

Abstract
Chronic hepatitis C infection leads to increased hepatocyte apoptosis. Because engulfment of apoptotic bodies (ABs) by hepatic stellate cells (HSC) is profibrogenic, we compared the effects of ABs derived from HCV-negative vs. HCV-infected (Con1+) Huh7 hepatoblastoma cells on fibrogenic and activation-related mRNA expression by a human hepatic stellate cell line (LX2). Uptake of Huh7Con1+ABs by LX2 cells dose-dependently upregulated profibrotic genes (COL1A1, TGFB1; TIMP1; TIMP2). When normalized to the apoptotic cytokeratin-18 M30 neoepitope, HCV+ ABs exhibited a more pronounced effect than HCV− ABs. In contrast, neither non-ingested ABs nor nucleic acids obtained from Huh7, Huh7Con1+, or HepG2 cells triggered those AB-dependent effects. Both the engulfment of Huh7Con1+ ABs and their effects were partially blocked by masking of phosphatidylserine with annexin V, and completely inhibited by the class-A scavenger receptor ligand, polyinosinic acid. Our findings demonstrate that AB uptake stimulates HSCs, and indicate that HCV infection leads to amplified fibrogenic mRNA expression and enhanced HSC activation.
Keywords: annexin V, Con1, Huh7 cells, LX2 cells, polyinosinic acid, profibrotic genes
Introduction
More than 170 million people may be infected with the hepatitis C virus worldwide (HCV) (1), (2), and more efficacious and tolerable treatments are urgently needed. Moreover, the majority of patients with chronic HCV infection remain untreated, which accounts for 25% of all liver cirrhosis and 27% of all hepatocellular carcinomas (3), (4). Cirrhosis results from ongoing liver injury and sustained fibrosis, with induction of a range of fibrogenic and proliferative cytokines, and enhanced deposition of extracellular matrix (5), (6).
Chronic HCV infection induces excessive hepatocyte apoptosis (7), (8). The resulting apoptotic bodies (ABs) trigger inflammation and fibrosis upon phagocytosis by Kupffer cells and hepatic stellate cells (HSCs) (9), (10). This process is mediated by recognition of the ABs’ surface phospatidylserine (PS) as a phagocytosis-inducing signal via cellular PS receptors (PS-R) and may further be facilitated by AB recognition through class-A scavenger receptors (11), (12). AB ingestion enhances expression of several death ligands, tumor necrosis factor-α (TNF-α) by Kupffer cells, and autocrine stimulation of HSCs by transforming growth factor-β (TGFB1) (9).
Since engulfment of ABs by HSCs acts profibrogenically (13), we compared the effects of ABs derived from HCV-negative vs. HCV-infected hepatocytes (HCs) on the expression of activation- (ACTA2, PDGFRB) and fibrosis-related (COL1A1, TGFB1, TIMP1 and TIMP2) mRNAs by HSCs, and examined whether these processes may be inhibited. To this end, we employed the Huh-7-derived clone, FCA-1, that harbors the HCV Con1 replicon representing HCV 1b (NS3-NS5b-3′ UTR) and encoding for the non-structural HCV proteins NS3, NS4a, NS4b, NS5a, and NS5b. HCV+ ABs generated from such HCV Con1+ Huh7 cells were then incubated with immortalized human HSCs (LX-2 cells). Hence, by utilizing cell lines that largely reflect the features of the cell species actually affected in vivo, this study focused on potential primary processes underlying profibrotic gene induction in HSCs.
MATERIALS AND METHODS
Culture of Huh-7, Huh-7Con1+ and LX-2 Cells
Human liver cell lines were maintained as described before (14). a) Huh-7 Cells: Human Huh-7 hepatoma cells were cultured at 37°C/5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) with 4.5 g/l glucose, 1% glutamine, 10% heat-inactivated fetal bovine serum (FBS) and 100 U/ml penicillin/100 μg/ml streptomycin (termed Huh-7 standard medium) (all antibiotics and media: PAA, Pasching, Austria). At confluence of >80%, i.e., approximately one week after seeding, adherent cells were passaged and seeded at 3.5 × 107 per 75-cm2 culture flask. b) Huh-7Con1+ Cells: The Huh-7Con1+ cell line was generated by transfecting Huh-7 cells with the Con1 replicon (15). These cells were cultured in Huh-7 medium plus 1% geneticin/G418 for selection (termed Huh-7Con1+ standard medium) and were otherwise kept as indicated for Huh-7 cells. c) LX-2 Cells: The human hepatic stellate cell line, LX-2, was maintained under conditions identical to those described above, while the culture medium contained 1% FBS only (termed LX-2 standard medium).
Generation of Apoptotic Bodies (ABs)
Huh-7 and Huh-7Con1+ cells were seeded at 3 × 108 cells per 25 ml of Huh-7 standard medium (without G418, so as to avoid toxic side effects on the LX-2 cells), and incubated for two days until approximately 80% confluence. Apoptosis was induced by irradiation with a UV cross-linker (SpectroLinker XL1000, Spectronics Corporation, Westbury, NY, USA) with 100 mJ/cm2 UV light (λ = 254 nm). The cells were cultured for another 24 h. Formation of ABs was verified by inverted phase contrast microscopy. AB-containing supernatants were removed without detachment of intact cells and were centrifuged for 10 min at 300 g, RT. AB pellets were suspended in LX-2 standard medium and analyzed by M30 ELISA (see below) to determine the AB concentration.
AB Quantitation and Normalization by M30 ELISA
Apoptotic cleavage of cytokeratin-18 by activated caspase-3 exposes a neoepitope termed M30. For select experiments, ABs were thus quantified via measuring M30 concentrations in AB lysates with the M30-specific sandwich ELISA (Peviva, Bromma, Sweden) according to the manufacturer’s instructions. Briefly, ABs were rinsed off the Petri dishes, centrifuged for 10 min at 300 g and RT, washed once with PBS, and then suspended in 1 ml of LX-2 standard medium. While the AB samples were kept incubated for a 3-h period, 50 μl of each sample were entered into the M30 ELISA. ABs were then normalized to the M30 neoepitope. To this end, determined concentrations were diluted to 540 U/l of M30 for both conditions. The samples were then applied in the AB uptake assay (see below) as triplicates at either 20 μl (set as 1x) or 200 μl (set as 10x) AB solution within total volumes of 2 ml per well in 6-well plates. Due to their identical treatment, the stabilities of cytokeratin-18 vs. caspase activities in both AB species were considered as comparable.
AB Uptake by LX-2 Cells
Live trypsinized and washed single LX-2 cells were labeled for 1 min with the Green Fluorescent Cell Linker Kit for General Cell Membrane Labeling (Sigma) according to the manufacturer’s instructions. Cells were then washed three times at 290 g, RT with PBS, resuspended in LX-2 standard medium, and preincubated for 24 h on 24 × 24 mm cover slips. ABs were labeled for 1 min with the Red Fluorescent Cell Linker Kit for General Cell Membrane Labeling (Sigma), washed three times, and resuspended in LX-2 standard medium. Subsequently, LX-2 cells were incubated with ABs for 4 h. ABs that had not bound or been internalized by the cells were removed by triple rinsing with PBS. Cover slips were then transferred onto slides, mounted with Antifade Gold medium (Invitrogen, Paisley, GB). Samples were evaluated by inverted fluorescence microscopy (Axiovert, Zeiss, Oberkochen, Germany) with a mercury vapor lamp and a multiple-band filter cube for simultaneous detection of green (λ = 515–530 nm) and red (λ = 580–630 nm) fluorescence. The rate of AB uptake or surface binding by LX-2 cells was determined by counting total cells (green) vs. cells displaying ABs (red). At least 400 total cells were counted in non-overlapping optical fields. Digital imaging was performed via the confocal LSM 510 LASER scanning module as combined with Axiovision 4 software (both by Zeiss).
Inhibition of AB Uptake by LX-2 Cells
Masking of ABs
ABs were pre-incubated for 30 min with 10 μg/ml human placenta-derived AnnV (A–V; Sigma) before application on the LX-2 cells. The substance masks phospatidylserine exposed on the outer AB membrane.
Blocking of Class-A Scavenger Receptors on LX-2 Cells
Thirty min before adding ABs, the LX-2 standard medium was supplemented with 100 μg/ml polyinosinic acid (Poly-I; Sigma). Not to be confused with the viral-type dsRNA TLR-3 agonist poly-I:C, poly-I acts as a ligand for class A scavenger receptors that specifically blocks scavenger receptors potentially involved in the uptake of ABs by HSCs. For example, poly-I was earlier employed in the delivery of adenoviral vectors (16) as well as for verifying the scavenger receptor A specificity of a targeted drug delivery system (17).
Incubation of LX-2 Cells with ABs and Inhibitors
LX-2 cells were incubated to approximately 70% confluence for 2 days in tissue-culture grade 6-well plates (Greiner) at a starting dose of 0.5 × 107 cells/well. At this time, parallel insets were either incubated with different concentrations of ABs only, or they additionally received AnnV or Poly-I (see above), respectively, to inhibit AB uptake. Resulting differences in volume were evened out with LX-2 standard medium. After 24 h, supernatant samples were kept at −20°C for later protein determination. Total cellular RNA batches were isolated as described below.
RNA Isolation from LX-2 Cells
Total RNA was isolated from LX-2 cells with the RNEasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s specifications. RNA concentrations and purities were determined photometrically (Biophotometer; Eppendorf, Hamburg, Germany) at 260 nm and 280 nm. RNA samples were adjusted to final concentrations of 0.125 μg/μl by adding RNAse-free water and kept at −80°C until further use.
Reverse Transcription
The QuantiTect Reverse Transcription Kit (Qiagen) was applied according to the manufacturer’s instructions, with 2-min pre-incubation at 42°C, reverse transcription for 20 min at 42°C and enzyme inactivation at 95°C for 2 min. The resulting cDNAs were dissolved in 100 μl Aq. bidest. and kept at −20°C until further use.
Polymerase Chain Reaction
Quantitative real-time polymerase chain reactions (qRT-PCR) were performed with the Quantitect SYBRGreen System (Qiagen). A single reaction consisted of 15 μl Master Mix, 5 μl cDNA, 1 μl specific forward and reverse primers (final concentration: 0.5 μM; for primer sequences see table 1), as well as 8 μl Aq. bidest. per well of 96-well qRT-PCR reaction plates (Nerbe Plus, Winsen/Luhe, Germany). PCR reaction was conducted in an iCycler IQ thermocycler (BioRad, Munich, Germany), with Taq polymerase activation at 95°C for 15 min and 40 cycles of melting (30 s; 95°C), primer attachment (30 s; 55°C), and amplification (72°C; 30 s). A melting curve was established with 80 cycles at 0.5°C temperature decrease, each, starting at 95°C.
Tab. 1.
PCR Primer Sequences.
| Name | Sequence | TM [°C] |
|---|---|---|
| SDHA_p | 5′-acacagacctggtggagacc-3′ | 52 |
| SDHA_m | 5′-caaaagggcttcttctgttgc-3′ | 52 |
| PDGFRB_p | 5′-gggtgagaacatcaccctca-3′ | 54 |
| PDGFRB_m | 5′-gatggagcggatgtggttaag-3′ | 54 |
| ACTA2_p | 5′-acactcttccagccctcctt-3′ | 54 |
| ACTA2_m | 5′-tgtcacacttcatgatgctattg-3′ | 54 |
| TGFB1_p | 5′-gtacctgaacccgtgttgct-3′ | 52 |
| TGFB1_m | 5′-gaacccgttgatgtccactt-3′ | 52 |
| COL1A1_p | 5′-aacagccgcttcacctacag-3′ | 52 |
| COL1A1_m | 5′-ggaggtcttggtggttttgt-3′ | 52 |
| TIMP1_p | 5′-acttccacaggtcccacaac-3′ | 54 |
| TIMP1_m | 5′-cactgtgcattcctcacagc-3′ | 54 |
| TIMP2_p | 5′-ccctctgtgacttcatcgtg-3′ | 54 |
| TIMP2_m | 5′-ggagatgtagcacgggatca-3′ | 54 |
Primers (Tab. 1) were generated as based on information provided by OMIM, ENTREZ, GeneBank, and the BLAST algorithms (all of which: National Center for Biotechnology Information, Bethesda, MD, USA) and Primer 3 (http://frodo.wi.mit.edu).
QRT-PCR data were recorded and analyzed with iCycler iQ Optical System Software 3.0a and subsequently further analyzed with the Gene Expression Analysis for iCycler iQ v1.10 Excel Macros (both by BioRad).
ELISAs for TIMP1 and TIMP2
LX-2 supernatants were tested for the presence of tissue inhibitors of metalloproteinases 1 and 2 (TIMP1; TIMP2). ELISAs were performed in 96-well microtiter plates, including appropriate protein standards, and according to the manufacturer’s instructions (R&D Systems, Wiesbaden, Germany). Optical densities were determined at λ = 450 nm with an ELISA reader (MWG, Ebersberg, Germany). Raw data were then processed by MS Excel 2003 (Microsoft, Redmond, WA, USA) as based on a formula calculated by two-fold regression of the standards’ concentrations that allowed upon interpolation of the TIMP1 and TIMP2 concentrations.
Statistics
Data shown are means ± standard deviations, if not stated otherwise and were evaluated using MS Excel 2003 (Microsoft) and GraphPad-Prism (GraphPad Software, La Jolla, CA, USA). Differences between experimental conditions were detected using the unpaired (two-tailed) t-test and subsequently controlled with the F-Test for excluding non-uniform variances. Statistical significance was inferred at p <0.05.
RESULTS
LX-2 HSCs Internalize ABs
Our initial experiments assessed the efficiency of uptake of ABs generated from HCV-negative or -positive cells, respectively. Uptake by LX-2 cells was similar, i.e., (a) of HCV-negative ABs generated from Huh-7 cells by 89.1 ± 4.2% of LX-2 cells and (b) of HCV-positive ABs from Huh-7Con1+ cells by 84.1 ± 3.7% of the LX-2 cell line (Fig. 1a). Representative photomicrographs are shown in Fig. 2a and 2b. LX-2 controls incubated under comparable conditions, but in the absence of ABs, did not display any inclusions (data not shown).
Fig. 1.

Uptake of apoptotic bodies (ABs) by LX-2 cells. LX-2 cells were labeled with green fluorescence and then incubated with red fluorescent ABs for 4 h. ABs were generated from Huh-7 cells or Con1-positive Huh-7 cells by UV-irradiation (100 mJ/cm2) for 24 h, followed by red fluorescence labeling. Numbers of total and double-stained cells were determined by fluorescence microscopy. (a) Without inhibitors; (b) After PS masking with 10 μg/ml annexin-V (AnnV); or (c) incubation with the scavenger receptor antagonist, poly-inosine (Poly-I) at 100 μg/ml. Bars depict percentages of double-stained cells ± standard deviations. Conditions with vs. without inhibitors: ***: p < 0.0001.
Fig. 2.
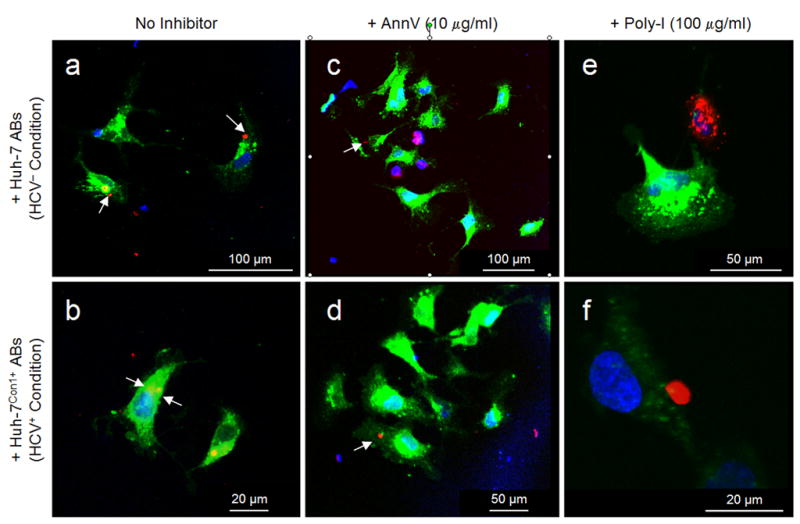
LX-2 cells ingest HCV− and HCV+ ABs. Representative photomicrographs depict green fluorescent cells that internalized red fluorescent ABs of HCV− Huh-7 cells (a, c, e) or HVC+ Huh-7Con1+ cells (b, d, f) upon incubation with ABs for 2 h, as visualized by confocal LASER scanning microscopy. Nuclei were counterstained with DAPI (blue). Cells were kept without inhibitors (a, b), or were incubated with AnnV (c, d) or Poly-I (e, f), respectively (cf. Fig. 1 for further detail). White arrows point at ABs (red) inside LX-2 cells (green). [Excitation wave lengths vs. detection ranges: blue fluorescence (λ = 364 nm vs. λ = 385–470 nm); green fluorescence (λ = 488 nm vs. λ = 505–530 nm); and red fluorescence (λ = 543 nm vs. λ = 560 nm)].
Annexin V and Polyinosinic Acid Decrease AB Uptake
We next determined the impact of annexin V and polyinosinic acid on AB uptake by HSCs. Under similar experimental conditions, but after an additional preincubation of ABs with 10μg/ml AnnV for 60 min, the uptake (a) of HCV-negative ABs generated from Huh-7 cells was decreased significantly to 26.4 ± 1.7% of LX-2 cells, and (b) of HCV-positive ABs from Huh-7Con1+ cells to 21.3 ± 0.6% of the LX-2 cell line (Fig. 1b; for representative photomicrographs, see Fig. 2c and 2d).
Blocking of class-A scavenger receptors by 30-min preincubation of LX-2 cells with 100 μg/ml Poly-I decreased the uptake (a) of HCV-negative ABs generated from Huh-7 cells significantly to 6.9 ± 0.9% and (b) of HCV-positive ABs from Huh-7Con1+ cells to 8.0 ± 1.1% (Fig. 1c). Representative photomicrographs are shown in Fig. 2e and 2f.
AB Ingestion Upregulates TGFB1 Expression
Uptake of ABs generated from Huh-7Con1− cells dose-dependently induced TGFB1 mRNA expression from 2.93 ± 0.9-fold for 1x the concentration; 3.36 ± 0.27-fold for 5x; 3.56 ± 0.56-fold for 10x and 7.46 ± 0.89-fold for the 20x concentration. Similarly, the uptake of ABs generated from Huh-7Con1+ cells dose-dependently induced TGFB1 mRNA expression from 1.23 ± 0.41-fold for the 1x concentration; 4.14 ± 0.41-fold for 5x; 6.13 ± 0.47-fold for 10x and 10.56 ± 0.89-fold for the 20x concentration (Fig. 3).
Fig. 3.
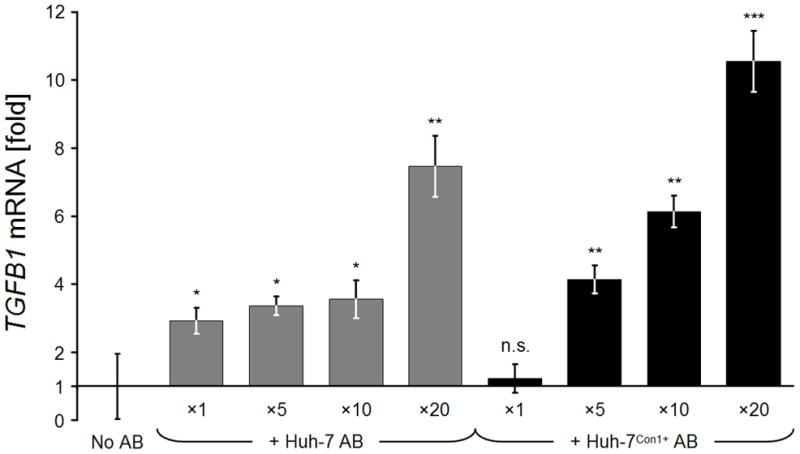
Relative TGFB1 gene expression rates of LX-2 cells after incubation with different AB concentrations. On day 1, cells received ×1 to ×20 concentrations of HCV− and HCV+ ABs. TGFB1 mRNA expression rates of treated cells were determined on day 2 and normalized against SDHA mRNA housekeeping gene expression, relative to non-AB-treated control LX-2 cells. Bars depict means of n = 3 determinations ± standard deviations. Two-tailed t-tests of treated vs. untreated conditions: *: p < 0.05; **: p < 0.01; ***: p < 0.001.
Effect of HCV− or HCV+ AB Uptake on Activation Marker Expression
AB concentrations were standardized to the M30 cytokeratin 18 neoepitope exposed upon apoptotic cleavage by caspase 3, thus allowing for normalization of the two AB species generated from Huh-7Con1− and Huh-7Con1+ cells. AB engulfment only slightly induced the mRNA expressions of PDGFRB and ACTA2 (i.e., α-SMA) as known HSC activation markers. The 10x concentration of HCV-negative ABs led to a 1.72 ± 0.58-fold induction of the PDFGRB gene, and of the ACTA2 gene by 1.35 ± 0.23-fold. Ten times HCV+ ABs led to 2.22 ± 0.71-fold expression of PDGFRB, and of 1.38 ± 0.14 fold of the ACTA2 gene (Fig. 4a and 4b).
Fig. 4.
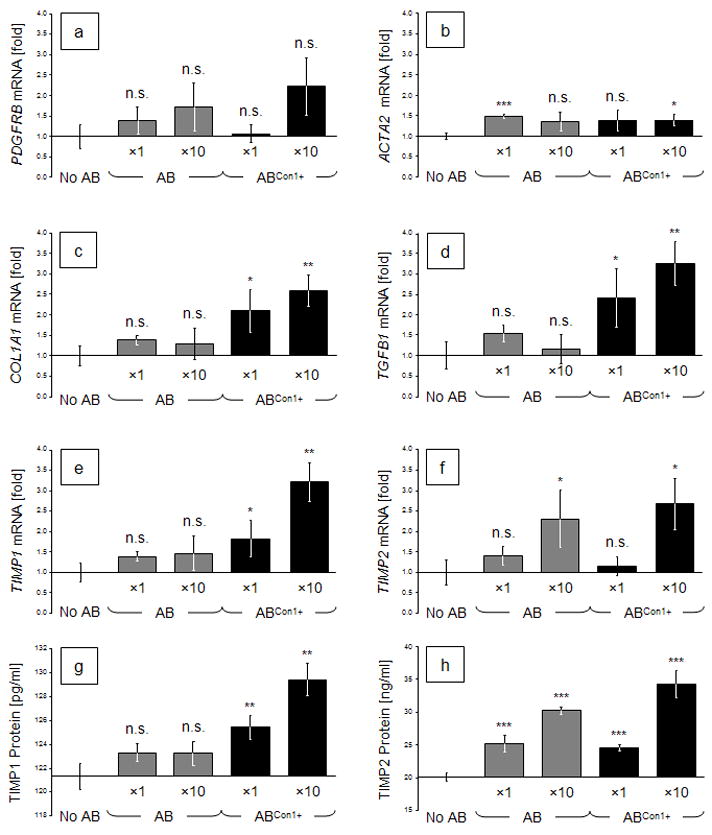
Relative expression rates of activation- and fibrosis-related genes in LX-2 cells after incubation with different concentrations of Con1− or Con1+ ABs. On day 1, cells were incubated with ×1 or ×10 concentrations of such ABs (normalized against apoptotically cleaved cytokeratin-18 neoepitope M30). (a) to (f): mRNA expression rates of treated cells were determined on day 2 and normalized against SDHA expression, relative to non-AB-treated controls. (g) and (h): TIMP1 and TIMP2 protein concentrations in LX-2 supernatants. Bars depict means (a–f: n = 3; g, h: n = 6) ± standard deviations. Two-tailed t-tests of treated vs. untreated conditions: *: p < 0.05; **: p < 0.01; or ***: p < 0.001.
HCV+ ABs Significantly Upregulate Profibrotic Gene Expression
In contrast to the examined activation markers, AB engulfment significantly induced profibrotic genes. Moreover, the AB-embedded HCV components also led to more pronounced gene inductions at similar AB concentrations. Expression of the COL1A1 gene by LX-2 cells after engulfment of 10x ABs generated from Huh7 cells led to a slight induction of 1.29 ± 0.38 fold (n.s.) while the expression was induced 2.58 ± 0.38-fold (p = 0.0037) at the same HCV+ AB concentration (Fig. 4c). Likewise, incubation with 10x HCV− ABs induced LX-2 cells to express TGFB1 by 1.16 ± 0.36-fold (n.s.), and HCV+ ABs led to a superinduction of 3.25 ± 0.53-fold (p = 0.0034) (Fig. 4d). Expression of the mRNAs encoding for tissue inhibitors of metalloproteinases (TIMP1 and TIMP2) by LX-2 cells were induced by the engulfment of 10x HCV− ABs by 1.46 ± 0.43-fold (n.s.), or by 2.30 ± 0.70-fold (p = 0.043), respectively. The same concentration of HCV+ ABs induced TIMP1 expression by 3.21 ± 0.47-fold (p = 0.0019), and TIMP2 2.67 ± 0.63-fold (p = 0.0147; Fig. 4e and 4f). Protein expression patterns for both TIMPs were confirmed by ELISA in the supernatants of identical samples (Fig. 4g and 4h).
AnnV and Poly-I Inhibit AB Uptake by LX-2 Cells and Profibrotic Gene Induction
We next determined the effect of masking phosphatidylserine with annexin V (AnnV) or of blocking class-A scavenger receptors with polyinosinic acid (Poly-I) on the uptake of ABs by HSCs as well as on the cells’ expression of profibrotic genes. Specifically, LX-2 cells were incubated with HCV− or HCV+ ABs with or without 10 μg/ml AnnV or 100 μg/ml Poly-I, respectively. After 24 h, gene expression rates were measured and normalized against SDHA mRNA housekeeping gene expression relative to untreated controls. Under identical conditions, we had already seen that cellular AB uptake is reduced in the presence of these inhibitors (cf. Figs. 1 and 2). Now we could demonstrate that the AB-dependent induction of profibrotic genes was reduced by blocking PS (Fig. 5, a to f) and even more so by masking class-A scavenger receptors (Fig. 5, e and f). These results reveal that the uptake of ABs is directly linked to the pathologic overexpression of these genes. Moreover, this experiment showed that agents inhibiting the uptake of disproportionately high AB concentrations resulting from excessive hepatocyte apoptosis in chronic hepatitis C may therapeutically alleviate liver fibrosis and, thus, the course of disease.
Fig. 5.
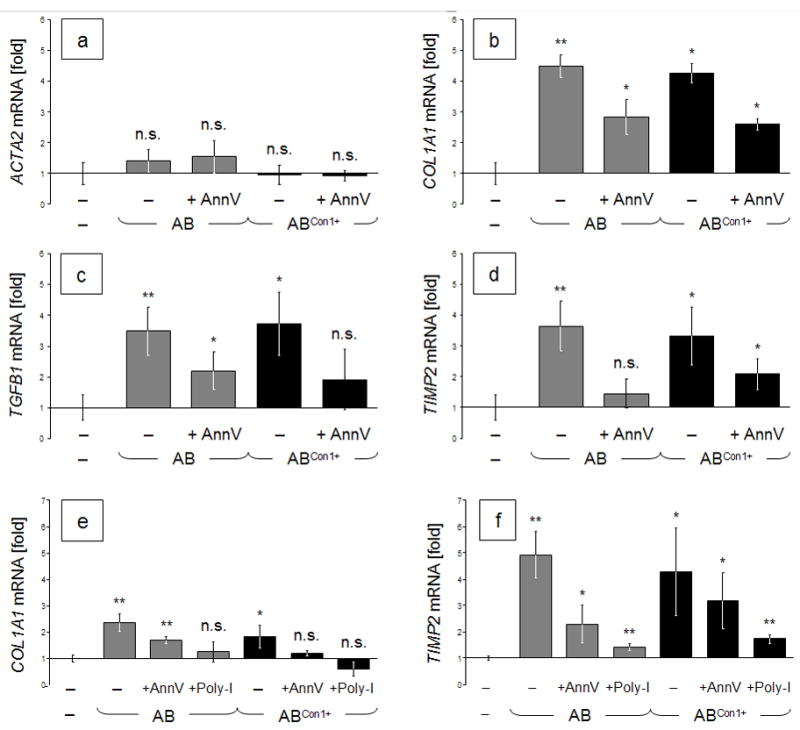
a–d: Lower AB uptake by LX-2 cells upon inhibition of uptake by AnnV reduces the induction of profibrotic genes. LX-2 cells were incubated with HCV− or HCV+ ABs in the presence or absence of AnnV (10 μg/ml). Gene expression rates were determined 24 h later and normalized against SDHA mRNA relative to untreated controls. e–f: AnnV- or Poly-I-dependent inhibition of profibrogenic gene expressions in LX-2 cells after incubation with ABs. Cells were treated with HCV− or HCV+ ABs. For blocking, cells received either 10 μg/ml AnnV or 100 μg/ml Poly-I. Gene expression rates were measured and normalized against SDHA housekeeping-gene expression, relative to untreated controls. Data are means of n = 3 ± standard deviations. Two-tailed t-tests of treated vs. untreated conditions: *: p < 0.05; or **: p < 0.01.
DISCUSSION
Hepatitis C is the most prevalent viral disease worldwide and significantly contributes to a rising incidence of cirrhosis-related deaths and of hepatocellular carcinoma. The mechanisms involved in HCV-induced liver injury are only poorly understood. Our results now establish a link between HCV-dependent hepatocyte apoptosis and the pathologic amplification of profibrotic HSC activation. Importantly, while HSCs engulf apoptotic bodies irrespective of whether or not being derived from HCV infected cells, the uptake of ABs embedding nonstructural HCV proteins further amplifies the profibrotic activity of HSCs.
In our LX-2 in-vitro model, the cells actively engulfed ABs derived from UV-irradiated Huh-7 cells as well as from HCV-replicon Con1-bearing Huh-7 hepatoblastoma cells. HCV+ ABs induced profibrotic genes more prominently than HCV− ABs. In this context, LX-2 cells engulfed both Huh-7 hepatoma cell-derived ABs and Huh-7Con1+-derived ABs to high degrees of 89.1% or 84.1%, respectively. This is in line with our earlier reporting of the uptake of ABs by the closely related LX-1 cell line (13). Phagocytosis of ABs by LX-2 cells had first been demonstrated in 2008 (10), with further electron-microscopic evidence of AB engulfment by HSCs (18). We had thus chosen this human in-vitro system due to its validity for the in-vivo situation.
As an important HSC activator, TGFβ1 is released by neighboring hepatocytes, sinusoidal endothelial cells, and platelets during HSC activation (19); the factor is also released in an autocrine manner by HSCs during their perpetuation phase (20), (21). In addition, Kupffer cells are stimulated to secrete TGFβ1 upon engulfing HC-derived ABs, thus activating HSCs via paracrine signaling (9). The induction of TGFB1 mRNA expression upon AB phagocytosis presented here suggested the presence of an additional autocrine stimulatory loop in preactivated HSCs, which could indeed be verified (see below).
In contrast to the fibrogenic genes, the HSC activation genes PDGFRB and ACTA2 were only slightly induced by AB engulfment. This further confirms that LX-2 cells are comparable to activated primary HSCs in vivo (22). In contrast however, phagocytosis of HCV+ positive ABs by LX-2 cells superinduced the profibrotic genes (e.g. COL1A1, TGFB1, TIMP1 and TIMP2) when compared to identical concentrations of HCV− ABs. Watanabe and colleagues earlier demonstrated that DNA from apoptotic hepatocytes TLR9-dependently upregulates TGFB1 and COL1A1 mRNAs in LX-2 cells and primary mouse HSCs (23). In contrast, we did not observe such mRNA-inducing effects when employing nucleic acids obtained from Huh7, Huh7Con1+, or HepG2 cells (data not shown), which suggests that the genes were in the present case induced by certain AB-encased non-structural HCV proteins. The fact that the HCV Con1 replicon encodes the proteins NS3, NS4a, NS4b, NS5a, and NS5b clearly paves the way for subsequent studies to define the specific non-structural HCV protein(s) responsible for this effect.
Activated HSCs contribute to fibrogenesis both by proliferating and by increasing their production of ECM components. In fibrosis, the most important scar constituent is COL1A1. Using LX-2 cells, Xu et al. found that COL1A1 mRNA expression was increased 3- to 4-fold upon TGFB1 stimulation (14). They also showed that LX-2 cells express TIMP2 and, to a lesser degree, TIMP1, which is in line with our gene expression results. Whether the described induction of other profibrotic genes is directly related to AB engulfment or results from the subsequent autocrine TGFβ1 stimulation loop remains to be determined.
Importantly, our results are in line with those of Bataller et al. in primary rat HSCs who had shown that expression of the non-structural HCV proteins NS3 to NS5 in an adenoviral transfection model induced the expression of TGF-β1 and pro-collagen-α1(I). They thus postulated a direct influence of HCV proteins on the transcriptional activity of HSCs (8). Similarly, we here demonstrated that the uptake of Con1+ ABs containing the specified nonstructural HCV proteins superinduced the expression of profibrotic genes in LX-2 cells when compared to HCV− ABs. This suggests that such proteins affect the profibrotic transcriptional activity of HSCs. The concrete HCV proteins responsible for this effect remain to be determined.
Phagocytosis of HCV proteins derived from replicon cells supports the relevance of our findings for patients suffering from chronic hepatitis C. Our findings strongly suggest that a liver microenvironment characterized by excessive HCV-dependent hepatocyte apoptosis may contribute to the pathogenesis of liver fibrosis. AB-encased delivery of HCV proteins thus continuously activates the HSCs’ transcriptional activity and promotes fibrosis.
In order to test this hypothesis, we inhibited the HSC pathways involved in the engulfment process. Blocking the “eat-me” signal PS located on the cells’ outer membrane by AnnV indeed significantly reduced the uptake of HCV− and HCV+ ABs. Even more impressively, competition for scavenger class-A receptors by Poly-I almost completely abrogated any AB uptake and, as a result, the AB-induced profibrogenic activity of HSCs. Our results may thus offer a rationale for early therapeutic intervention to prevent the deleterious induction of profibrotic gene expressions by HSCs in patients with chronic HCV infection.
Acknowledgments
We are most grateful to R. Bartenschlager (Dept. of Molecular Virology, University of Heidelberg), J. F. Schlaak (Dept. of Gastroenterology and Hepatology, University Hospital Essen), and their colleagues for their generous gift of the Huh-7 and Huh-7Con1+ cell lines. This work was supported by the Deutsche Forschungsgemeinschaft (DFG) CA (grant 267/6-1); the German Competence Network for Viral Hepatitis (Hep-Net) funded by the German Ministry of Education and Research (BMBF; grant No. 16,4); and the Wilhelm Laupitz Foundation.
ABBREVIATIONS
- ACTA2
smooth muscle α2 actin
- AnnV
annexin V
- AB
apoptotic body
- COL1A1
collagen type I, α1
- HC
hepatocyte
- HCV
hepatitis C virus
- HSC
hepatic stellate cell
- Huh7
a hepatoblastoma cell line
- LX-2 cells
a hepatic stellate-cell line
- M30
a caspase-3 exposed cytokeratin-18 neoepitope
- n.s.
not significant
- PDGFRB
platelet-derived growth factor receptor β
- Poly-I
polyinosinic acid
- PS
phospatidylserine
- PS-R
PS receptor
- qRT-PCR
quantitative real-time polymerase chain reaction
- SDHA
succinate dehydrogenase complex subunit A
- TGFB1
transforming growth factor β1
- TNF-α
tumor necrosis factor-α
- TIMP
tissue inhibitor of metalloproteinases
Footnotes
DISCLOSURE/CONFLICT OF INTEREST: The authors declare no conflict of interest.
References
- 1.Terrault NA, Berenguer M. Treating hepatitis C infection in liver transplant recipients. Liver Transpl. 2006 Aug;12(8):1192–1204. doi: 10.1002/lt.20865. [DOI] [PubMed] [Google Scholar]
- 2.Kikuchi K, Umehara T, Fukuda K, Kuno A, Hasegawa T, Nishikawa S. A hepatitis C virus (HCV) internal ribosome entry site (IRES) domain III–IV-targeted aptamer inhibits translation by binding to an apical loop of domain IIId. Nucleic Acids Res. 2005;33(2):683–692. doi: 10.1093/nar/gki215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoofnagle JH. Course and outcome of hepatitis C. Hepatology. 2002 Nov;36(5 Suppl 1):S21–9. doi: 10.1053/jhep.2002.36227. [DOI] [PubMed] [Google Scholar]
- 4.Timm J, Roggendorf M. Sequence diversity of hepatitis C virus: implications for immune control and therapy. World J Gastroenterol. 2007 Sep 28;13(36):4808–17. doi: 10.3748/wjg.v13.i36.4808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Friedman SL. Liver fibrosis -- from bench to bedside. J Hepatol. 2003;38( Suppl 1):S38–53. doi: 10.1016/s0168-8278(02)00429-4. [DOI] [PubMed] [Google Scholar]
- 6.Guyot C, Lepreux S, Combe C, Doudnikoff E, Bioulac-Sage P, Balabaud C, et al. Hepatic fibrosis and cirrhosis: the (myo)fibroblastic cell subpopulations involved. Int J Biochem Cell Biol. 2006 Feb;38(2):135–51. doi: 10.1016/j.biocel.2005.08.021. [DOI] [PubMed] [Google Scholar]
- 7.Canbay A, Friedman S, Gores GJ. Apoptosis: the nexus of liver injury and fibrosis. Hepatology. 2004 Feb;39(2):273–8. doi: 10.1002/hep.20051. [DOI] [PubMed] [Google Scholar]
- 8.Bantel H, Lügering A, Heidemann J, Volkmann X, Poremba C, Strassburg CP, et al. Detection of apoptotic caspase activation in sera from patients with chronic HCV infection is associated with fibrotic liver injury. Hepatology. 2004 Nov;40(5):1078–1087. doi: 10.1002/hep.20411. [DOI] [PubMed] [Google Scholar]
- 9.Canbay A, Feldstein AE, Higuchi H, Werneburg N, Grambihler A, Bronk SF, et al. Kupffer cell engulfment of apoptotic bodies stimulates death ligand and cytokine expression. Hepatology. 2003 Nov;38(5):1188–98. doi: 10.1053/jhep.2003.50472. [DOI] [PubMed] [Google Scholar]
- 10.Jiang JX, Mikami K, Shah VH, Torok NJ. Leptin induces phagocytosis of apoptotic bodies by hepatic stellate cells via a Rho guanosine triphosphatase-dependent mechanism. Hepatology. 2008 Nov;48(5):1497–505. doi: 10.1002/hep.22515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hengartner MO. The biochemistry of apoptosis. Nature. 2000 Oct 12;407(6805):770–6. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 12.Henson PM, Bratton DL, Fadok VA. The phosphatidylserine receptor: a crucial molecular switch? Nat Rev Mol Cell Biol. 2001 Aug;2(8):627–33. doi: 10.1038/35085094. [DOI] [PubMed] [Google Scholar]
- 13.Canbay A, Taimr P, Torok N, Higuchi H, Friedman S, Gores GJ. Apoptotic body engulfment by a human stellate cell line is profibrogenic. Lab Invest. 2003 May;83(5):655–63. doi: 10.1097/01.lab.0000069036.63405.5c. [DOI] [PubMed] [Google Scholar]
- 14.Xu L, Hui AY, Albanis E, Arthur MJ, O’Byrne SM, Blaner WS, et al. Human hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis of hepatic fibrosis. Gut. 2005 Jan;54(1):142–51. doi: 10.1136/gut.2004.042127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu Z, Choi J, Yen TS, Lu W, Strohecker A, Govindarajan S, et al. Synthesis of a novel hepatitis C virus protein by ribosomal frameshift. EMBO J. 2001 Jul 16;20(14):3840–8. doi: 10.1093/emboj/20.14.3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haisma HJ, Kamps JAAM, Kamps GK, Plantinga JA, Rots MG, Bellu AR. Polyinosinic acid enhances delivery of adenovirus vectors in vivo by preventing sequestration in liver macrophages. J Gen Virol. 2008 May;89(Pt 5):1097–1105. doi: 10.1099/vir.0.83495-0. [DOI] [PubMed] [Google Scholar]
- 17.Rensen PCN, Gras JCE, Lindfors EK, van Dijk KW, Jukema JW, van Berkel TJC, et al. Selective targeting of liposomes to macrophages using a ligand with high affinity for the macrophage scavenger receptor class A. Curr Drug Discov Technol. 2006 Jun;3(2):135–144. doi: 10.2174/157016306778108893. [DOI] [PubMed] [Google Scholar]
- 18.Zhan S, Jiang JX, Wu J, Halsted C, Friedman SL, Zern MA, et al. Phagocytosis of apoptotic bodies by hepatic stellate cells induces NADPH oxidase and is associated with liver fibrosis in vivo. Hepatology. 2006 Mar;43(3):435–43. doi: 10.1002/hep.21093. [DOI] [PubMed] [Google Scholar]
- 19.Bilzer M, Roggel F, Gerbes AL. Role of Kupffer cells in host defense and liver disease. Liver Int. 2006 Dec;26(10):1175–86. doi: 10.1111/j.1478-3231.2006.01342.x. [DOI] [PubMed] [Google Scholar]
- 20.Breitkopf K, Godoy P, Ciuclan L, Singer MV, Dooley S. TGF-beta/Smad signaling in the injured liver. Z Gastroenterol. 2006 Jan;44(1):57–66. doi: 10.1055/s-2005-858989. [DOI] [PubMed] [Google Scholar]
- 21.Inagaki Y, Okazaki I. Emerging insights into Transforming growth factor beta Smad signal in hepatic fibrogenesis. Gut. 2007 Feb;56(2):284–92. doi: 10.1136/gut.2005.088690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herrmann J, Gressner AM, Weiskirchen R. Immortal hepatic stellate cell lines: useful tools to study hepatic stellate cell biology and function? J Cell Mol Med. 11(4):704–22. doi: 10.1111/j.1582-4934.2007.00060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Watanabe A, Hashmi A, Gomes DA, Town T, Badou A, Flavell RA, et al. Apoptotic hepatocyte DNA inhibits hepatic stellate cell chemotaxis via toll-like receptor 9. Hepatology. 2007 Nov;46(5):1509–1518. doi: 10.1002/hep.21867. [DOI] [PubMed] [Google Scholar]