4E-BP1 antibody, or rabbit anti-eIF4G antibody.
To maintain potential protein-protein interactions, fresh muscle was also homogenized in CHAPS buffer (pH 7.5) composed of the following(in millimolars): 40 HEPES, 120 NaCl, 1 EDTA, 10 pyrophosphate, 10 β-glycerophosphate, 50 NaF, 1.5 sodium vanadate, 0.3% CHAPS, and one protease inhibitor cocktail tablet (11, 16). The homogenate was clarified by centrifugation, and an aliquot of the supernatant was combined with either anti-TSC2, anti-mTOR, anti-raptor, or anti-eIF3b antibody, and immune complexes were isolated with goat anti– rabbit BioMag IgG (PerSeptive Diagnostics, Boston, Mass) beads. The beads were collected, washed with CHAPS buffer, precipitated by centrifugation, and subjected to SDS-PAGE as described above. All blots were then developed with enhanced chemiluminescence, and the autoradiographs were quantified as above.
Statistical analysis
Experimental data for each condition are summarized as means ± SE, where the number of animals in each treatment group is indicated in the legend to the figure or table. Statistical evaluation of the data was performed using either Student t test for two-group comparisons or ANOVA followed post hoc by Student-Neuman-Keuls test for multiple group comparisons (GraphPad InStat, version 3.05; GraphPad, La Jolla, Calif). Differences between the groups were considered significant when P < 0.05.
RESULTS
Sepsis impairs muscle protein synthesis, mTOR kinase activity, and formation of eIF4F complex
Approximately 24 h after induction of sepsis, translational efficiency of gastrocnemius was assessed in vivo and found to be reduced 37% in septic rats compared with time-matched control rats (control = 114 ±9 vs septic = 72 ±10 nmol Phe incorporated/mg RNA per hour; n = 8 per group; P < 0.05). Note that values are normalized to the total RNA content, which provides an estimate of ribosomal number in muscle. Therefore, the data indicate that the sepsis-induced reduction in muscle protein synthesis was due to impaired translation per se and not a decrease in the relative abundance of ribosomes.
Representative Western blots of muscle homogenates for total and phosphorylated proteins playing a central role in regulating protein translation are presented in Figure 2A. Sepsis decreased 4E-BP1 phosphorylation as well as the phosphorylation of mTOR at Ser2448 and Ser2481, collectively indicating a decrement in mTOR kinase activity. The phosphorylation of S6K1 tended to be reduced in muscle from septic rats (data not shown), but definitive changes were difficult to document because of the relatively low constitutive phosphorylation of S6K1 in the basal fasted state in vivo. However, both Ser1108-phosphorylated eIF4G and Ser240/244-phosphorylated ribosomal protein S6, both S6K1 substrates and rapamycin-sensitive (e.g., mTOR-dependent), were decreased in muscle from septic rats. These sepsis-induced decreases in phosphorylation were independent of a change in the total amount of 4E-BP1, S6, mTOR, or eIF4G. Finally, when the same amount of eIF4E was immunoprecipitated from both groups, the amount of 4E-BP1 bound to eIF4E (e.g., inactive complex) was increased, and, conversely, the amount of eIF4G bound to eIF4E (e.g., active complex) was decreased in muscle from septic rats, compared with control values (Fig. 2B). These data indicate that sepsis decreases functional eIF4F complex formation. Quantitation for the immunoblots in Figure 2 is not presented because comparable changes in these end points have been previously reported by our laboratory (2, 5, 9). These data are presented to confirm the fidelity of the septic model and provide the necessary background for the remainder of the investigation.
Fig. 2.
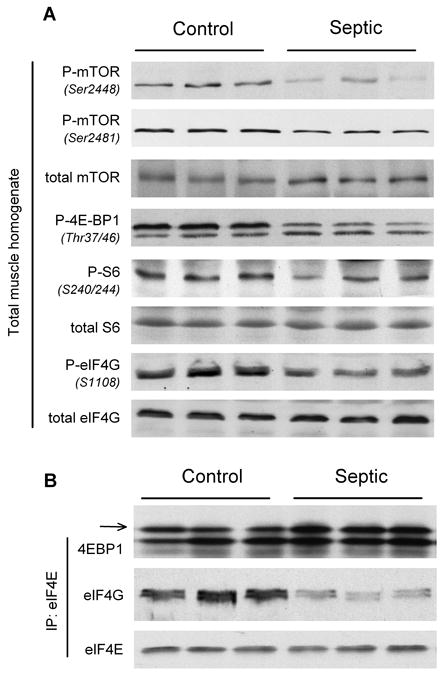
Effect of sepsis on the relative content of total and phosphorylated proteins regulating translation initiation in skeletal muscle. Gastrocnemius was sampled 24 h after CLP or from time-matched pair-fed sham control rats. A, Muscle homogenates were processed, and representative Western blots for phosphorylated mTOR, 4E-BP1, S6, and eIF4G are presented. B, eIF4E was immunoprecipitated (IP) from muscle homogenates and immunoblotted for either 4E-BP1, eIF4G, or eIF4E, and representative blots shown. Arrow refers to the most heavily phosphorylated form of 4E-BP1 and hence most active form of the protein. Sample size was 7 to 10 rats per group for each protein, and representative immunoblots are presented.
Sepsis alters upstream regulators of mTOR
The overall energy status of muscle is sensed by AMPK, which is activated by phosphorylation in various catabolic conditions (17). AMPK Thr172 phosphorylation in gastrocnemius from CLP-treated rats was increased by 65%, compared with time-matched control values (Fig. 3, A and C). The phosphorylation of ACC, a downstream substrate for AMPK, was increased 60%, and this change was independent of a change in total ACC protein (Fig. 3, A and E). AMPK is an LKB1 substrate, and increased LKB1 Ser428 phosphorylation is important in activating AMPK (18). Sepsis increased Ser428-phosphorylated LBK1 greater than 50% (Fig. 3, B and D). REDD1 is a novel stress-response gene, which is upregulated by a diverse array of catabolic insults in an AMPK-dependent manner (19) and decreases mTOR activity (20). Muscle from septic rats had a greater than 8-fold increase in REDD1 protein, compared with control values (Fig. 3, B and F).
Fig. 3.
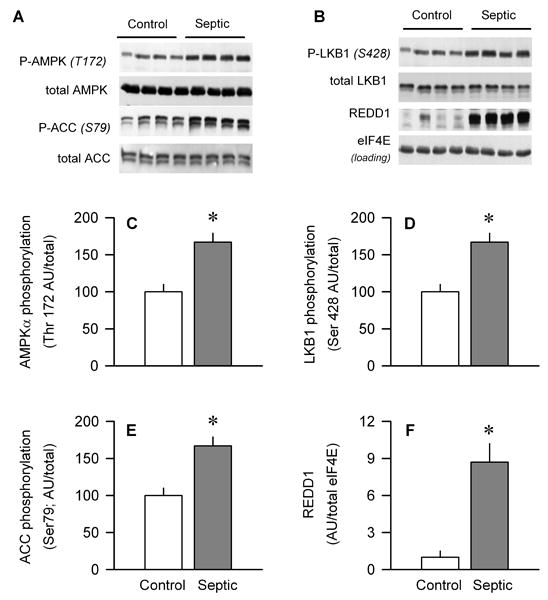
Effect of sepsis on the total amount and phosphorylation of AMPK, ACC, and LKB1 as well as total REDD1 protein in gastrocnemius. A and B, Representative Western blots for total and phosphorylated proteins. Bar graphs, quantitation of all Western blot data for Thr172-phosphorylated AMPK (C), Ser428-phosphorylated LKB1 (D), Ser79-phosphorylated ACC (E), and total REDD1 (F) normalized to the total amount of the respective protein or loading control, and the control value set at 100 arbitrary units (AUs). Values are means ± SEM; n = 7–10 rats each. *P < 0.05, compared with time-matched pair-fed control values.
As Akt is upstream of mTOR and modulates its Ser/Thr kinase activity, we assessed Akt phosphorylation on two sites thought to be necessary for its full activation (7). Sepsis coordinately decreased both Thr308-phosphorylated (e.g., phosphatidylinositol-32032 kinase [PI3K]-dependent) and Ser473-phosphorylated (e.g., mTORC2-dependent) Akt in muscle by approximately 40% (Fig. 4, A, C, and E). However, there was a discordant sepsis-induced change in the phosphorylation of two downstream Akt substrates. That is, whereas sepsis decreased PRAS40 phosphorylation (Thr246) by 40% (Fig. 4, B and D), no change in Ser21/9-phosphorylated GSKα/β was detected in gastrocnemius (Fig. 4, B and F).
Fig. 4.
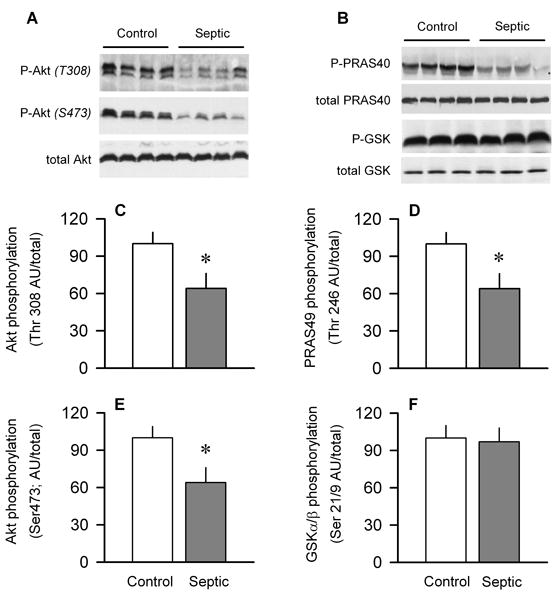
Effect of sepsis on phosphorylation of Akt and downstream target proteins PRAS40 and GSK in gastrocnemius. A and B are representative Western blots of phosphorylated and total Akt, PRAS40, and GSK. C and E, Quantitation of all Western blot data for Thr308- and Ser473-phosphorylated Akt, respectively, normalized to total Akt. D and F, Quantitation of all Western blot data for Thr246-phosphorylated PRAS40 and Ser21/9-phosphorylated GSKα/β, respectively, normalized to total protein. Values for control animals were set at 100 AU. Values are means ± SEM; n = 7–10 rats each. *P < 0.05, compared with time-matched pair-fed control values.
Activated Akt directly phosphorylates TSC2 on Thr1462 and ultimately relieves the inhibitory action of the TSC1 00B7 TSC2 complex on mTOR activity (21). AMPK and REDD1 also alter mTOR kinase activity at least in part by modulation of TSC2 phosphorylation and TSC1 00B7 TSC2 complex formation. However, the total amount of TSC1, TSC2, Thr1462-phosphorylated TSC2 and the amount of the TSC1 00B7 TSC2 heterodimer in muscle did not differ between control and septic rats (Fig. 5).
Fig. 5.
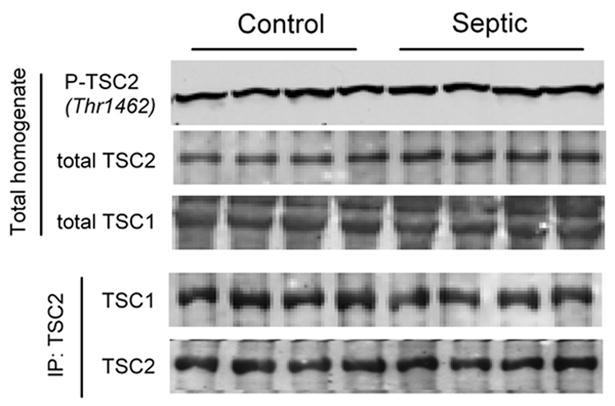
Effect of sepsis on TSC in gastrocnemius. Top three panels, Representative Western blots from Thr1462-phosphorylated TSC2 and total TSC2, and total TSC1, respectively. Bottom two panels, Representative immunoblots of TSC1 and TSC2 performed after immunoprecipitation (IP) of TSC2 from muscle homogenates. Statistical analysis of data from 7 to 10 rats per group indicate no statistical significance for any end point between control and septic rats (data not shown).
Sepsis-induced change in mTORC1
mTORC1 is a multiprotein complex regulating protein synthesis and consisting of mTOR, raptor, GβL, PRAS40, and DEPTOR (7, 11, 22). Of these proteins, the total amount of mTOR (Fig. 1A), PRAS40 (Fig. 3B), GβL (Fig. 6A), and raptor (Fig. 6A) did not differ between control and septic rats. However, sepsis increased raptor phosphorylation by almost 90% (Fig. 6B). Finally, sepsis increased the total DEPTOR, an mTOR-interacting protein believed to be a negative regulator of mTOR (23), in septic muscle by 70%, compared with control values (Fig. 6C).
Fig. 6.
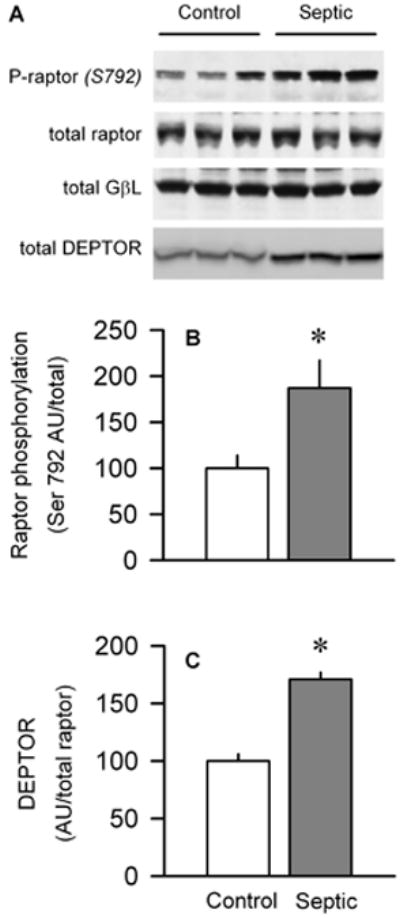
Effect of sepsis on raptor phosphorylation and total DEPTOR. A, Representative Western blots for Ser792-phosphorylated raptor as well as total raptor, GβL, and DEPTOR , respectively. Statistical analysis of all data indicated that there was no significant difference in the amount of total raptor or GβL between muscle from control and septic rats (data not shown). B and C, Quantitation of Western blot data for phosphorylated raptor and total DEPTOR, respectively, normalized to loading protein and control value set at 100 AU. Values are means ± SEM; n = 5–10 rats each. *P < 0.05, compared with time-matched pair-fed control values.
Raptor functions as a scaffold protein recruiting substrates to mTORC1 via short TOS (mTOR signaling) motifs in its substrates (11, 22). Therefore, raptor was immunoprecipitated and then immunoblotted for PRAS40, 4E-BP1, S6K1, and GβL. Although sepsis did not alter the association of raptor with PRAS40 or GβL (Fig. 7A), it did increase both 4E-BP1 (Fig. 7B) and S6K1 (Fig. 7C) binding to raptor by 70%.
Fig. 7.
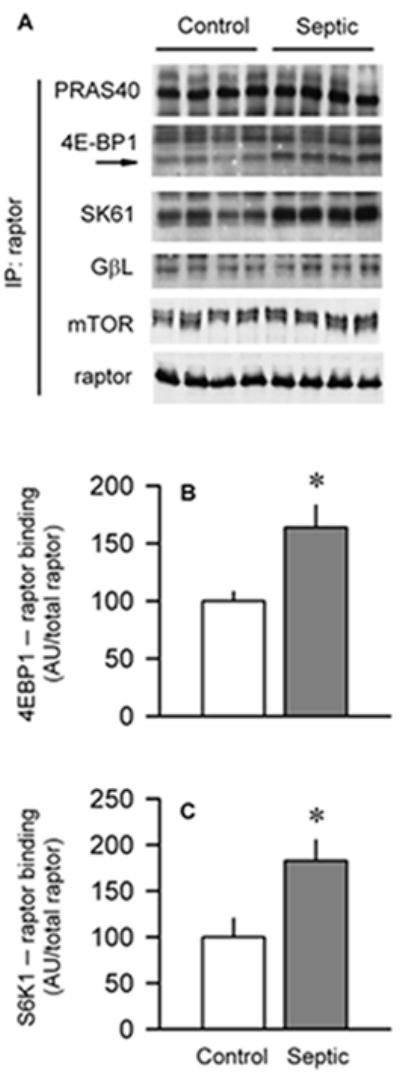
Effect of sepsis on binding of raptor to various partner proteins in gastrocnemius. A, Raptor was immunoprecipitated (IP) and then immunoblotted for PRAS40, 4E-BP1, S6K1, GβL, mTOR, or raptor. B and C, Quantitation of Western blot data normalized to amount of raptor in the IP, with control value set at 100 AU. Values are means ± SEM; n = 6 rats each. *P < 0.05, compared with time-matched pair-fed control values.
In addition, when raptor was immunoprecipitated, the amount of mTOR bound to raptor did not differ between control and septic rats (Fig. 7A). Although this finding was unexpected, it was confirmed by performing the reverse immunoprecipitation procedure (data not shown). However, we did detect a greater than 90% reduction in mTOR 00B7 raptor interaction in rats treated with rapamycin (data not shown), indicating our ability to detect a change in this protein-protein complex.
With regard to mTORC2, there was no sepsis-induced change in the total amount of rictor in muscle homogenates or the amount of rictor bound to an mTOR immunoprecipitate (data not shown).
Sepsis-induced change in eIF3
The largest translation initiation factor, eIF3, is composed of 13 nonidentical subunits (24). Although the physiological function of each subunit is not known, it is clear that eIF3 must bind to mTORC1 for optimal mTOR kinase activity (25). We assessed the total amount of two of the subunits, eIF3-b (e.g., one of the five conserved core polypeptides essential for translation) and eIF3-f (e.g., the subunit shown to be rapidly degraded by the ubiquitin-proteasome). Our results indicate that the total content of eIF3b and eIF3f in muscle homogenates did not differ between control and septic rats (Fig. 8A). Moreover, there was no sepsis-induced change in the amount of total or phosphorylated eIF4B, total eIF4A1, or total PABP—all proteins implicated in regulating mRNA protein translation (Fig. 8A). Finally, we immunoprecipitated eIF3b from muscle and determined the amount of bound raptor. Data in Figure 8C indicate that sepsis decreased the amount of the eIF3 00B7 raptor complex by 40%.
Fig. 8.
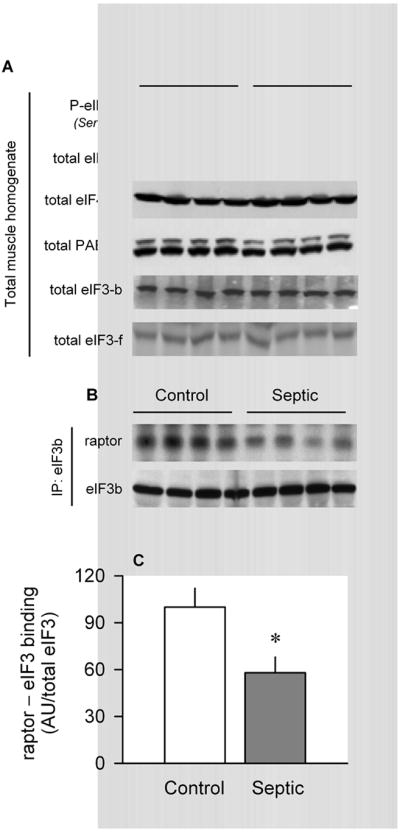
Effect of sepsis on total eIF3 and eIF3 00B7 raptor in muscle. A, Representative Western blots of total and Ser422-phosphorylated eIF4B, eIF4A1, PABP, eIF3-b, and eIF3-f. For each of these proteins, there was no statistical difference in the relative amount of the protein in muscle between control and septic rats (quantitative data not shown). B, eIF3b was immunoprecipitated (IP) from muscle and immunoblotting performed for both raptor and eIF3b. C, Bar graph, quantitation of Western blot data of eIF3 00B7 raptor binding normalized to the amount of eIF3b in the IP, where control value was set at 100 AU. Values are means ± SEM; n = 5 rats each. *P < 0.05, compared with time-matched pair-fed control values.
Leucine-induced changes in mTORC1
In the second study, leucine was orally gavaged approximately 24 h after induction of sepsis or sham surgery. Leucine administration to control rats increased muscle protein synthesis by 37%, and this anabolic response was completely absent in septic rats (Fig. 9A). This leucine resistance was not due to sepsis- or leucine-induced changes in mTOR 00B7 raptor binding (Fig. 9B), but it was associated with concordant changes in raptor binding to eIF3 (Fig. 9C). The phosphorylation of both 4E-BP1 (Fig. 9D) and S6K1 (data not shown) was also increased by leucine in control but not septic rats (Fig. 9D), and such a response is consistent with the change in mTOR kinase activity anticipated by previously mentioned changes in raptor 00B7 eIF3b binding. The differential ability of leucine to alter raptor 00B7 eIF3 binding could not be attributed to altered raptor Ser792 phosphorylation (Fig. 9E) or PRAS40 Ser246 phosphorylation (Fig. 9F). Acute leucine stimulation did not alter basal AMPKα or Akt phosphorylation in control rats or the sepsis-induced change in phosphorylation for these two proteins (data not shown). Finally, none of the observed differences between control and septic rats could be attributed to differences in the plasma leucine either under basal conditions (control = 112 ±28 μmol/L and septic = 133 ± 19 μmol/L; not statistically significant) or in response to the leucine gavage (control = 967 ±34 μmol/L and septic = 1002 ± 55 μmol/L; not statistically significant).
Fig. 9.

Leucine-induced changes in protein synthesis and mTORC1 in skeletal muscle from control and septic rats. Gastrocnemius was sampled 30 min after oral administration of a maximally stimulating dose of the branched-chain amino acid leucine. In vivo muscle protein synthesis was determined as described in Materials and Methods (A). Bar graphs are quantitation of immunoblots after immunoprecipitation (IP) of either raptor (B) or eIF3b (C). Western blot data from whole muscle tissue homogenates have also been quantitated (D, E, and F). There was no sepsis or leucine effect on the total amount of raptor, mTOR, eIF3b, 4E-BP1, or PRAS40 (data not shown). Saline-treated control values were arbitrarily set to 1.0 AU. Values are means ± SEM; n = 7–9 rats per group. Values with different letters are statistically different (P < 0.05).
DISCUSSION
mTOR signaling occupies a central role in integrating nutrient, energy, and growth stimuli to regulate muscle protein synthesis (7) (Fig. 1). Sepsis and endotoxin impair mTOR kinase activity in muscle as evidenced by the reduction in mTOR autophosphorylation as well as decreased phosphorylation of downstream substrates 4E-BP1 and S6K1 (8, 9). The data in the present study confirm these observations. Although we acknowledge that this study is largely descriptive in nature, our data do suggest potential mechanisms by which sepsis impairs mTOR kinase. Of the two mTOR-containing protein complexes, mTORC1 seems primarily involved in regulating protein synthesis (11, 22). However, our data indicate that, under basal postabsorptive conditions, sepsis does not alter the assembly of the mTOR 00B7 raptor complex in skeletal muscle where protein synthesis and translational efficiency were simultaneously ascertained to be reduced. These findings in septic rats differ from those reported in alcohol-treated rats where impaired muscle protein synthesis and mTOR kinase activity were associated with an increased mTOR 00B7 raptor binding (16). The reason for the difference in mTOR 00B7 raptor binding between these two catabolic conditions is not evident, and little insight is gained from in vitro studies in nonmuscle cells where the mTOR 00B7 raptor interaction to the withdrawal of serum (e.g., growth factors) and/or amino acids has also been shown to be divergent (11, 22, 26, 27). We speculate that such divergent results suggest that other protein-binding partners are more critically involved in regulating mTORC1 activity under catabolic conditions than the simple association of mTOR and raptor.
Cellular stresses inhibit mTORC1 through a diverse array of potential mechanisms. For example, AMPK activation in response to energy stress inhibits protein synthesis in cultured myocytes and in vivo muscle (28). Our data demonstrate an LKB1-dependent sepsis-induced increase in AMPK activation, as evidenced by the enhanced phosphorylation of AMPK and ACC. In turn, AMPK phosphorylates multiple targets. One mechanism whereby AMPK decreases protein synthesis is via the TSC complex, as increased TSC2 phosphorylation as well as enhanced formation of the TSC1 00B7 TSC2 heterodimer can negatively regulate mTOR activity (29). However, although AMPK in muscle was activated by sepsis, this mechanism does not seem operational because TSC1 00B7 TSC2 association and the AMPK-sensitive Thr1462 phosphorylation of TSC2 did not differ between control and septic rats. AMPK may also inhibit mTOR kinase activity in a TSC-independent manner as raptor Ser792 phosphorylation facilitates recruitment of the 14-3-3 protein (29). Such a sepsis-induced increase in raptor phosphorylation was detected and is consistent with the increased raptor phosphorylation observed in rats administered the AMPK activator AICAR (5-aminoimidazole-4-carboxamide-1β-d-ribonucleoside) (28) and in cultured myocytes incubated with endotoxin (30). Collectively, these data suggest that AMPK “senses” an energy deficit (e.g., increased AMP/ATP ratio) in muscle, presumably as a result of mitochondrial dysfunction, which may mediate part of the sepsis-induced increased decrease in protein synthesis in this tissue. Such a sepsis-induced defect in mitochondrial oxidative phosphorylation has been reported in cardiac muscle (31).
Our data indicate that sepsis also upregulates the REDD1 protein in muscle, a response comparable to that observed after AMPK activation by AICAR (32). The ability of REDD1 to negatively regulate mTOR seems to proceed via the binding of REDD1 to 14-3-3, the latter of which normally binds to and inhibits TSC2 activity (19). Because such a redistribution of 14-3-3 from TSC2 to REDD1 would not be expected to disrupt the TSC1 00B7 TSC2 complex, our data cannot exclude this as a possible mechanism for the sepsis-induced inhibition of mTOR activity.
PRAS40 also binds to raptor and is believed to be a translational repressor, so its overexpression decreases mTOR activity (33). Upon phosphorylation induced by insulin and other growth factors, PRAS40 is released from raptor and thereby enhances protein synthesis by facilitating the engagement of 4E-BP1 and S6K1 with the limiting amount of raptor. Multiple lines of evidence indicate that PRAS40 Thr246 phosphorylation is Akt-dependent (34), and our current data showing a sepsis-induced decrease in both Akt activation and PRAS40 phosphorylation support such a mechanism. Moreover, Akt may directly phosphorylate mTOR (35). Collectively, these results are also consistent with the endotoxin-induced reduction in Akt, mTOR, and PRAS40 phosphorylation seen in cultured myotubes (30). Therefore, the lack of a significant difference in the amount of the PRAS40 00B7 raptor complex in muscle from septic rats was unexpected. Although PRAS40 preferentially binds to raptor, it can also interact with the kinase domain of mTOR (33); however, no difference in the amount of the PRAS40 00B7 mTOR complex was detected in muscle of control and septic rats (unpublished observation).
In contrast, we did detect a sepsis-induced increase in the amount of 4E-BP1 00B7 raptor and S6K1 00B7 raptor complex in skeletal muscle (Fig. 10). Recruitment of these mTOR substrates to mTORC1 is necessary for their canonical phosphorylation by mTOR kinase, and raptor is known to preferentially bind the nonphosphorylated substrates (11). We interpret the increased complex formation in raptor immunoprecipitates, in conjunction with the reduced phosphorylation of 4E-BP1 and S6K1 in total tissue homogenates, to indicate there is no sepsis-induced defect in the 4E-BP1 or S6K1 binding to raptor per se, but that their subsequent phosphorylation and release from mTORC1 are impaired. This in vivo scenario differs from that seen in vitro where cells cultured under the catabolic conditions produced by the absence of amino acids and/or growth factors demonstrate a reduced formation of 4E-BP1 00B7 raptor (27, 36). It is noteworthy that the sepsis-induced change in mTOR kinase activity was associated with a marked increase in DEPTOR, a negative regulator of mTORC1 activity (23), and the importance of this increase warrants investigation.
Fig. 10.

Schematic for sepsis-induced changes in mTORC1. Under basal conditions, S6K1 is associated with the eIF3 at the PIC (preinitiation complex). In addition, 4E-BP1 is bound to the eIF4E, which in turn is bound to the 7m-GTP cap of the mRNA at the 52032 end. Under inhibitory conditions (as in rapamycin-treated), mTOR/raptor complex is not part of this mRNA PIC. Upon stimulation (as in after anabolic stimulation), mTOR/raptor complex is recruited to the eIF3 complex. Activated mTOR then phosphorylates both S6K1 (at T389) and 4E-BP1 (at several sites in a hierarchy manner). Upon phosphorylation, these mTOR substrates (S6K1 and 4E-BP1) dissociate from the eIF3. The unbound dissociated (phosphorylated) S6K1 can be further phosphorylated by PDK1 (3-phosphoinositide–dependent kinase 1) at T229; this latter secondary phosphorylation is believed to be required for the full activation of the S6K which is capable of phosphorylating more than 10 known substrates. All abbreviations are defined in the text and in Figure 1.
eIF3 is a multisubunit protein complex that serves as a docking site for the binding of several components of the translational machinery (24). Incubation of myotubes with various catabolic agents has been reported to decrease eIF3f content, and eIF3f knockdown decreases protein synthesis (37) and produces muscle atrophy (38). However, our data demonstrate that the total eIF3f content did not differ in muscle of control and septic rats. Moreover, the interaction of eIF3 with the eIF4F 00B7 mRNA complex facilitates the binding of mRNA to the 43S ribosomal complex and formation of the 48S complex. S6K1 dissociates from eIF3 upon activation (25), and therefore, catabolic states would be expected to increase the association of hypophosphorylated (e.g., inactive) S6K1 with eIF3. Conversely, the binding of mTORC1 to eIF3 would be expected to decrease under conditions where protein synthesis is inhibited. Consistent with this expectation, our current data indicate that sepsis decreased the binding of raptor in mTORC1 to eIF3b. Finally, eIF4B is an ancillary factor for optimal ATPase/RNA helicase activity of eIF4A in the functional eIF4F complex. Phosphorylation of eIF4B enhances its association with eIF3 and promotes translation. Although eIF4B associates with eIF3 under insulin-stimulated conditions, but not serum-starved conditions (25), this association was not investigated in the current study because a significant sepsis-induced change in Ser422-phosphorylated eIF4B in whole muscle homogenate was not detected. The absence of a decrease in eIF4B phosphorylation was unexpected as eIF4B is a known substrate for S6K1 and Akt, and the kinase activity of both proteins was concomitantly downregulated by sepsis (3, 5, 9).
In our second study, we used acute in vivo leucine stimulation to perturb protein balance as a physiological approach to identify potential intracellular signaling intermediates contributing to the sepsis-induced decrease in muscle protein synthesis. In this regard, we confirmed that muscle from septic rats is essentially unresponsive to the ability of leucine to enhance protein synthesis and stimulate mTOR kinase activity (5). As discussed above in the basal state, the sepsis-induced leucine resistance could not be attributed to altered mTOR 00B7 raptor binding, but it was consistent with the inability of leucine to increase raptor 00B7 eIF3 binding as observed in control muscle. Furthermore, this defect in raptor 00B7 eIF3 binding and mTOR kinase activity could not be explained by differential raptor phosphorylation, at least at residue Ser792, as seen in other conditions (29). As AMPKα directly phosphorylates the Ser792 residue of raptor (29), these data suggest that the sepsis-induced increase in AMPKα activity is an unlikely mediator of leucine resistance. Finally, PRAS40 phosphorylation is necessary for leucine-stimulated mTOR kinase activity (e.g., S6K1 phosphorylation) in cardiac muscle (39). Our data support this conclusion in skeletal muscle of control rats. However, in contrast, the leucine-induced increment in PRAS40 phosphorylation was comparable in both control and septic rats, suggesting that PRAS40 phosphorylation may not be causative in the sepsis-induced leucine resistance. The systematic study of other mechanisms by which sepsis impairs the anabolic effect of leucine is beyond the scope of the current article, but it might involve the G protein Rag (40) or Vps34 (41). In conclusion, the multitude of signaling events described herein provides a conceptual framework regarding the ability of sepsis to impair muscle protein synthesis via altering protein-protein interactions within mTOR1, specifically the binding of 4E-BP1 and eIF3 with raptor, which may impair both basal and nutrient-stimulated increases in muscle protein synthesis (Fig. 10).
Acknowledgments
The authors thank Drs. Jefferson and Kimball from the Penn State College of Medicine for the generous gift of eIF4E antibody.
This work was supported in part by National Institutes of Health grant GM38032 (to C.H.L.) and the Pennsylvania Department of Health using Tobacco Settlement Funds (to A.A.K.).
Footnotes
The Department of Health specifically disclaims responsibility for any analyses, interpretations, or conclusions.
References
- 1.Long CL, Jeevanandam M, Kim BM, Kinney JM. Whole body protein synthesis and catabolism in septic man. Am J Clin Nutr. 1977;30:1340–1344. doi: 10.1093/ajcn/30.8.1340. [DOI] [PubMed] [Google Scholar]
- 2.Lang CH, Frost RA. Sepsis-induced suppression of skeletal muscle translation initiation mediated by tumor necrosis factor alpha. Metabolism. 2007;56:49–57. doi: 10.1016/j.metabol.2006.08.025. [DOI] [PubMed] [Google Scholar]
- 3.Lang CH, Frost RA, Vary TC. Regulation of muscle protein synthesis during sepsis and inflammation. Am J Physiol Endocrinol Metab. 2007;293:E453–E459. doi: 10.1152/ajpendo.00204.2007. [DOI] [PubMed] [Google Scholar]
- 4.Hinton TM, Coldwell MJ, Carpenter GA, Morley SJ, Pain VM. Functional analysis of individual binding activities of the scaffold protein eIF4G. J Biol Chem. 2007;282:1695–1708. doi: 10.1074/jbc.M602780200. [DOI] [PubMed] [Google Scholar]
- 5.Lang CH, Frost RA. Glucocorticoids and TNFalpha interact cooperatively to mediate sepsis-induced leucine resistance in skeletal muscle. Mol Med. 2006;12:291–299. doi: 10.2119/2006-00071.Lang. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hara K, Yonezawa K, Kozlowski MT, Sugimoto T, Andrabi K, Weng QP, Kasuga M, Nishimoto I, Avruch J. Regulation of eIF-4E BP1 phosphorylation by mTOR. J Biol Chem. 1997;272:26457–26463. doi: 10.1074/jbc.272.42.26457. [DOI] [PubMed] [Google Scholar]
- 7.Laplante M. Sabatini DM: mTOR signaling at a glance. J Cell Sci. 2009;122:3589–3594. doi: 10.1242/jcs.051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lang CH, Frost RA. Endotoxin disrupts the leucine-signaling pathway involving phosphorylation of mTOR, 4E-BP1, and S6K1 in skeletal muscle. J Cell Physiol. 2005;203:144–155. doi: 10.1002/jcp.20207. [DOI] [PubMed] [Google Scholar]
- 9.Lang CH, Frost RA. Differential effect of sepsis on ability of leucine and IGF-I to stimulate muscle translation initiation. Am J Physiol Endocrinol Metab. 2004;287:E721–E730. doi: 10.1152/ajpendo.00132.2004. [DOI] [PubMed] [Google Scholar]
- 10.Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A, Hall MN. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6:1122–1128. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- 11.Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, Tokunaga C, Avruch J, Yonezawa K. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. 2002;110:177–189. doi: 10.1016/s0092-8674(02)00833-4. [DOI] [PubMed] [Google Scholar]
- 12.Bentzinger CF, Romanino K, Cloetta D, Lin S, Mascarenhas JB, Oliveri F, Xia J, Casanova E, Costa CF, Brink M, et al. Skeletal muscle-specific ablation of raptor, but not of rictor, causes metabolic changes and results in muscle dystrophy. Cell Metab. 2008;8:411–424. doi: 10.1016/j.cmet.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 13.Ruvinsky I, Meyuhas O. Ribosomal protein S6 phosphorylation: from protein synthesis to cell size. Trends Biochem Sci. 2006;31:342–348. doi: 10.1016/j.tibs.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 14.Crozier SJ, Kimball SR, Emmert SW, Anthony JC, Jefferson LS. Oral leucine administration stimulates protein synthesis in rat skeletal muscle. J Nutr. 2005;135:376–382. doi: 10.1093/jn/135.3.376. [DOI] [PubMed] [Google Scholar]
- 15.Vary TC, Lang CH. Assessing effects of alcohol consumption on protein synthesis in striated muscles. Methods Mol Biol. 2008;447:343–355. doi: 10.1007/978-1-59745-242-7_22. [DOI] [PubMed] [Google Scholar]
- 16.Lang CH, Pruznak AM, Nystrom GJ, Vary TC. Alcohol-induced decrease in muscle protein synthesis associated with increased binding of mTOR and raptor: comparable effects in young and mature rats. Nutr Metab (Lond) 2009;6:4. doi: 10.1186/1743-7075-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- 18.Xie Z, Dong Y, Scholz R, Neumann D, Zou MH. Phosphorylation of LKB1 at serine 428 by protein kinase C-zeta is required for metformin-enhanced activation of the AMP-activated protein kinase in endothelial cells. Circulation. 2008;117:952–962. doi: 10.1161/CIRCULATIONAHA.107.744490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DeYoung MP, Horak P, Sofer A, Sgroi D, Ellisen LW. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev. 2008;22:239–251. doi: 10.1101/gad.1617608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kimball SR, Do AN, Kutzler L, Cavener DR, Jefferson LS. Rapid turnover of the mTOR complex 1 (mTORC1) repressor REDD1 and activation of mTORC1 signaling following inhibition of protein synthesis. J Biol Chem. 2008;283:3465–3475. doi: 10.1074/jbc.M706643200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tee AR, Blenis J, Proud CG. Analysis of mTOR signaling by the small G-proteins, Rheb and RhebL1. FEBS Lett. 2005;579:4763–4768. doi: 10.1016/j.febslet.2005.07.054. [DOI] [PubMed] [Google Scholar]
- 22.Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 23.Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, Gray NS, Sabatini DM. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137:873–886. doi: 10.1016/j.cell.2009.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hinnebusch AG. eIF3: a versatile scaffold for translation initiation complexes. Trends Biochem Sci. 2006;31:553–562. doi: 10.1016/j.tibs.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 25.Holz MK, Ballif BA, Gygi SP, Blenis J. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell. 2005;123:569–580. doi: 10.1016/j.cell.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 26.Oshiro N, Yoshino K, Hidayat S, Tokunaga C, Hara K, Eguchi S, Avruch J, Yonezawa K. Dissociation of raptor from mTOR is a mechanism of rapamycin-induced inhibition of mTOR function. Genes Cells. 2004;9:359–366. doi: 10.1111/j.1356-9597.2004.00727.x. [DOI] [PubMed] [Google Scholar]
- 27.Wang L, Harris TE, Roth RA, Lawrence JC., Jr PRAS40 regulates mTORC1 kinase activity by functioning as a direct inhibitor of substrate binding. J Biol Chem. 2007;282:20036–20044. doi: 10.1074/jbc.M702376200. [DOI] [PubMed] [Google Scholar]
- 28.Pruznak AM, Kazi AA, Frost RA, Vary TC, Lang CH. Activation of AMP-activated protein kinase by 5-aminoimidazole-4-carboxamide-1-beta-d-ribonucleoside prevents leucine-stimulated protein synthesis in rat skeletal muscle. J Nutr. 2008;138:1887–1894. doi: 10.1093/jn/138.10.1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frost RA, Nystrom GJ, Lang CH. Endotoxin and interferon-gamma inhibit translation in skeletal muscle cells by stimulating nitric oxide synthase activity. Shock. 2009;32:416–426. doi: 10.1097/SHK.0b013e3181a034d2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levy RJ, Vijayasarathy C, Raj NR, Avadhani NG, Deutschman CS. Competitive and noncompetitive inhibition of myocardial cytochrome C oxidase in sepsis. Shock. 2004;21:110–114. doi: 10.1097/01.shk.0000108400.56565.ab. [DOI] [PubMed] [Google Scholar]
- 32.Frost RA, Huber D, Pruznak A, Lang CH. Regulation of REDD1 by insulin-like growth factor-I in skeletal muscle and myotubes. J Cell Biochem. 2009;108:1192–1202. doi: 10.1002/jcb.22349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, Carr SA, Sabatini DM. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007;25:903–915. doi: 10.1016/j.molcel.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 34.Nascimento EB, Ouwens DM. PRAS40: target or modulator of mTORC1 signalling and insulin action? Arch Physiol Biochem. 2009;115:163–175. doi: 10.1080/13813450902988580. [DOI] [PubMed] [Google Scholar]
- 35.Nave BT, Ouwens M, Withers DJ, Alessi DR, Shepherd PR. Mammalian target of rapamycin is a direct target for protein kinase B: identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem J. 1999;344(pt 2):427–431. [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang L, Kimball SR, Jefferson LS, Shenberger JS. Hydrogen peroxide impairs insulin-stimulated assembly of mTORC1. Free Radic Biol Med. 2009;46:1500–1509. doi: 10.1016/j.freeradbiomed.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou C, Arslan F, Wee S, Krishnan S, Ivanov AR, Oliva A, Leatherwood J, Wolf DA. PCI proteins eIF3e and eIF3m define distinct translation initiation factor 3 complexes. BMC Biol. 2005;3:14. doi: 10.1186/1741-7007-3-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lagirand-Cantaloube J, Offner N, Csibi A, Leibovitch MP, Batonnet-Pichon S, Tintignac LA, Segura CT, Leibovitch SA. The initiation factor eIF3-f is a major target for atrogin1/MAFbx function in skeletal muscle atrophy. EMBO J. 2008;27:1266–1276. doi: 10.1038/emboj.2008.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sanchez CC, Demeulder B, Ginion A, Bayascas JR, Balligand JL, Alessi DR, Vanoverschelde JL, Beauloye C, Hue L, Bertrand L. Activation of the cardiac mTOR/p70(S6K) pathway by leucine requires PDK1 and correlates with PRAS40 phosphorylation. Am J Physiol Endocrinol Metab. 2010;298:E761–E769. doi: 10.1152/ajpendo.00421.2009. [DOI] [PubMed] [Google Scholar]
- 40.Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320:1496–1501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Backer JM. The regulation and function of Class III PI3Ks: novel roles for Vps34. Biochem J. 2008;410:1–17. doi: 10.1042/BJ20071427. [DOI] [PubMed] [Google Scholar]