

Abstract
Inactivation of von Hippel-Lindau tumor suppressor protein (pVHL) is associated with von Hippel-Lindau disease, an inherited cancer syndrome, as well as the majority of patients with sporadic clear cell renal carcinoma (RCC). While the involvement of pVHL in oxygen sensing through targeting HIFα subunits to ubiquitin-dependent proteolysis has been well documented, less is known about pVHL regulation under both normoxic and hypoxic conditions. We found that pVHL levels decreased in hypoxia and that hypoxia-induced cell cycle arrest is associated with pVHL expression in RCC cells. pVHL levels fluctuate during the cell cycle, paralleling cyclin B1 levels, with decreased levels in mitosis and G1. pVHL contains consensus Destruction box sequences, and pVHL associates with Cdh1, an activator of the anaphase promoting complex/cyclosome (APC/C) E3 ubiquitin ligase. We show that pVHL has a decreased half-life in G1, Cdh1 downregulation results in increased pVHL expression, while Cdh1 overexpression results in decreased pVHL expression. Taken together these results suggest that pVHL is a novel substrate of APC/CCdh1. Destruction box-independent pVHL degradation was also detected, indicating that other ubiquitin ligases are also activated for pVHL degradation.
Keywords: Von Hippel-Lindau protein, renal cell carcinoma, anaphase promoting complex/cyclosome, hypoxia, cell cycle
Introduction
Germline inactivation of the von Hippel-Lindau tumor suppressor protein (pVHL) is associated with von Hippel-Lindau disease, an inherited cancer syndrome in which patients are predisposed to develop various vascular tumors and clear cell renal cell carcinomas (ccRCC) (Kaelin, 2007a). pVHL is also inactivated in the majority of patients with sporadic ccRCC (Kaelin, 2007b). The VHL protein is expressed in two forms of 25 kDa and 19 kDa due to the use of alternative translation initiation codons (Schoenfeld et al., 1998) and is the substrate recognition component of a cullin-RING ubiquitin ligase complex (CRL) that includes cullin-2, Rbx 1, and elongins B and C (Deshaies and Joazeiro, 2009; Kaelin, 2008). The VHL protein contains two structural domains: an α domain that interacts directly with elongin C and a β domain that interacts with target substrates (Stebbins et al., 1999). The best understood ubiquitylation substrates of the pVHL CRL complex are the proline hydroxylated alpha subunits of hypoxia-inducible factor (HIFα) (Kaelin, 2005; Semenza, 2009). It is not clear whether other reported pVHL interacting proteins, such as p53, JADE1, fibronectin, or collagen IV, are pVHL CRL substrates (Kaelin, 2007a). However, it is apparent that these pVHL-interacting proteins collectively play a role in cell growth control and that in the context of pVHL inactivation in VHL disease or ccRCC are associated with tumor progression.
Hypoxia, defined as reduced O2 levels (≤2% O2), occurs in a variety of pathological conditions, including stroke, tissue ischemia, inflammation, and the growth of solid tumors (Semenza, 2009). Acute hypoxia inhibits cell proliferation by mediating cell differentiation, quiescence, and apoptosis or necrosis. In contrast, chronic hypoxia may induce cell proliferation by stimulating adaptive responses through the activation of HIFα. Numerous recent studies have detailed the upregulation of gene transcription primarily through HIF activation in response to hypoxia (Chi et al., 2006; Lendahl et al., 2009). Hypoxia also induces a global suppression of translation, due at least in part to the shift from oxidative phosphorylation to anaerobic glycolysis and the resulting decrease in ATP generation (Fähling, 2009). However, translation of some proteins, such as HIFα and VEGF, may occur in hypoxic cells through a cap-independent translation initiation mechanism (Liu and Simon, 2004).
Anaphase-promoting complex/cyclosome (APC/C) is a multi-subunit E3 ubiquitin ligase that plays a critical role in cell cycle regulation, the DNA damage response, TGF β-initiated signal transduction, and tumorigenesis (Liu et al., 2008; Liu et al., 2007; Sudo et al., 2001; Wan et al., 2001; Wasch et al., 2009). APC/C is a target of the mitotic spindle checkpoint; it is active for maintenance of a stable G1 phase and functions as a regulator of the G1/S transition (Baker et al., 2007; Jackson, 2004; van Leuken et al., 2008; Wasch et al., 2009). The ubiquitin ligase activity and substrate specificity of APC/C are controlled by two subunits: Cdc20 and Cdh1 (Baker et al., 2007; Jackson, 2004; van Leuken et al., 2008; Wasch et al., 2009). During the cell cycle, Cdc20 binding with APC/C results in the initiation of anaphase by targeting different substrates for proteolysis, whereas Cdh1-activated APC leads to the exit of mitosis and maintenance of a stable G1 phase. Recognition of substrates by Cdc20 and Cdh1 is mediated through consensus sequences including the Destruction (D) box (RXXLXXXXN/D), the KEN box, the A Box, and the GXEN box (Wasch et al., 2009).
While it is established that pVHL is involved in the cellular response to hypoxia, factors that regulate pVHL abundance and function in the cell are less well defined. We show here that VHL protein levels were decreased in hypoxic cells through a post-transcriptional mechanism. Hypoxia-associated cell cycle arrest was dependent on pVHL-expression in RCC cells, and pVHL levels fluctuated during the cell cycle. In addition, pVHL stably associated with Cdh1 and appeared to be a ubiquitylation substrate for APC/CCdh1. pVHL was also targeted for degradation by other as-yet to be determined ubiquitin ligases. Our results suggest a novel aspect of the hypoxic response in which reduced pVHL levels are associated with increased expression of hypoxia-induced gene products and that pVHL may play a role in cell cycle progression.
Results
Post-transcriptional control of pVHL levels in hypoxia
We examined pVHL expression levels in various cell lines that were cultured either under normoxic (room air, 21% oxygen) or hypoxic (1% oxygen) conditions. Since pVHL levels may be affected by cell density (Pause et al., 1998), all cells were seeded at low density and harvested at less than 80% confluency. When cells were transferred from normoxia to hypoxia endogenous pVHL levels decreased in 293T cells and HeLa cells with a significant decline between 2 to 4 h after transfer (Figure 1a and 1b). Endogenous pVHL in 293T cells and HeLa cells was expressed predominantly if not exclusively as the 19 kDa form, which is active (Iliopoulos et al., 1998; Schoenfeld et al., 1998). Exogenously-expressed pVHL in 786-O G7F RCC cells showed a similar hypoxia-associated decrease (Figure 1c). In these cells both the p25 and p19 pVHL isoforms were expressed, and while both isoforms were reduced in hypoxia, p25 VHL was reduced at an accelerated rate compared to p19 VHL (Figure 1c). As pVHL levels decreased in hypoxia we saw a reciprocal increase in the abundance of the glucose transporter, GLUT1, a hypoxia-induced protein (Figure 1d). These results suggest a novel hypoxia response mechanism where reduction of pVHL levels is associated with reciprocal increases in hypoxia-responsive gene products.
Figure 1. Hypoxia-induced downregulation of pVHL via the ubiquitin-proteasome pathway.
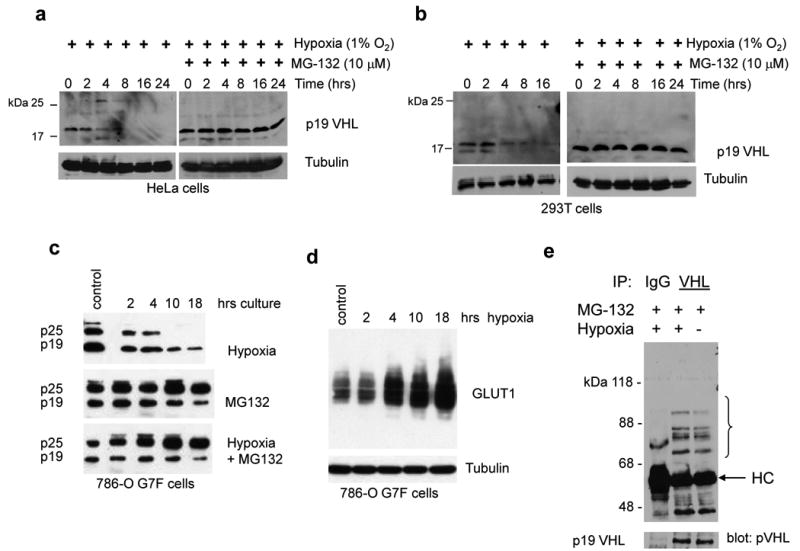
(a) HeLa cells, (b) 293T cells, and (c) 786-O G7F RCC cells were cultured in hypoxia in the absence or presence of 10 μM MG132 for the indicated time intervals. The 0 time point represents lysates from normoxic cultures that were prepared at the time of transfer to hypoxia. pVHL expression was detected by Ig32 anti-pVHL western blotting, and blots were probed with tubulin antibody to demonstrate equal loading. (d) Increasing GLUT1 expression levels in 786-O G7F cells that were cultured in hypoxia. (e) 293T cells were treated with 10 μM MG132 and cultured under normoxic or hypoxic conditions for 24 h. Whole cell lysates were prepared with RIPA buffer, and immunoprecipitation and western blotting were performed with anti-VHL (Ig32) and anti-ubiquitin antibody, respectively. HC, immunoglobulin heavy chain.
Hypoxia-associated reduction in pVHL levels was blocked by the proteasome inhibitor MG132 in all cell lines tested (Figures 1a-c), suggesting dependence on the ubiquitin-proteasome pathway. Anti-pVHL immunoprecipitations followed by anti-ubiquitin western immunoblotting showed that ubiquitylated pVHL was detected in 293T cells that were cultured in either normoxia or hypoxia (Figure 1e). Similar experiments using transient overexpression of HA-ubiquitin revealed additional high molecular weight polyubiquitylated pVHL species with a slight increase in polyubiquitylated pVHL in cells that were cultured in hypoxia (Supplemental Figure 1). These results show that pVHL turnover in either normoxia or hypoxia is mediated through ubiquitylation and proteasomal degradation.
Pulse-chase studies were performed to address whether the observed reduction in pVHL levels in hypoxia was due to increased turnover or decreased synthesis. We found that pVHL turnover was similar in cells cultured under either normoxic or hypoxic conditions and that the half-life of p25 VHL was between 1 and 2 h (Figure 2a). pVHL species of approximately 18 - 20 kDa were detected at the 1 and 2 h chase time points that could represent p25 VHL degradation intermediates or modified pVHL species. In addition, the intensity of p19 VHL appeared to increase at the 1 – 2 h time points. While p19 VHL is expected to be expressed through alternative translation initiation of the VHL mRNA, it is possible that the increase in p19 VHL seen also represents a stable p25 VHL degradation intermediate. We did not detect accumulation of higher molecular weight species that might be indicative of ubiquitylation, since these experiments were not performed in the presence of proteasome inhibitors.
Figure 2. Post-transcriptional regulation of VHL expression.
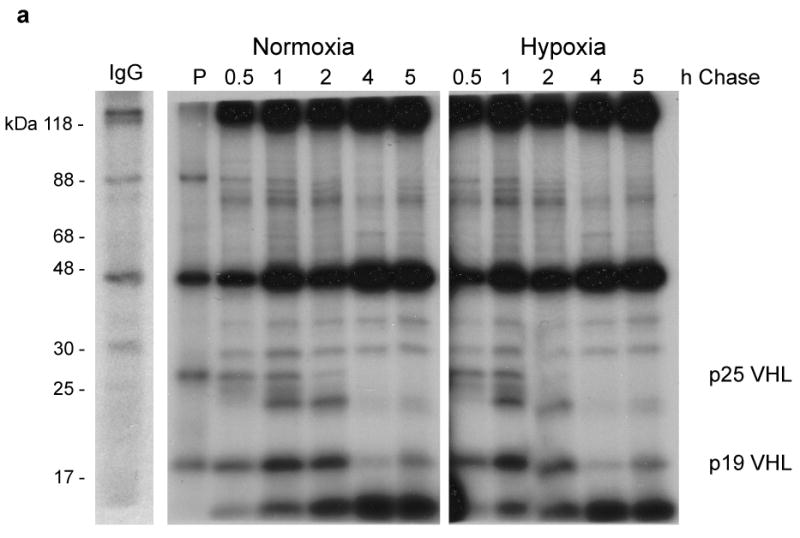

(a) 786-O G7F RCC cells were pulse labeled with 35S-Methionine for 30 min (P) at which point labeling medium was washed away and replaced with medium containing cold methionine. RIPA lysates were prepared from chase cultures at the indicated time points. A representative control IgG immunoprecipitation from a pulse-labeled lysate is shown. (b) 293T cells were cultured in normoxia (N) or in hypoxia for 6 or 16 h, and VHL and VEGF mRNA expression levels were determined by quantitative real time RT-PCR. Relative quantitation (RQ) was calculated using 18S rRNA amplification as reference. Amplification of cDNA from hypoxic cells is expressed relative to amplification of normoxic cells (N), which was given a value of 1. The data shown here are representative of three independent experiments.
We also examined the effects of hypoxia on VHL mRNA levels in 293T cells. VHL mRNA levels were found to be modestly increased in hypoxia (approximately 3-fold), while for comparison VEGF mRNA levels increased more than 20-fold in the same experiment (Figure 2b). Since pVHL did not appear to turn over at a greater rate in hypoxia as compared to normoxia (Figure 2a) and VHL mRNA levels increased slightly in hypoxia (Figure 2b), our results suggest that hypoxia-associated reduction in pVHL occurs at a post-transcriptional level, and probably at the level of translation initiation as has been described previously (Fähling, 2009).
pVHL expression and the cell cycle
A consequence of the shift from culture in normoxia to hypoxia in many cell types is cell cycle arrest (Gardner et al., 2001; Schmaltz et al., 1998). Cell cycle analyses were performed on RCC cells that were cultured under hypoxic conditions. While parental 786-O RCC cells or 786-O vector control cells exhibited no apparent effects of hypoxia on cell cycle progression (Figure 3a), pVHL-expressing 786-O G7F cells exhibited an accumulation of cells in G1 phase of the cell cycle after 16 h of hypoxia, indicative of G1 arrest (Figure 3b). Cell cycle arrest was reversible with reoxygenation (Figure 3c) as was pVHL expression (Figure 3d). These results suggest that hypoxia-associated cell cycle arrest appears to be dependent on pVHL expression in RCC cells. However, G1 cell cycle arrest of 786-O G7F cells did not occur to a significant degree between 4h to 8h of culture in hypoxia (Figure 3b), time points when pVHL levels are decreased (Figure 1). How pVHL expression and loss in hypoxia is associated with G1 cell cycle arrest in hypoxia remains to be determined.
Figure 3. Hypoxia-induced cell cycle arrest and pVHL downregulation.

RCC cells were cultured in normoxia or hypoxia for the indicated times and cells were harvested for (a - c) cell cycle analysis by propidium iodide staining and flow cytometry or (d) western blot analysis. Representatives of multiple experiments are shown here. Overall there was less than 10% variance in cell numbers at the G1, S, and G2/M phases of the cell cycle among the various experiments. Western blots were probed with Ig32 pVHL antibody or tubulin antibody to demonstrate equal loading.
pVHL levels during cell cycle progression were determined in cultures in which cell cycle arrest was induced at mitosis or G1/S. Endogenous pVHL levels in HeLa cells and exogenous pVHL levels in 786-O G7F RCC cells were similarly downregulated during the interval between 2 and 8-9 h after release from G2/M nocodazole block (Figure 4a, lanes 5-10 for HeLa cells and lanes 4-10 for 786-O G7F cells). This time period is consistent with cells passing through mitosis and entering the G1 phase of the cell cycle (Figure 4b). We asked whether pVHL downregulation in G1 phase was due to increased protein turnover. Pulse-chase experiments were performed on cells in G1 and S phases of the cell cycle. pVHL half-life in G1 cells was approximately 30 min, while pVHL half-life was greater than 1 h in S phase cells (Figure 4c). Therefore, pVHL downregulation in G1 phase appears to be associated with an increase in the rate of protein degradation.
Figure 4. pVHL levels fluctuate through the cell cycle.
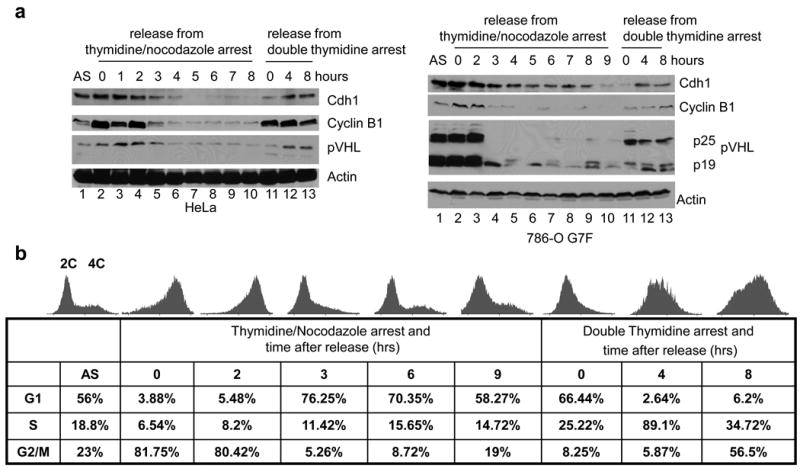

Cell cycle synchronization of HeLa and 786-O G7F RCC cells was performed using double thymidine block or thymidine plus nocodazole block as described in the Materials and Methods. Cells were released from synchronization and harvested at the indicated times. (a) Expression of the indicated proteins in HeLa cells or 786-O G7F RCC cells was examined by western blotting, and β-actin detection was used to demonstrate equal loading. (b) Analyses of HeLa cells were performed to confirm cell cycle arrest and release using propidium iodide staining and flow cytometry. Histograms are shown as well as the corresponding numbers of cells in G1, S, and the G2/M phases. The 0 time point indicates cells arrested with the indicated agents but not released from block. A representative experiment of three performed is shown. Similar results were obtained using 786-O G7F RCC cells (data not shown). (c) HeLa cells were synchronized by thymidine plus nocodazole block. Three hrs after release pulse-chase studies were performed as described in Figure 2 and the Materials and Methods.
Targeting of pVHL by APC/CCdh1
Cyclin B1 protein levels, which have been shown to be degraded in metaphase and not re-expressed until late G1 (Baker et al., 2007), closely paralleled pVHL levels during progression through mitosis and through entry of the cells into S phase (Figure 4a). Cyclin B1 is targeted for degradation by the APC/C E3 ubiquitin ligase, and cyclin B re-expression in late G1 is a consequence of the phosphorylation and inactivation of APC/C (Baker et al., 2007). Since the expression profile of pVHL in our experiments was similar to that of cyclin B1, this suggested that pVHL may be a substrate for APC/C.
Cdh1 is an activating subunit that confers substrate specificity for APC/C. APC/CCdh1 is active from late mitosis to early G1 phases of the cell cycle. Cdh1 protein levels were low at time points corresponding to late G1 phase (Figure 4a, lanes 7-10 for HeLa cells and lanes 10-11 for 786-O G7F cells), as previously reported (Listovsky et al., 2004; Liu et al., 2008). Cdh1 and pVHL were co-expressed 4 to 8 h after release from double thymidine block (Figure 4a, lanes 12-13) as well as 0 to 2 h after nocodazole block, (Figure 4a, lanes 2-3) in both HeLa and 786-O G7F cells. Thus, pVHL is co-expressed with Cdh1 in late G1 and early mitosis, when APC/CCdh1 would not be expected to be active.
To further define the relationship between Cdh1 and pVHL we performed co-immunoprecipitation experiments using lysates from 293T cells. Anti-Cdh1 antibody was able to co-immunoprecipitate endogenous p19 VHL (Figure 5a). The anti-Cdh1 and anti-pVHL antibodies that were used here are both derived from mice, which resulted in high-background western blots. Since Cdh1 is ∼55 kDa, we could not detect reciprocal co-immunoprecipitation of Cdh1 with anti-pVHL antibody because of interfering immunoglobulin heavy chain. To address this we performed co-transfection experiments of plasmids that expressed HA-tagged Cdh1 and full-length myc/His-tagged VHL. We isolated pVHL with His-targeted metal affinity resin and found that Cdh1 co-purified with pVHL-myc/His (Figure 5b, lane 4). The VHL-myc/His construct that was used here had an optimized Kozak consensus sequence surrounding codon 1, and the p25 form was overexpressed relative to p19, accounting for the relatively low levels of p19 VHL-myc/His isolated in the metal affinity pulldown studies. Cdh1 also was co-immunoprecipitated with FLAG-tagged pVHL and HA-tagged pVHL using either mouse monoclonal or rabbit polyclonal antibodies (Figure 5c and d). Taken together the results shown in Figure 5 demonstrate that Cdh1 and pVHL physically associate.
Figure 5. pVHL interacts with Cdh1.

(a) Endogenous pVHL was co-immunoprecipitated with anti-Cdh1 antibody from 293T cell extracts. Western blots were probed with Ig32 anti-pVHL antibody. (b) 293T cells were transfected with vectors expressing HA-Cdh1 and pVHL-myc/His alone or together and complexes were isolated using metal affinity resin. Western blots were probed with either anti-HA (top panel) or anti-myc (bottom panel). Cdh1 was co-immunoprecipitated with FL181 anti-pVHL antibody from pVHL-expressing (c) 786-O G7F RCC cells or (d) WT7 RC cells but not from pVHL-negative 786-O RCC cells. In panels c and d western blots were probed with either anti-Cdh1 (top panel) or anti-pVHL (Ig32) (bottom panel).
A search of the human pVHL sequence identified two putative Destruction boxes at residues 60 to 67 and 82 to 90 in human pVHL, which are potential APC/C recognition elements (Wasch et al. 2009). These sequences, termed D1 and D2, respectively (Figure 6a) are completely conserved among human, mouse, and rat sequences (Figure 6b). As a first step in evaluating the importance of potential D box sequences on pVHL function, we queried the VHL gene mutation database, a compilation of published VHL gene mutations (http://www.umd.be) (Beroud et al., 2000), for DNA point mutations that occur within the consensus sequences. No point mutations involving nucleotides encoding the D1 box consensus amino acids, R60, L63 or N67 were listed, although several point mutations at residues other than the key Destruction box consensus residues were identified (Figure 6c). Conversely, the D2 box is more frequently mutated, with multiple mutations identified encoding each of the consensus amino acids, R82, L85, and N90 (Figure 6c). Since it appears that no naturally occurring mutations have been identified involving pVHL amino acids R60, L63, and N67, and there are no VHL gene polymorphisms found in this region, this suggested to us that if mutation of these residues did occur it may not be associated with loss of pVHL function (and therefore not predisposing to tumorigenesis). Alternatively, mutations of these residues may have adverse effects on cell viability and/or normal development.
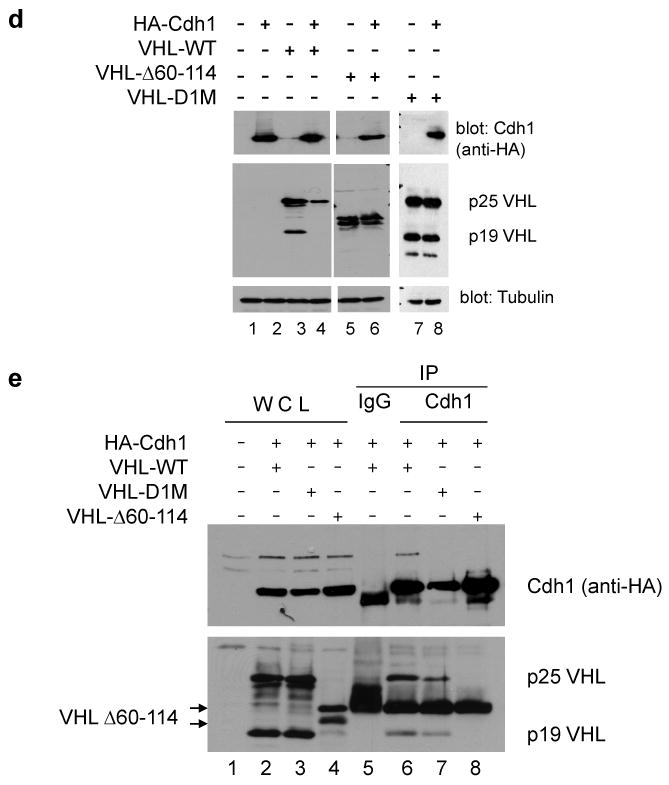
Figure 6. pVHL contains Cdh1 recognition sequences and is a novel substrate of APC/CCdh1.

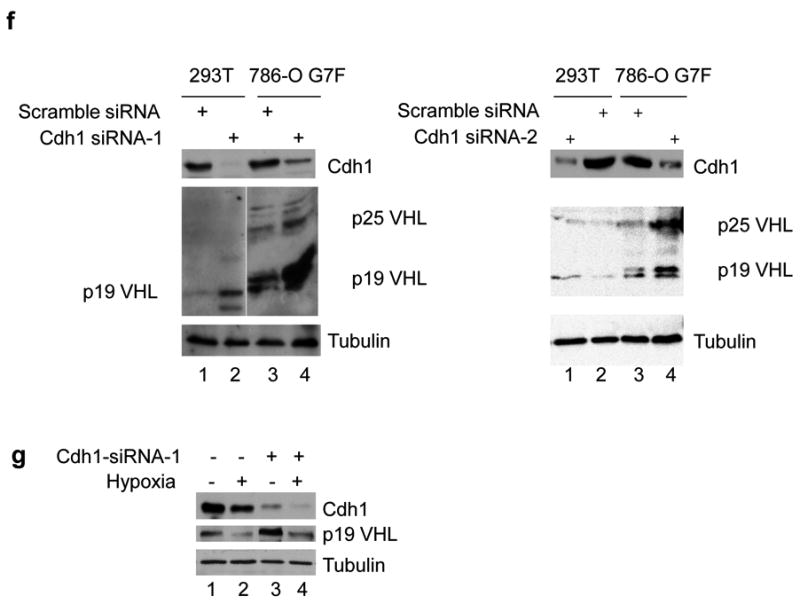
(a) The pVHL protein sequence contains two consensus Destruction boxes (D1 and D2) that are similar to those found in known APC/CCdh1 target proteins. (b) The pVHL D1 and D2 sequences are evolutionarily conserved. Sequence alignment and generation of a consensus sequence was performed using Vector NTI. (c) VHL gene point mutations that have been identified in the D1 (codons 60-67) and D2 (codons 82-90) sequences were obtained from the VHL gene mutation database (http://www.umd.be). X signifies a nonsense mutation. (d) 293T cells were transfected with vectors expressing myc/His-tagged wild type pVHL or Destruction box mutants alone or together with HA-Cdh1. RIPA extracts were prepared and protein expression levels were determined by western blotting. (e) 293T cells were co-transfected with vectors expressing HA-Cdh1 and myc/His-tagged VHL constructs. Lysates were prepared in NP40 lysis buffer and co-immunoprecipitations were performed with anti-Cdh1 antibody. Western blots were probed with anti-HA (top) or anti-myc (bottom) antibodies. (f) 293T and 786-O G7F cells were transfected with two different siRNA's targeting Cdh1 or a scrambled control as described in the Materials and Methods. Cdh1 and pVHL expression were detected by western blot with anti-Cdh1 (top panel) or Ig32 pVHL (middle panel) antibodies. (g) 293T cells were cultured in normoxia or hypoxia in the presence of Cdh1 siRNA or control siRNA, and Cdh1 and pVHL (Ig32) expression levels were detected by western blotting. Western blots were probed with tubulin antibody to demonstrate equal loading.
To determine the importance of the D1 box in the regulation of pVHL stability, site-directed mutagenesis was performed to create simultaneous alanine mutants at all three key D1 box residues (termed D1M, R63A+L63A+N67A). The D1M construct or a pVHL mutant with a deletion of residues 60 to 114 (including the putative D1 and D2 boxes) were transfected alone or in combination with HA-Cdh1 into 293T cells, and pVHL expression was detected by western blotting. Co-expression with HA-Cdh1 resulted in a significantly reduced expression of wild type pVHL while expression levels of the D1M or Δ60-114 mutants were unchanged by Cdh1 co-transfection (Figure 6d), suggesting that the D1 box may play a critical role in Cdh1 mediated pVHL degradation. Regulation of pVHL levels by Cdh1 was dependent on physical association between these proteins. Cdh1 antibody was able to co-immunoprecipitate wild type pVHL (Figure 6e, lane 6), however, co-immunoprecipitation of pVHL-D1M or pVHL-Δ60-114 was diminished or lost, respectively (Figure 6e, lanes 7 and 8). While Cdh1 overexpression was associated with decreased pVHL levels, Cdh1 knockdown resulted in increased pVHL levels (Figure 6f). The observed effects of Cdh1 overexpression and knockdown suggest that pVHL is recognized and targeted for destruction by APC/CCdh1 through the pVHL D1 box.
Multiple E3 ligases including APC/CCdh1 may be involved in the reduction of pVHL levels in hypoxia
We also evaluated the effects of Cdh1 knockdown in 293T cells that were cultured in hypoxia. As expected p19 VHL levels were decreased with hypoxia treatment (Figure 6g, lane 2) and increased with Cdh1 knockdown in normoxia (Figure 6g, lane 3). However, the combination of culture in hypoxia and Cdh1 knockdown resulted in intermediate p19 VHL levels (Figure 6g, compare lanes 3 and 4), suggesting that other ubiquitin ligase(s), in addition to APC/CCdh1, may contribute to hypoxia-induced pVHL degradation.
Our data indicate that in similar populations of cells pVHL degradation occurred as rapidly as within 4 h after a shift to hypoxia, while hypoxia-induced G1 phase cell cycle arrest took at least 16 h to occur. This discrepancy led us to compare hypoxia-associated pVHL decreases with cell cycle-associated pVHL fluctuation. HeLa cell extracts were prepared from cells in the G1 phase of the cell cycle or from cells that were cultured in hypoxia for 24 h. Radiolabeled wild type pVHL or pVHL mutants were used for in vitro protein degradation assays using these extracts. We found that when incubated in G1 HeLa cell exacts, wild type pVHL rapidly decayed, while both the D1M and the pVHL 60-114 deletion mutants were stable over the time course of the experiment (Figures 7a), suggesting that pVHL decay in these experiments was Destruction box-dependent. To confirm that APC/CCdh1 mediated pVHL decay, G1 phase HeLa extracts were immunodepleted of APC/C using anti-cdc27 (Supplemental Figure 2). A consequence of anti-cdc27 immunodepletion was depletion of Cdh1 as well (Supplemental Figure 2). We found that pVHL was not subject to decay when incubated in APC/C Cdh1-depleted G1 phase HeLa extracts (Figure 7b), indicating that APC/C targets pVHL for degradation. When incubated in the presence of hypoxic HeLa cell extracts, wild type pVHL and the pVHL mutants that were tested all exhibited decay, although the pVHL D1M mutant decayed at a slower rate (Figure 7c). These results suggest that pVHL decay in hypoxia may not be solely Destruction box-dependent and that additional E3 ubiquitin ligase(s) may be responsible for pVHL degradation.
Figure 7. Destruction box-dependent and –independent degradation of pVHL.

HeLa cells were (a, b) synchronized in G1 phase or (c) cultured in hypoxia for 24 h, followed by hypotonic lysis as described in the Materials and methods. APC/C depletion was performed by immunoprecipitation with anti-cdc27 antibody (see Supplementary Figure 2). The indicated wild type or mutant pVHL proteins were radiolabeled through in vitro protein synthesis, incubated in the HeLa cell extracts for the indicated time intervals, and reactions were stopped by removing an aliquot, mixing with Laemmli protein sample buffer, and heating to 95°C. The 0 time points indicate the addition of radiolabeled protein to the HeLa extracts and immediate removal of an aliquot. The band densities of input radiolabeled pVHL were analyzed using Bio-Rad Quantity One imaging software.
Discussion
We demonstrated that pVHL levels are decreased in hypoxia through a post-transcriptional mechanism that is dependent on the ubiquitin-proteasome system. This suggests not only a novel mechanism for pVHL inactivation, but also a novel component of the cellular adaptive response to hypoxia. In the presence of oxygen HIFα subunits are hydroxylated on conserved proline residues in the oxygen-dependent degradation domain and are targeted by the pVHL CRL for polyubiquitylation and proteasomal degradation (Kaelin, 2005). HIFα hydroxylation is mediated by proline hydroxylases (PHD) and the reaction is dependent on oxygen, 2-oxoglutarate, and ascorbate. In hypoxia PHD activity is inhibited, HIFα subunits are not hydroxylated, and are stabilized. Decreases in pVHL levels in hypoxia provide a complementary mechanism to further facilitate maximal HIF activity, while pVHL re-expression upon re-oxygenation would then serve to suppress HIF activity.
While pVHL-expressing RCC cells exhibited reversible cell cycle arrest in response to hypoxia, pVHL-negative RCC cells did not (Figure 3), suggesting that pVHL may be important for regulation of cell cycle regulation in hypoxia. Since pVHL expression is downregulated in hypoxia, it suggests that a pVHL target may be involved in arrest. Our results differ from another study using similar RCC cells that showed antagonistic effects of HIF-1α and HIF-2α on cell cycle progression in hypoxia (Gordan et al., 2007). They and others (Koshiji et al., 2004) showed that hypoxia-induced HIF-1α suppresses c-myc activity, resulting in cell cycle arrest (Gordan et al., 2007). On the other hand hypoxic induction of HIF-2α promoted cell cycle progression by stimulating c-myc activity (Gordan et al., 2007). In contrast to the results of Gordan et al. (2007), however, Hackenbeck et al. (2009) showed that both HIF-1α and HIF-2α independently induced cell cycle arrest in hypoxia in NIH3T3 cells. Thus, the role for HIF-2α in cell cycle regulation in hypoxia is controversial.
The RCC cells used in our present study were pVHL-expressing derivatives of the 786-O RCC cell line, which express only HIF-2α. Gordan et al. (2007) also studied a 786-O derivative, WT8, which expresses HA-tagged pVHL. While it might be expected that different pVHL-expressing 786-O cell lines should exhibit similar activities in similar experiments, there are some differences between 786-O G7F cells and WT8 cells. The WT8 cell line is a clone expressing HA-tagged pVHL (Iliopoulos et al., 1995), while 786-O G7F cells are a bulk population produced through retroviral transduction. Stickle et al. (2005) showed that WT8 cells have a single mutated p53 allele (R248W), which results in a stable p53 protein that is defective in transactivation of target promoters. Stickle et al. (2005) speculated that the p53 mutation in WT8 cells was acquired some time after their initial characterization, although it is not clear when this may have occurred or what clonal variation may exist among the many laboratories that study this cell line. If the WT8 cells used by Gordan et al. (2007) have a p53 mutation, then those cells might not exhibit appropriate cell cycle control in hypoxia. There are other possibilities to account for observed differences in cell cycle regulation by hypoxia. Warnecke et al (2008) showed that culture conditions and/or serum factors affected expression of HIF-2α targets in the presence of a hypoxia mimetic. Gordon et al (2007) cultured cells in 0.5% oxygen, while we cultured cells in 1% oxygen (see also Hackenbeck et al. (2009)). More severe levels of hypoxia or differing culture conditions may result in differential effects on cell cycle progression. In addition, as discussed by Wycoff et al (2004) expression levels of the pVHL transgene could influence experimental results. They observed that overexpression of exogenous pVHL may have an inhibitory effect on hypoxia-mediated changes in gene expression. HA-pVHL in WT8 cells is significantly overexpressed in comparison to pVHL-FLAG in 786-O G7F cells (Supplemental Figure 3). If pVHL overexpression inhibits expression of some hypoxia-induced genes, then this could also affect cell cycle regulation. Further studies will be needed to address the pVHL functional differences among these reports.
pVHL levels were found to fluctuate during the cell cycle in normoxia (Figure 4). Two pieces of evidence have previously linked pVHL activity to cell cycle control. Pause et al (1998) showed that RCC cells re-expressing wild type pVHL exited the cell cycle in response to serum starvation, while pVHL-negative RCC cells continued through the cell cycle in the absence of serum. The ability of RCC to proliferate in the absence of serum was later linked to autocrine/paracrine growth stimulation by TGF-α (de Paulsen et al., 2001). More recently it was shown that VHL gene inactivation in mouse embryo fibroblasts resulted in senescence, which was dependent on pRb and p400 activity (Young et al., 2008). The studies may explain why pVHL inactivation is seen only in RCC and not in other tumor types: pVHL loss in non-renal cells results in senescence instead of a proliferative pathway. Considering our results together with these studies suggests that pVHL promotes cell cycle progression under normal growth conditions and blocks cell cycle progression under conditions of stress.
Consensus Destruction box sequences in pVHL provided target recognition specificity by APC/CCdh1. Knock down of Cdh1 using siRNA or over expression of Cdh1 resulted in increased or decreased pVHL levels, respectively, and this regulation was dependent on physical association through the D1 sequences in pVHL. We were able to co-immunoprecipitate pVHL-Cdh1 complexes from cells in the absence of proteasome inhibitors, suggesting that the pVHL-Cdh1 association may be stable and/or that pVHL degradation by APC/CCdh1 may be regulated. APC/CCdh1 is active in the G1 phase of the cell cycle and pVHL half-life is decreased in cells in G1 cells relative to cells in S phase (Figure 4c) or asynchronous cells (Figure 2a). In addition, immunodepletion of APC/CCdh1 from cell extracts resulted in pVHL stability (Figure 7b). Taken together our data support the hypothesis that pVHL is bound and degraded by APC/CCdh1. Based on the data presented in Figure 7 it is clear, however, that APC/CCdh1 is not the only ubiquitin ligase that targets pVHL for degradation. The ubiquitin conjugating protein, E2-EPF, has also been shown to target pVHL for degradation in hypoxia (Jung et al., 2006), and it is likely that this enzyme is also playing a role in pVHL turnover in our studies. Our data support the model presented in Figure 8 in which hypoxia induces pVHL degradation through D box-dependent and D box-independent mechanisms.
Figure 8. Model for pVHL degradation in hypoxia.

Destruction box-dependent pVHL degradation by APC/CCdh1 is proposed to occur as a result of hypoxia-induced cell cycle arrest at G1 phase, while Destruction box-independent pVHL degradation may result through the action of additional ubiquitin ligases.
We predicted that the D1 sequence was a more likely targeting motif for pVHL by APC/CCdh1 complex than the D2 sequence (Figure 6). One reason for this prediction was that an examination of the pVHL crystal structure in the β domain indicated that D1 sequences are exposed on the surface of pVHL while the D2 sequences are not (Molecular Modeling Database ID #13080; www.ncbi.nlm.nih.gov/Structure/mmdb/mmdbsrv.cgi?uid=13080). We expected that the most accessible residues would be the most likely targets. No mutations of residues R60, L63, and N67 have been reported to date in the VHL gene mutation database, suggesting that recognition of pVHL D1 sequences by APC/CCdh1 and subsequent pVHL turnover may be required for cell cycle progression. Recent studies have shown that pVHL loss is associated with microtubule instability and mitotic spindle misorientation, potentially leading to the formation of renal cysts, as well as a weakened spindle mitotic checkpoint, leading to chromosomal instability (Hergovich et al., 2003; Thoma et al., 2007; Thoma et al., 2009). Since APC/C is a target of the spindle checkpoint (Musacchio and Salmon, 2007), our results suggest that there may be a relationship between pVHL CRL activity and APC/C activity at this checkpoint. While we show here that APC/CCdh1 targets pVHL for degradation, the possibility that pVHL CRL may have an effect on Cdh1 and/or APC/C remains to be explored.
Materials and Methods
Reagents, antibodies, and chemicals
MG132 and anti-Cdh1 (CC43) were purchased from Calbiochem. Thymidine, nocodazole and anti-β-tubulin antibody were from Sigma. Anti-VHL (Ig32) was purchased from BD Pharmingen, anti-HA (Y-11), anti-cyclin B1, anti-cdc27 and anti-VHL (FL-181) antibodies were purchased from Santa Cruz Biotechnology, and anti-myc (9E10) antibody was purchased from Covance. The HA-tagged Cdh1 expression vector was Addgene plasmid 11596 deposited by Dr Marc Kirschner (Pfleger et al., 2001).
Cell culture, transfections, and RNA interference
Methodology is provided in Supplementary Information.
Western blotting and real time RT-PCR
Methodology is provided in Supplementary Information.
Cell cycle synchronization
Cells were synchronized at G1/S or G2/M phases of the cell cycle by double thymidine or thymidine/nocodazole treatment, respectively, as described previously (Harper, 2005). Cell cycle status of treated cultures was determined by flow cytometry. Cells were fixed with 70% ethanol at −20°C overnight, treated with 10 μg/ml RNase, and stained with 100 μg/ml propidium iodide. Cellular DNA content was examined using an Epics XL (Beckman Coulter) flow cytometer and data were analyzed using the Summit software package.
Protein-protein interactions
Methodology is provided in Supplementary Information.
In vitro protein degradation
HeLa cell extracts were prepared with hypotonic buffer (10 mM Tris-HCl [pH 7.5], 10 mM KCl, 10 mM MgCl2, 1 mM dithiothreitol) containing protease inhibitors cocktail (Roche) (Liu et al., 2007). VHL proteins were synthesized using the Quick-Coupled TnT expression system (Promega) in the presence of 35S-methionine. Approximately 10 ng of 35S-labeled pVHL or pVHL mutants was added to 20 μl fresh HeLa cell extracts supplemented with degradation cocktail (1.25 mg/ml ubiquitin, 1× ATP regeneration system, and 0.1 mg/ml cycloheximide). Aliquots were removed at the indicated time points and analyzed by SDS-PAGE and autoradiography.
Supplementary Material
Acknowledgments
We thank Dr. Arthur Haas for support and discussions, Eric Hittle, Kimberlee Rankin, and Miki Katuwal for technical assistance, and the Cancer Association of Greater New Orleans for support to HX. This work was supported by the LSUHSC-NO Dean's Research Fund and NIH grant CA125930. David T Eckert was supported by T32DK007774.
References
- Baker DJ, Dawlaty MM, Galardy P, van Deursen JM. Mitotic regulation of the anaphase-promoting complex. Cellular & Molecular Life Sciences. 2007;64:589–600. doi: 10.1007/s00018-007-6443-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beroud C, Collod-Beroud G, Boileau C, Soussi T, Junien C. UMD (Universal mutation database): a generic software to build and analyze locus-specific databases. Hum Mutat. 2000;15:86–94. doi: 10.1002/(SICI)1098-1004(200001)15:1<86::AID-HUMU16>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Chi JT, Wang Z, Nuyten DS, Rodriguez EH, Schaner ME, Salim A, et al. Gene expression programs in response to hypoxia: cell type specificity and prognostic significance in human cancers. PLoS Med. 2006;3:e47. doi: 10.1371/journal.pmed.0030047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Paulsen N, Brychzy A, Fournier MC, Klausner RD, Gnarra JR, Pause A, et al. Role of transforming growth factor-alpha in von Hippel--Lindau (VHL)(-/-) clear cell renal carcinoma cell proliferation: a possible mechanism coupling VHL tumor suppressor inactivation and tumorigenesis. Proc Natl Acad Sci U S A. 2001;98:1387–92. doi: 10.1073/pnas.031587498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- Fähling M. Cellular oxygen sensing, signalling and how to survive translational arrest in hypoxia. Acta Physiologica. 2009;195:205–230. doi: 10.1111/j.1748-1716.2008.01894.x. [DOI] [PubMed] [Google Scholar]
- Gardner LB, Li Q, Park MS, Flanagan WM, Semenza GL, Dang CV. Hypoxia inhibits G1/S transition through regulation of p27 expression. J Biol Chem. 2001;276:7919–26. doi: 10.1074/jbc.M010189200. [DOI] [PubMed] [Google Scholar]
- Gordan JD, Bertout JA, Hu CJ, Diehl JA, Simon MC. HIF-2alpha promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell. 2007;11:335–47. doi: 10.1016/j.ccr.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackenbeck T, Knaup KX, Schietke R, Schödel J, Willam C, Wu X, et al. HIF-1 or HIF-2 induction is sufficient to achieve cell cycle arrest in NIH3T3 mouse fibroblasts independent from hypoxia. Cell Cycle. 2009;8:1386–1395. doi: 10.4161/cc.8.9.8306. [DOI] [PubMed] [Google Scholar]
- Harper JV. Synchronization of cell populations in G1/S and G2/M phases of the cell cycle. Methods Mol Biol. 2005;296:157–66. doi: 10.1385/1-59259-857-9:157. [DOI] [PubMed] [Google Scholar]
- Hergovich A, Lisztwan J, Barry R, Ballschmieter P, Krek W. Regulation of microtubule stability by the von Hippel-Lindau tumour suppressor protein pVHL. Nat Cell Biol. 2003;5:64–70. doi: 10.1038/ncb899. [DOI] [PubMed] [Google Scholar]
- Iliopoulos O, Kibel A, Gray S, Kaelin WG. Tumor suppression by the human von Hippel-Lindau gene product. Nat Med. 1995;1:822–6. doi: 10.1038/nm0895-822. [DOI] [PubMed] [Google Scholar]
- Iliopoulos O, Ohh M, Kaelin WG., Jr pVHL19 is a biologically active product of the von Hippel-Lindau gene arising from internal translation initiation. Proc Natl Acad Sci U S A. 1998;95:11661–6. doi: 10.1073/pnas.95.20.11661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson PK. Linking tumor suppression, DNA damage and the anaphase-promoting complex. Trends in Cell Biology. 2004;14:331–4. doi: 10.1016/j.tcb.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Jung CR, Hwang KS, Yoo J, Cho WK, Kim JM, Kim WH, et al. E2-EPF UCP targets pVHL for degradation and associates with tumor growth and metastasis. Nat Med. 2006;12:809–16. doi: 10.1038/nm1440. [DOI] [PubMed] [Google Scholar]
- Kaelin WG. Proline hydroxylation and gene expression. Annu Rev Biochem. 2005;74:115–28. doi: 10.1146/annurev.biochem.74.082803.133142. [DOI] [PubMed] [Google Scholar]
- Kaelin WG. Von Hippel-Lindau disease. Annu Rev Pathol. 2007a;2:145–73. doi: 10.1146/annurev.pathol.2.010506.092049. [DOI] [PubMed] [Google Scholar]
- Kaelin WG., Jr The von Hippel-Lindau tumor suppressor protein and clear cell renal carcinoma. Clin Cancer Res. 2007b;13:680s–684s. doi: 10.1158/1078-0432.CCR-06-1865. [DOI] [PubMed] [Google Scholar]
- Kaelin WG., Jr The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nat Rev Cancer. 2008;8:865–73. doi: 10.1038/nrc2502. [DOI] [PubMed] [Google Scholar]
- Koshiji M, Kageyama Y, Pete EA, Horikawa I, Barrett JC, Huang LE. HIF-1alpha induces cell cycle arrest by functionally counteracting Myc. EMBO J. 2004;23:1949–56. doi: 10.1038/sj.emboj.7600196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lendahl U, Lee KL, Yang H, Poellinger L. Generating specificity and diversity in the transcriptional response to hypoxia. Nat Rev Genet. 2009;10:821–832. doi: 10.1038/nrg2665. [DOI] [PubMed] [Google Scholar]
- Listovsky T, Oren YS, Yudkovsky Y, Mahbubani HM, Weiss AM, Lebendiker M, et al. Mammalian Cdh1/Fzr mediates its own degradation. EMBO J. 2004;23:1619–26. doi: 10.1038/sj.emboj.7600149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Simon MC. Regulation of transcription and translation by hypoxia. Cancer Biol Ther. 2004;3:492–7. doi: 10.4161/cbt.3.6.1010. [DOI] [PubMed] [Google Scholar]
- Liu W, Li W, Fujita T, Yang Q, Wan Y. Proteolysis of CDH1 enhances susceptibility to UV radiation-induced apoptosis. Carcinogenesis. 2008;29:263–72. doi: 10.1093/carcin/bgm251. [DOI] [PubMed] [Google Scholar]
- Liu W, Wu G, Li W, Lobur D, Wan Y. Cdh1-anaphase-promoting complex targets Skp2 for destruction in transforming growth factor beta-induced growth inhibition. Molecular & Cellular Biology. 2007;27:2967–79. doi: 10.1128/MCB.01830-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol. 2007;8:379–93. doi: 10.1038/nrm2163. [DOI] [PubMed] [Google Scholar]
- Pause A, Lee S, Lonergan KM, Klausner RD. The von Hippel-Lindau tumor suppressor gene is required for cell cycle exit upon serum withdrawal. Proc Natl Acad Sci U S A. 1998;95:993–8. doi: 10.1073/pnas.95.3.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfleger CM, Lee E, Kirschner MW. Substrate recognition by the Cdc20 and Cdh1 components of the anaphase-promoting complex. Genes Dev. 2001;15:2396–407. doi: 10.1101/gad.918201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmaltz C, Hardenbergh PH, Wells A, Fisher DE. Regulation of proliferation-survival decisions during tumor cell hypoxia. Mol Cell Biol. 1998;18:2845–54. doi: 10.1128/mcb.18.5.2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenfeld A, Davidowitz EJ, Burk RD. A second major native von Hippel-Lindau gene product, initiated from an internal translation start site, functions as a tumor suppressor. Proc Natl Acad Sci U S A. 1998;95:8817–22. doi: 10.1073/pnas.95.15.8817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. Involvement of oxygen-sensing pathways in physiologic and pathologic erythropoiesis. Blood. 2009;114:2015–9. doi: 10.1182/blood-2009-05-189985. [DOI] [PubMed] [Google Scholar]
- Stebbins CE, Kaelin WG, Jr, Pavletich NP. Structure of the VHL-ElonginC-ElonginB complex: implications for VHL tumor suppressor function. Science. 1999;284:455–61. doi: 10.1126/science.284.5413.455. [DOI] [PubMed] [Google Scholar]
- Stickle NH, Cheng LS, Watson IR, Alon N, Malkin D, Irwin MS, et al. Expression of p53 in renal carcinoma cells is independent of pVHL. Mut Res. 2005;578:23–32. doi: 10.1016/j.mrfmmm.2005.02.016. [DOI] [PubMed] [Google Scholar]
- Sudo T, Ota Y, Kotani S, Nakao M, Takami Y, Takeda S, et al. Activation of Cdh1-dependent APC is required for G1 cell cycle arrest and DNA damage-induced G2 checkpoint in vertebrate cells. EMBO J. 2001;20:6499–508. doi: 10.1093/emboj/20.22.6499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoma CR, Frew IJ, Hoerner CR, Montani M, Moch H, Krek W. pVHL and GSK3beta are components of a primary cilium-maintenance signalling network. Nat Cell Biol. 2007;9:588–95. doi: 10.1038/ncb1579. [DOI] [PubMed] [Google Scholar]
- Thoma CR, Toso A, Gutbrodt KL, Reggi SP, Frew IJ, Schraml P, et al. VHL loss causes spindle misorientation and chromosome instability. Nat Cell Biol. 2009;11:994–1001. doi: 10.1038/ncb1912. [DOI] [PubMed] [Google Scholar]
- van Leuken R, Clijsters L, Wolthuis R. To cell cycle, swing the APC/C. Biochimica et Biophysica Acta. 2008;1786:49–59. doi: 10.1016/j.bbcan.2008.05.002. [DOI] [PubMed] [Google Scholar]
- Wan Y, Liu X, Kirschner MW. The anaphase-promoting complex mediates TGF-beta signaling by targeting SnoN for destruction. Mol Cell. 2001;8:1027–39. doi: 10.1016/s1097-2765(01)00382-3. [DOI] [PubMed] [Google Scholar]
- Warnecke C, Weidemann A, Volke M, Schietke R, Wu X, Knaup KX, et al. The specific contribution of hypoxia-inducible factor-2alpha to hypoxic gene expression in vitro is limited and modulated by cell type-specific and exogenous factors. Exp Cell Res. 2008;314:2016–27. doi: 10.1016/j.yexcr.2008.03.003. [DOI] [PubMed] [Google Scholar]
- Wasch R, Robbins JA, Cross FR. The emerging role of APC/C(Cdh1) in controlling differentiation, genomic stability and tumor suppression. Oncogene. 2010;29:1–10. doi: 10.1038/onc.2009.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Ayad NG, Wan Y, Zhang GJ, Kirschner MW, Kaelin WG., Jr Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature. 2004;428:194–8. doi: 10.1038/nature02381. [DOI] [PubMed] [Google Scholar]
- Wykoff CC, Sotiriou C, Cockman ME, Ratcliffe PJ, Maxwell P, Liu E, et al. Gene array of VHL mutation and hypoxia shows novel hypoxia-induced genes and that cyclin D1 is a VHL target gene. Br J Cancer. 2004;90:1235–43. doi: 10.1038/sj.bjc.6601657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young AP, Schlisio S, Minamishima YA, Zhang Q, Li L, Grisanzio C, et al. VHL loss actuates a HIF-independent senescence programme mediated by Rb and p400. Nature Cell Biology. 2008;10:361–9. doi: 10.1038/ncb1699. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.