

Abstract
Protein–protein interactions between members of the Myc transcription factor network are potential targets of small molecule inhibitors and stabilizers. Diverse screening strategies—including fluorescence resonance energy transfer, fluorescence polarization, 2-hybrid, and protein complementation assays—have identified several lead compounds that inhibit Myc–Max dimerization and one compound that stabilizes the Max homodimer. Representative compounds interfere with Myc-induced transcriptional activation, Myc-mediated oncogenic transformation, Myc-driven cellular replication, and DNA binding of Myc. For the best-characterized compounds, specific binding sites have been determined, and molecular mechanisms of action have been documented. This knowledge of small molecule–protein interaction is currently applied to highly targeted approaches to identify novel compounds with improved potency.
Keywords: Max, Mad, ID proteins, 10058-F4
The concept that the abnormal proliferation and metabolism of cancer cells can be pharmacologically exploited is quite old and represents the foundation upon which chemotherapeutic approaches have rested for a half century.1–4 Only recently have we begun to realize its true potential as a result of an improved understanding of the molecular underpinnings responsible for the cancer cell’s replicative and survival advantages. This has led to the deliberate design of various inhibitors that attack tumor cells with a much higher degree of specificity and with fewer side effects than previously attainable.5–7 Despite the often stunning success of this approach in previously refractory diseases, many molecular targets have remained frustratingly immune to attack. These include transcription factors whose protein–protein or protein–DNA interactions have been considered intrinsically resistant to small molecules.8–10 Here we review recent progress in therapies designed to target c-Myc (Myc). We weigh the arguments for and against targeting this ubiquitously expressed and rarely mutated oncoprotein, discuss the various approaches currently being examined and their limitations, and propose how some of the therapeutic bottlenecks might be overcome. Finally, we evaluate the possibility for the use of Myc inhibitors in nonmalignant states associated with cellular hyperproliferation.
Arguments Against Therapeutic Targeting of Myc
A priori, there are numerous reasons to view Myc as a challenging therapeutic target. Among the practical and theoretical arguments typically encountered are the following.
First, Myc is rarely mutated in cancer despite its high level of expression.11,12 Exceptions do occur in Burkitt and AIDS-related lymphomas where approximately 30% of primary tumors carry amino acid substitutions in Myc.13–17 The majority of these cluster around or directly affect Thr58 and affect Myc’s half-life by interfering with its ubiquitination-mediated proteasomal degradation.13,18 However, the overall paucity of mutations dictates that the design of therapeutics with widespread potential cannot be predicated on paradigms that have been successfully used to develop inhibitors of cancer-associated protein kinases, which typically possess gain-of-function mutations that distinguish them from their normal counterparts.19–22 The development of such agents is guided by the long-established principles of enzyme inhibitor design and will have limited applicability to nonenzymatic proteins such as Myc.23,24
Second, Myc expression is a nearly universal property of all proliferating cells, and its inhibition might be associated with unacceptable toxicities.
Third, Myc inhibitor design will be difficult. The most obvious approaches—for instance, targeting the association between Myc and Max or other essential cofactors, such as TRRAP25—involve the disruption of protein–protein interactions. The surfaces at which these occur tend to be large, flat, and relatively featureless, and they often lack recognizable motifs or clefts, such as those found in enzymes. The disruption of protein–protein interactions must also overcome a large free energy of association from the interacting protein moieties.26–28
Arguments for Therapeutic Targeting of Myc
Despite the foregoing objections, there are numerous reasons to believe that the general strategy of inhibiting Myc is reasonable and attractive and that the potential benefits for pursuing this approach outweigh the concerns and disadvantages. These arguments serve to balance the list of disadvantages in the preceding section.
First, despite the general lack of Myc protein mutations, most tumors are Myc dependent to varying degrees. For example, in a recent survey of more than 20 human cancer lines, short hairpin RNA–mediated depletion of Myc led to a permanent proliferative arrest in every case examined.29 Consistent with this observation are the findings obtained with conditional in vivo models indicating that continuous Myc expression is required to sustain tumor proliferation and viability30–32 and that, in at least some cases, its reinstatement may actually lead to a seemingly paradoxical apoptotic response.32 This finding is not universal, however, and certain types of tumors appear to lose their dependence on deregulated Myc.33,34 However, in none of these cases has their lack of dependence on endogenous Myc been established. Taken together, these studies suggest that at some level—whether it be normally expressed and regulated Myc or the deregulation that accompanies many tumors—Myc inhibition is likely to inhibit tumor progression and/or survival. The possibility that tumor growth might be further impaired by the proapoptotic reestablishment of Myc expression suggests that long-term therapeutic suppression of Myc may at least in some circumstances be neither warranted nor even desirable.
Second, Myc expression by normal cells might not necessarily limit the use of Myc-based therapies. At any given time, most normal cells are quiescent and express little, if any, Myc; thus, they might not be subject to the effects of Myc inhibitors. At the whole organism level, this could mean that the associated toxicities of such therapies would mimic those of more standard, nontargeted agents and include primarily hematopoietic and gastrointestinal effects. Consistent with this idea are the findings of Soucek et al., who suppressed endogenous Myc via the tetracycline-regulatable conditional expression of a dominant-negative (DN) Myc protein in transgenic mice that were engineered to develop K-rasG12D-driven lung adenomas.35 Although the anticipated pancytopenia, epidermal thinning, and intestinal villus attrition did occur quite quickly upon expression of the DN-Myc, these effects were well tolerated, unassociated with overt toxicity, and largely reversible, even when DN-Myc expression was allowed to persist. Unexpectedly, the mice showed a reduced incidence of adenomas, consistent with the concept that most, if not all, oncogenic signaling/proliferative pathways are likely to converge upon Myc and that its inhibition results in nearly universal proliferative arrest.36,37
Third, the notion that protein–protein interactions might be refractory to small molecule inhibitors has gradually yielded to experimental evidence to the contrary. Indeed, that single amino acid substitutions in the bHLH-ZIP dimerization domain of Myc could abolish its interaction with Max and abrogate its transcriptional activation function and biological properties38,39 speaks to this point and so provided the impetus and rationale for Myc inhibitor discovery by one of our groups.40 It is also now appreciated that the interaction between the flat surfaces of interacting domains is initiated by a limited number high-affinity interactions, which can sometimes account for the majority of the free energy of binding.41 This implies that a small molecule capable of recognizing such a site has the potential for exerting a disproportionate effect on protein–protein interaction. Further evidence by analogy for the success of the small molecule approach has now been seen in other areas, including the disruption of TP53–HDM2 interactions by nutlins10 and the inhibition of Bcl-2, Bcl-XL, and Bcl-w by small-molecule BH3 mimetics such as ABT-737.42,43
Finally, Myc-based therapeutics rely on a novel molecular target. They should therefore be compatible and largely non-cross-resistant with many preexisting chemotherapeutic agents.
Identification of Inhibitors of Myc-Max Dimerization (Myc Inhibitors)
Several in vitro and cell-based methods can be used in screens to identify inhibitors of Myc–Max dimerization. A simple and straightforward in vitro technique is based on fluorescence resonance energy transfer (FRET). In this procedure, the basic helix-loop-helix leucine-zipper (bHLH-ZIP) domain of Myc is fused to cyan fluorescent protein (CFP), and the bHLH-ZIP domain of Max is fused to yellow fluorescent protein (YFP). The 2 proteins are allowed to dimerize, as followed by excitation of CFP at the wavelength of 433 nm. Dimerization generates a FRET spectrum characterized by a strong emission signal of YFP at 525 nm and a weaker emission signal of CFP at 475 nm. The ratio of fluorescence intensity at 525 nm over 475 nm is typically 1.7 at complete dimerization of MycCFP with MaxYFP. The intensity ratio is 0.4 for the monomeric state of MycCFP. Inhibitors of Myc–Max dimerization reduce the ratio of fluorescence at 475 nm over that at 525 nm.44 This in vitro technique measures the Myc–Max interaction specifically and directly.
A second in vitro technique that has been used in screens for inhibitors of Myc–Max dimerization is based on fluorescence polarization.45,46 An oligonucleotide containing several iterations of the Myc–Max consensus-binding sequence (E-box) is labeled with a fluorophore (5-carboxyfluorescein, Alexa Fluor 633). This indicator is excited with plane polarized light. The polarization of the emitted light depends on the mobility of the molecule, which is in turn a function of molecular size. Binding of the fluorescent indicator to the Myc–Max dimer increases this size and enhances the fluorescence polarization. Among the advantages of this technique is simplicity; only one of the interacting components needs to be tagged with fluorophore. However, the technique does not discriminate between inhibitors of Myc–Max dimerization and inhibitors of Myc–Max DNA binding and will therefore result in some false positives that have to be eliminated by a different method.
Among the cell-based techniques, the 2-hybrid method has been successfully used to isolate inhibitors of Myc–Max dimerization and is described later.40
A second in vivo technique is the protein complementation assay (PCA). The strategy of the PCA is to mediate the complementation of 2 β-lactamase fragments by 2 interacting proteins that are conjugated to the enzyme fragments. Animal cells are devoid of endogenous β-lactamase. Two separate β-lactamase fragments are conjugated to the bHLH-ZIP domains of Myc and Max.47,48 A flexible linker between Myc or Max and lactamase fragments facilitates reassociation of the 2 enzyme fragments when Myc and Max dimerize. We have generated HEK293 cells stably transfected with these chimeric constructs. For the PCA, 1 M CCF2/AM is added to the cultures. CCF2/AM diffuses across the cell membrane and is hydrolyzed by cytoplasmic esterases, generating the β-lactamase substrate CCF2. Complementing fragments of β-lactamase hydrolyze CCF2, generating a coumarin fragment that emits blue fluorescence at 447 nm. The nonhydrolyzed CCF2 emits in the green spectrum at 530 nm. Controls include single transfectants (no lactamase), double transfectants without test compound (active lactamase), and double transfectants with a known Myc–Max inhibitor (reduced lactamase activity). FACS sorting at 447 nm is used for a quantitative determination of inhibitor potency, which is reflected by the ratio of nonfluorescing cells to fluorescing cells.49
The FRET screening technique has been used in conjunction with 2 chemical libraries. One is a peptidometic library generated by solution phase synthesis.50 The second library, the “credit card” library, uses a planar aromatic core further functionalized by the addition of highly diverse motifs.51 These libraries yielded several inhibitors of Myc–Max dimerization. The lead compounds also interfere with the binding of Myc–Max to DNA in electrophoretic mobility shift assays (EMSAs).44,51 They are active in reporter assays, inhibiting Myc-mediated but not N-Myc-mediated transcriptional activation.51,52 For some of these compounds, interference with Myc–Max dimerization was documented in ELISA assays that measure binding of GFP-coupled Myc to immobilized Max.44 Oncogenic transformation induced in cultures of primary chicken embryonic cells by Myc is strongly inhibited by most but not all the identified compounds. The half maximal inhibitory concentration (IC50) of the Myc–Max inhibitors in all these assays is between 10 and 50 µM. At higher inhibitor concentrations, cell growth is also negatively affected. The inhibition of oncogenic focus formation extends to Jun- but not Src-mediated transformation. Jun and Myc use leucine zippers to dimerize, and the cross-reaction of the inhibitors may reflect structurally similar motifs in Jun and in Myc that interact with the inhibitors. However, there is at present no evidence for this supposition, and more work is required to characterize the interaction between the compounds and their targets. Src-mediated transformation requires Myc function, and inhibitors of Myc are expected to affect the transforming activity of Src.53 However, under the conditions of the focus assay, the inhibitors never completely block Myc function. The residual Myc activity may be sufficient to support transformation by Src. Several important questions about these inhibitors of Myc–Max dimerization remain to be answered. It is not known whether these inhibitors bind to Myc, Max, or both proteins. Binding presumably targets the monomeric form, but the actual binding sites of the inhibitors on Myc or Max have not been determined.
Greater molecular details of the mechanism of inhibitor action have been worked out by Yin et al., who utilized a different approach for the identification of candidate compounds.40 They developed a yeast 2-hybrid-based approach in which heterodimerization between the bHLH-ZIP domains of Myc and Max would reconstitute a bipartite but functional Gal4 transcription factor whose activity could easily be quantified by a β-galactosidase (β-gal) assay. They then screened about 10,000 small molecules for those that selectively reduced β-gal activity while having no effect on an identical yeast strain expressing Gal4 fusions of the Id2 and E47 HLH dimerization domains. This latter control allowed for the rapid elimination of compounds with nonspecific effects on yeast growth, β-gal enzyme activity, or target gene binding by Gal4. Ultimately, 7 compounds were identified whose specificity for Myc–Max inhibition was greater than 90% as judged by their limited inhibition of more than 30 tested HLH, ZIP, and HLH–ZIP interactions in the same yeast background. A direct effect on Myc–Max association and DNA binding was demonstrated by glutathione–agarose pulldown experiments and EMSAs. Similar experiments demonstrated that the compounds inhibited the formation of new heterodimers and disrupted preexisting ones. Cells treated with these compounds also showed reduced expression of 2 Myc-responsive reporters. Importantly, normal fibroblasts and those transformed as a result of deregulated Myc expression were growth inhibited, whereas myc−/− fibroblasts were not.54 Transformed fibroblasts also demonstrated markedly reduced tumorigenicity following exposure to the compounds.
The Mechanisms of Myc Compound Binding
The above studies, including those of Berg et al.,44 raised several important and ultimately related questions. The first was whether Myc compounds were binding to Myc, Max, or the Myc–Max heterodimer. The second was whether the compounds were binding to identical or distinct sites. Finally, could Myc compounds be used in combination to provide additive or synergistic effect?
With regard to the first question, the original yeast 2-hybrid studies indirectly implicated the Myc monomer as the site of compound binding given that they minimally affected Max homodimerization or Max–Mad member heterodimerization.40 Monomeric binding was also supported by the finding that the compounds prevented the de novo formation of Myc–Max heterodimers in vitro. The issue was definitively settled by Wang et al.,37 Follis et al.,55 and Hammoudeh et al.,56 who used a combination of fluorescence polarization and circular dichroism (CD) to provide direct physical evidence that the recombinant Myc bHLH-ZIP domain but not the comparable domain of Max bound every one of the 7 compounds originally identified by Yin et al.40 With regard to the second question, the possibility that Myc compounds were occupying different sites was suggested by their diverse chemical structures. Implying this as well were the different patterns of nonspecific interactions of these compounds with the numerous other HLH, ZIP, and HLH-ZIP yeast 2-hybrid partners used as controls.40 The tools used to establish that all the compounds were binding to monomeric Myc were used to localize their exact binding sites via a series of highly purified recombinant Myc bHLH-ZIP deletion and point mutants. Distinct binding sites were identified for 3 of the original parental compounds: residues 363–381 for 10074-G5, residues 370–407 for 10074-A4, and residues 402–412 for 10058-F4. The first site corresponds to the junction of the basic domain and helix 1; the second site corresponds to the N-terminus of helix 1; and the third site corresponds to the junction of helix 2 and the ZIP domain. The remaining 4 compounds bound to either the first site (10050-C10) or the third site (10075-G5, 1009-G9, and 10031-B8). Final confirmation of binding site assignments came from experiments with synthetic peptides whose calculated Kd’s for the compounds were in good agreement with those obtained using the longer recombinant bHLH-ZIP proteins (Figure 1).37,55,56 It will be of interest to determine whether other previously identified Myc inhibitors44–46 or those identified in the future will show similar properties. The intrinsically disordered (ID) nature of these 3 sites suggests that they may be particularly well adapted as targets for many, if not all, small molecule Myc inhibitors.
Figure 1.
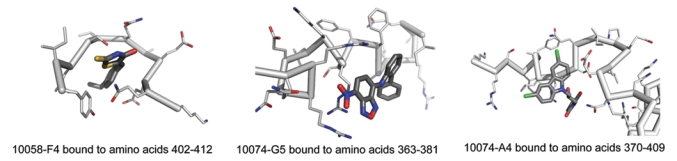
NMR models of Myc compounds bound to their cognate sites in the Myc monomer. Synthetic peptides encompassing each of the previously determined binding sites were used to obtain NMR models. Because of the intrinsically disordered nature of each binding site, the images shown do not represent actual structures; rather, they are composites of the ensembles of structures representing the most likely bound conformation for each site.56 (© 2009, American Chemical Society. Reprinted with permission.)
Regarding whether individual compounds could be used more effectively in combination, we observed little more than an additive effect when those with distinct binding-site preferences were tested (Wang and Prochownik, unpublished). However, as described in the next section, much different results have been obtained when these compounds were joined via chemical cross-linking to create bivalent compounds.
Improving Myc Compound Potency
None of the originally identified Myc compounds bound its cognate site with particularly high affinity. For example, the parental compounds 10058-F4 and 10074-G5 showed Kd’s of approximately 2.5 µM and 4.4 µM, respectively, as determined by fluorescence polarization.37,55 Similarly, binding of the nonfluorescent compound 10074-A4 to its cognate site, which was determined by CD, was calculated to be about 21 µM.56 These values were generally somewhat lower than the empirically determined IC50 values in several cell types, which ranged from approximately 20 to 50 µM.37,40 Given that compounds with such low affinities are unlikely to be viable therapeutic candidates, considerable effort has gone into deriving more potent analogs. Three distinct approaches have been taken.
The Random Analog Approach
Initially lacking any knowledge of the structure of these compounds in association with their Myc-binding sites, a random analog screen was conducted to identify more potent structural relatives of the parental compound 10058-F4, which is chemically the simplest of the original 7 parental Myc compounds.40 The 10058-F4 compound contains a 6-member ethylbenzylidine ring and a 5-member thioxothiazolidin-4-one, or rhodanine, ring. 48 second-generation analogs containing alterations of only the first ring and 15 analogs with alterations of only the rhodanine ring were screened with a number of independent assays, including the aforementioned fluorescence polarization tests, EMSAs, coimmunoprecipitations from compound-treated cells, and cell growth inhibition assays. The best 6-member ring analogs were then combined with the best 5-member ring analogs to generate third-generation compounds with alterations in each ring. In all, 80 chemically distinct 10058-F4 analogs were analyzed.
Despite some fairly radical departures from the base 10058-F4 structure,37 a surprisingly large number of analogs retained activity in all assays. However, only 4 analogs had Ka’s for recombinant Myc bHLH-ZIP domain that were significantly better than that of 10058-F4, and the best of these was only 4-fold better. Moreover, binding affinities did not necessarily correlate with the ability of the compounds to inhibit Myc–Max heterodimerization in intact cells and block proliferation. For example, the 6-member-ring substituted analog with the best affinity for Myc (28Rh, Ka = 1.0 µM) had an IC50 in HL60 promyelocytic leukemia cells that was only modestly better than that of 10058-F4 (36 µM versus 49 µM).37 In contrast, No. 764, a 5-member ring analog with the best IC50 (6.5 µM), actually had a slightly inferior binding affinity relative to that of 10058-F4. The imperfect correlation between compound binding and growth inhibition probably reflects the differences in biological behaviors among the various analogs, including rates of influx and efflux, metabolism, and intracellular trafficking and nonspecific protein binding.
Pharmacophore Model Screens
Subsequent to the above-described work, NMR models were generated of representative parental compounds (10058-F4, 10074-G5, and 10074-A4) bound to their cognate synthetic peptides or a short recombinant segment of the Myc bHLH-ZIP domain.55,56 The ID nature of all 3 binding sites does not allow these to be represented as actual structures; rather, they serve as the best average approximations of the ensemble of dynamic structures most likely to exist at any given time (Fig. 1).
The previous studies yielded a large amount of structure–activity relationships for the active and inactive analogs of 10058-F4.37,40 These were used to generate a molecule-derived pharmacophore model that incorporated these relationships using the GALAHAD program (Genetic Algorithm with Linear Assignment for Hypermolecular Alignment of Datasets)57,58 and were further refined using the Tuplets model in SYBYL 8.0 (http://www.tripos.com). In silico screening of the ZINC database of approximately 5 × 106 compounds59 initially identified a large number of structurally diverse molecules, as determined by their Tanimoto scores of 0.5. This group was filtered down to a set of 30 compounds with the most desirable ADME properties and then further reduced to 9, which retained their structural diversity and were viewed as poor substrates for the cytochrome P450 isoform CYP3A4, the major human enzyme responsible for xenobiotic metabolism.60 Of the 9 compounds, 7 showed activity at high concentrations when tested against preformed Myc–Max heterodimers using CD. The 4 best compounds competing for Max in this assay were generally 2- to 10-fold better than 10058-F4. They also competed 10058-F4 from its binding site, as determined by fluorescence polarization,37,55 and were able to inhibit DNA binding by Myc–Max heterodimers in an EMSA. However, the calculated affinities of the compounds for Myc in these latter 2 assays were not significantly greater than those for 10058-F4. Finally, when tested in cell proliferation assays, none of the tested compounds were more than 2-fold better than 10058-F4. As with the 10058-F4-derived random analog compounds,37 this likely reflected the more complex nature of the biologically based assay where inhibition of cell proliferation reflects the balance of many factors other than compound binding, which is the only parameter evaluated in simple 2-component-based CD and fluorescence polarization assays.
It seems likely that the failure of the pharmacophore model–based approach to identify molecules with greater biological activity is a measure of the limited number of compounds interrogated and the imperfections of the model itself. Improving the model by incorporating new and structurally diverse compounds as they become available will likely increase the rate at which more potent inhibitors are detected. Despite these shortcomings, there have been 2 dividends of this approach, particularly when considered in light of the random analog approach. The first is the simple demonstration that the overall pharmacophore model for identifying Myc compounds is viable and reasonably robust. The second is the surprising structural diversity of Myc inhibitors (Figure 2).
Figure 2.
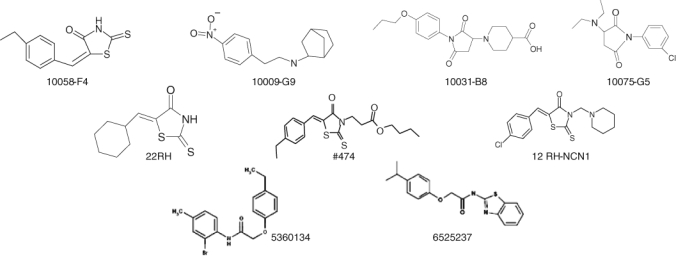
Examples of the structural diversity of active Myc compounds identified by different methods. All the compounds shown bind to the same site on Myc, namely residues 402–412.37,55,56 Furthermore, 10058-F4, 10075-G5, 1009-G9, and 10031-B8 were among the original parental Myc compounds identified by Yin et al.40 using the yeast 2-hybrid-based approach. In addition, 22RH, 474, and 12RH-NCN1 are 10058-F4 analogs identified by the random analog approach described by Wang et al.37 Relative to 10058-F4, 22RH, 474, and 12RH-NCN1 contain structural modifications of the 6-member ring only, the 5-member ring only, and both rings, respectively. Finally, 5360134 and 6525237 are molecules from the ZINC library, as identified by pharmacophore model screening.88
Link Compounds
The revelation that many, if not all, Myc compounds bind to monomeric Myc has allowed for a novel approach to identify more potent Myc compounds. This involved the linking of 2 small molecules with different binding sites to create a bivalent molecule.61,62 That such an approach is feasible was supported by the fact that the closely related monomeric bHLH-ZIP domain of v-Myc (and, presumably, c-Myc) is relatively unstructured in solution63 and assumes a rigid conformation only upon associating with Max.63,64 The lack of a well-defined structure of the monomeric Myc bHLH-ZIP domain would be expected to provide sufficient conformational freedom to allow it to readily interact with even the most rigid bivalent molecule.
To this end, a series of so-called Link compounds has recently been generated in which parental Myc inhibitors 10058-F4 and 10074-G5 have been joined at various sites by flexible aliphatic linkers of variable composition and length (Follis et al., in preparation). As expected from the model of the unstructured Myc bHLH-ZIP domain, all Link compounds have demonstrated impressive increases in activity relative to either of the individual monovalent components. In the best case, up to 5,000-fold improvements in binding affinity have been observed, representing more than 1,000-fold better affinity for Myc than that of Max for Myc. There are at least 2 potential and nonmutually exclusive explanations for these remarkable increases in binding affinities. First, the binding of either moiety to its cognate site on Myc increases the local concentration and the dwell time of the second linked moiety near its binding site. Second, the off-rate of either moiety is countered by virtue of being tethered to its still-bound companion moiety. Link compounds provide an immediate solution to the potency problem because high pM–low nM affinities are now readily achievable. Preliminary results indicate that cellular uptake still remains disproportionately poor but is nevertheless up to 100-fold better than that of even those most potent monovalent compounds thus far discovered. To date, Link compounds have been prepared only with parental forms of Myc compounds.40 Further improvements can be expected when higher-affinity monovalent binders are used as Link substrates.
Myc and Intrinsic Disorder
That the structures of many effective Myc compounds deviated significantly from that of the original parental compound 10058-F4 (Figure 2) was initially confusing but ultimately understandable in light of the ID nature of all 3 Myc-binding sites.56 ID regions have been defined as highly unstructured, dynamic, and constantly shifting segments of amino acids that assume an ordered state only upon binding to specific targets, which can themselves be of variable structure. That the binding of ID regions can alter biological function conflicts with prevailing concepts of structure–function paradigms.65 ID regions are common and appear to be abundant in signaling molecules and transcription factors, where they have been proposed to play important roles in regulating their activity.66,67 The mapping of Myc compound–binding sites to ID regions, with the frequency with which such sites appear in many transcription factors,66 suggests that the design of small molecule inhibitors of transcription factors will be able to take advantage of ID regions and that the presence of such regions will serve as guides to future drug discovery efforts.
In Vivo Myc Compound Efficacy and Metabolism
Thus far, only a single study has assessed the metabolism and/or efficacy of small molecule Myc compounds in vivo. Guo et al. studied 10058-F4 in SCID mice bearing xenografts of human PC3 and DU145 prostate cancer cell lines.68 Although the compound exhibited a high maximally-tolerated dose, it had a short terminal half-life, approximately 1 hour, and showed rapid breakdown into at least 8 metabolites. Highest concentrations of the compound were seen in liver, lung, kidney, and fat, and peak tumor concentrations were at least 10-fold lower than those achieved in serum. No significant effects on tumor growth were observed over a 2-week period of 5 times per week with intraperitoneal treatment. The lack of efficacy was attributed to a combination of rapid metabolism and a failure to reach adequate intratumoral levels. Preliminary studies with the compound 10074-G5 have shown similar rapid metabolism (Liggett et al., in preparation).
The development of more potent Myc compounds, whether they be new analogs of monovalent compounds or new Link molecules, will require eventual pharmacologic testing that is likely to represent a significant bottleneck to the development of these agents for clinical use. Alternate means of delivery, such as encapsulation in liposomes, may help to circumvent some of these bottlenecks. Alternatively, direct local delivery, perhaps at a surgical site of excised tumor and perhaps embedded in a biodegradable matrix, may provide a more protected environment, allow a higher concentration of compound to be attained, and extend compound life span to achieve more pronounced antiproliferative effects.
The Potential for Myc Inhibitors in Nonneoplastic Settings
Interest in Myc inhibition is clearly driven by the potential for its clinical application in the oncologic setting. However, based on current understanding of Myc action and the known mechanisms of the various Myc inhibitors discussed above, there is reason to believe that these agents might prove equally attractive for the treatment of certain nonneoplastic disease states associated with cellular hyperproliferation. Arterial restenosis and proliferative retinopathy are common conditions that might prove amenable to Myc inhibitors. In each case, effective targeting would involve action of the compounds working in confined spaces and/or over short distances, thus allowing much smaller doses of the inhibitor to be delivered and eliminating or greatly reducing some of the potential side effects and metabolism issues associated with the more systemic delivery approaches.68
Balloon angioplasty with the concurrent placement of drug-eluting stents (DES) has emerged as the nonsurgical treatment of choice for arterial stenosis.69 The purpose of DES is to prevent the neointimal hyperplasia and inflammatory response that frequently occur as a result of local injury induced by the angioplasty itself and by the placement and/or continued presence of the stent. Currently, the most commonly employed drugs in DES include paclitaxel and sirolimus, with some advantage to the latter having recently been documented.70 Animal models of restenosis have shown that locally administered Myc antisense oligonucleotides can lead to improvements in restenosis rates despite achieving only modest reductions in Myc levels.71,72 Preliminary results in humans have also shown promise.73 The results are encouraging enough to consider the far less costly use of small molecule Myc inhibitors that could replace or be used in conjunction with any of these other approaches and would likely achieve superior levels of Myc inhibition.
Age-related macular degeneration and diabetic retinopathy are the most common causes of blindness in the adult population and are associated with a high degree of endothelial proliferation.74,75 Anti–vascular endothelial growth factor therapies are effective forms of therapy but are expensive, require frequent intravitreal injection, and can be associated with complications such as retinal detachment.76 The direct delivery of low–molecular weight Myc inhibitors would be significantly less expensive. Intravitreal compounds might also show significantly longer half-lives by virtue of their being protected from the rapid metabolic breakdown that has thus far been seen with systemic administration.
Stabilizers of the Max Homodimer
Unlike Myc, the Max protein can homodimerize in vitro and in vivo.77,78 Max homodimers are less stable than Max heterodimers.63 The reduced stability of the Max homodimer reflects a packing defect at its protein–protein interface.64 Max homodimers do not affect transcription except if they are overexpressed with the help of exogenous vectors.79 Overexpressed Max can then repress transcription and interfere with Myc-induced oncogenesis.80–82
The Max–Max homodimers function as the cellular repository for Max, which is the essential and universal partner for all other proteins of the Myc network. Because of the relative instability of the Max–Max homodimer and because Max levels usually exceed those of its partners, Max is readily captured by its heterodimer partners into more stable complexes that function in transcriptional control. Among these partners, Myc is the one that can be highly overexpressed, notably in cancer cells. High levels of Myc require an abundance of Max to become functional. Stabilization of the Max–Max homodimer could therefore preferentially affect such overexpressed Myc and attenuate its oncogenic effects while still allowing life-sustaining functions of other Myc network heterodimers.
Max–Max stabilizers have been identified by virtual ligand screening with the AutoDock program.83 Virtual ligand screening has been used to predict inhibitors of protein–protein interactions.84–87 This type of screening depends on structural information for the target molecules. The monomeric form of Myc is only partially structured, and this is probably true for Max.63 In contrast, the Myc–Max and Max–Max dimers are highly structured and are therefore suitable targets for in silico docking screens.64 Such screens with the AutoDock suite canvassed about 1,700 compounds from the NCI Diversity Set. Potential ligands were identified by their low docking energies and were clustered according to 3 predicted binding sites: a site between the DNA-binding helices of the Max–Max dimer, the basic and adjacent neutral HLH region, and the intersection of the leucine zipper and the HLH region. The third cluster yielded ligands that showed specificity for the Max–Max homodimer and did not interact with the Myc–Max heterodimer. One of the compounds identified by virtual ligand screening, NSC13728, is an effective stabilizer of Max–Max as documented by FRET. Stabilization is independent of interaction with DNA. In ELISAs and surface plasmon resonance assays, the stabilizer inhibits the binding of MycCFP to immobilized Max. Analytical ultracentrifugation suggests that the stabilizer strongly reduces the Kd of the dimer, confirming stabilization. Compound NSC13728 also strongly interferes with Myc-mediated oncogenic transformation in cell culture while not affecting transformation induced by Jun, Src, or PI3K. Myc-induced transcriptional activation of specific target genes is also inhibited. Important questions about the mechanism of action of the Max–Max stabilizer remain to be answered. Although virtual ligand screening suggests a binding site for the stabilizer, that site has not been experimentally verified. A priori reasoning suggests that the binding of the stabilizer is to the dimer, not the monomer, a view supported by the structural differences between dimer and monomer.64
Conclusion
The inhibitors of Myc–Max and the stabilizers of Max–Max constitute a promising beginning. These small molecules can interfere with all Myc functions, with the stimulation of cell replication, with DNA binding, with transcriptional activation, and with oncogenic transformation. For some of these compounds, binding sites have been determined, and probable mechanisms of action have been established. Promising pharmacophores have been identified. The stage is set for further progress, which will concentrate on enhanced potency and greater mechanistic understanding. The ultimate goal remains that of compounds that are therapeutically effective in clinical situations that involve gain of function in Myc.
Acknowledgments
E.V.P. is grateful to his collaborators on the work mentioned in this review—notably, Drs Steven J. Metallo, Arielle Follis, and Dhalia Hammoudeh of Georgetown University and Huabo Wang, John Lazo, Ivet Bahar, Julie Eiseman, and Gabriela Mustata of the University of Pittsburgh Medical Center. Their thoughtful comments and corrections on this article are also highly appreciated. E.V.P.’s work and that of the aforementioned individuals is supported by grant RO1 CA140624-01 from the National Cancer Institute. P.K.V. is grateful to his collaborators on the Myc project: Drs Thorsten Berg, Dale L. Boger, Steven B. Cohen, Joel Desharnais, Joel Goldberg, Kim D. Janda, Daniel J. Maslyar, Jason A. Moss, Jin Shi, Corinna Sonderegger, James S. Stover, Landon R. Whitby, Yang Xu, and Noboru Yamamoto.
Footnotes
P.K.V.’s work is supported by grants from the National Institutes of Health. This is manuscript 20742 of The Scripps Research Institute.
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
- 1. Brues AM, Jacobson LO. Comparative therapeutic effects of radioactive and chemical agents in neoplastic diseases of the hemopoietic system. Am J Roentgenol Radium Ther 1947;58:774-82 [PubMed] [Google Scholar]
- 2. Farber S, Diamond LK. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. N Engl J Med 1948;238:787-93 [DOI] [PubMed] [Google Scholar]
- 3. Huggins C. Endocrine control of prostatic cancer. Science 1943;97:541-4 [DOI] [PubMed] [Google Scholar]
- 4. Mashburn LT, Wriston JC., Jr Tumor inhibitory effect of L-asparaginase from Escherichia coli. Arch Biochem Biophys 1964;105:450-2 [DOI] [PubMed] [Google Scholar]
- 5. Dhomen N, Marais R. BRAF signaling and targeted therapies in melanoma. Hematol Oncol Clin North Am 2009;23:529-45 [DOI] [PubMed] [Google Scholar]
- 6. Druker BJ. Translation of the Philadelphia chromosome into therapy for CML. Blood 2008;112:4808-17 [DOI] [PubMed] [Google Scholar]
- 7. Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001;344:783-92 [DOI] [PubMed] [Google Scholar]
- 8. Deng J, Grande F, Neamati N. Small molecule inhibitors of Stat3 signaling pathway. Curr Cancer Drug Targets 2007;7:91-107 [DOI] [PubMed] [Google Scholar]
- 9. Diaz-Gonzalez JA, Russell J, Rouzaut A, Gil-Bazo I, Montuenga L. Targeting hypoxia and angiogenesis through HIF-1alpha inhibition. Cancer Biol Ther 2005;4:1055-62 [DOI] [PubMed] [Google Scholar]
- 10. Patel S, Player MR. Small-molecule inhibitors of the p53-HDM2 interaction for the treatment of cancer. Expert Opin Investig Drugs 2008;17:1865-82 [DOI] [PubMed] [Google Scholar]
- 11. Lee JW, Soung YH, Kim SY, et al. Mutational analysis of MYC in common epithelial cancers and acute leukemias. APMIS 2006;114:436-9 [DOI] [PubMed] [Google Scholar]
- 12. Nesbit CE, Tersak JM, Prochownik EV. MYC oncogenes and human neoplastic disease. Oncogene 1999;18:3004-16 [DOI] [PubMed] [Google Scholar]
- 13. Bahram F, von der Lehr N, Cetinkaya C, Larsson LG. c-Myc hot spot mutations in lymphomas result in inefficient ubiquitination and decreased proteasome-mediated turnover. Blood 2000;95:2104-10 [PubMed] [Google Scholar]
- 14. Bhatia K, Huppi K, Spangler G, Siwarski D, Iyer R, Magrath I. Point mutations in the c-Myc transactivation domain are common in Burkitt’s lymphoma and mouse plasmacytomas. Nat Genet 1993;5:56-61 [DOI] [PubMed] [Google Scholar]
- 15. Clark HM, Yano T, Otsuki T, Jaffe ES, Shibata D, Raffeld M. Mutations in the coding region of c-MYC in AIDS-associated and other aggressive lymphomas. Cancer Res 1994;54:3383-6 [PubMed] [Google Scholar]
- 16. Pulverer BJ, Fisher C, Vousden K, Littlewood T, Evan G, Woodgett JR. Site-specific modulation of c-Myc cotransformation by residues phosphorylated in vivo. Oncogene 1994;9:59-70 [PubMed] [Google Scholar]
- 17. Yano T, Sander CA, Clark HM, Dolezal MV, Jaffe ES, Raffeld M. Clustered mutations in the second exon of the MYC gene in sporadic Burkitt’s lymphoma. Oncogene 1993;8:2741-8 [PubMed] [Google Scholar]
- 18. Gregory MA, Hann SR. c-Myc proteolysis by the ubiquitin-proteasome pathway: stabilization of c-Myc in Burkitt’s lymphoma cells. Mol Cell Biol 2000;20:2423-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer 2009;9:550-62 [DOI] [PubMed] [Google Scholar]
- 20. Kantarjian HM, Cortes J, La Rosee P, Hochhaus A. Optimizing therapy for patients with chronic myelogenous leukemia in chronic phase. Cancer 2010;116:1419-30 [DOI] [PubMed] [Google Scholar]
- 21. McCubrey JA, Steelman LS, Abrams SL, et al. Emerging Raf inhibitors. Expert Opin Emerg Drugs 2009;14:633-48 [DOI] [PubMed] [Google Scholar]
- 22. Vogt PK, Gymnopoulos M, Hart JR. PI 3-kinase and cancer: changing accents. Curr Opin Genet Dev 2009;19:12-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ross DM, Hughes TP. Cancer treatment with kinase inhibitors: what have we learnt from imatinib? Br J Cancer 2004;90:12-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Thaimattam R, Banerjee R, Miglani R, Iqbal J. Protein kinase inhibitors: structural insights into selectivity. Curr Pharm Des 2007;13:2751-65 [DOI] [PubMed] [Google Scholar]
- 25. Cowling VH, Cole MD. Mechanism of transcriptional activation by the Myc oncoproteins. Semin Cancer Biol 2006;16:242-52 [DOI] [PubMed] [Google Scholar]
- 26. Huang Z. Structural chemistry and therapeutic intervention of protein-protein interactions in immune response, human immunodeficiency virus entry, and apoptosis. Pharmacol Ther 2000;86:201-15 [DOI] [PubMed] [Google Scholar]
- 27. Jones S, Thornton JM. Principles of protein–protein interactions. Proc Natl Acad Sci U S A 1996;93:13-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zutshi R, Brickner M, Chmielewski J. Inhibiting the assembly of protein–protein interfaces. Curr Opin Chem Biol 1998;2:62-6 [DOI] [PubMed] [Google Scholar]
- 29. Wang H, Mannava S, Grachtchouk V, et al. c-Myc depletion inhibits proliferation of human tumor cells at various stages of the cell cycle. Oncogene 2008;27:1905-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Felsher DW. Reversibility of oncogene-induced cancer. Curr Opin Genet Dev 2004;14:37-42 [DOI] [PubMed] [Google Scholar]
- 31. Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell 1999;4:199-207 [DOI] [PubMed] [Google Scholar]
- 32. Jain M, Arvanitis C, Chu K, et al. Sustained loss of a neoplastic phenotype by brief inactivation of MYC. Science 2002;297:102-4 [DOI] [PubMed] [Google Scholar]
- 33. Boxer RB, Jang JW, Sintasath L, Chodosh LA. Lack of sustained regression of c-MYC-induced mammary adenocarcinomas following brief or prolonged MYC inactivation. Cancer Cell 2004;6:577-86 [DOI] [PubMed] [Google Scholar]
- 34. Karlsson A, Giuriato S, Tang F, Fung-Weier J, Levan G, Felsher DW. Genomically complex lymphomas undergo sustained tumor regression upon MYC inactivation unless they acquire novel chromosomal translocations. Blood 2003;101:2797-803 [DOI] [PubMed] [Google Scholar]
- 35. Soucek L, Whitfield J, Martins CP, et al. Modelling Myc inhibition as a cancer therapy. Nature 2008;455:679-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bazarov AV, Adachi S, Li SF, Mateyak MK, Wei S, Sedivy JM. A modest reduction in c-myc expression has minimal effects on cell growth and apoptosis but dramatically reduces susceptibility to Ras and Raf transformation. Cancer Res 2001;61:1178-86 [PubMed] [Google Scholar]
- 37. Wang H, Hammoudeh DI, Follis AV, et al. Improved low molecular weight Myc–Max inhibitors. Mol Cancer Ther 2007;6:2399-408 [DOI] [PubMed] [Google Scholar]
- 38. Bello-Fernandez C, Packham G, Cleveland JL. The ornithine decarboxylase gene is a transcriptional target of c-Myc. Proc Natl Acad Sci U S A 1993;90:7804-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Smith MJ, Charron-Prochownik DC, Prochownik EV. The leucine zipper of c-Myc is required for full inhibition of erythroleukemia differentiation. Mol Cell Biol 1990;10:5333-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yin X, Giap C, Lazo JS, Prochownik EV. Low molecular weight inhibitors of Myc–Max interaction and function. Oncogene 2003;22:6151-9 [DOI] [PubMed] [Google Scholar]
- 41. Clackson T, Wells JA. A hot spot of binding energy in a hormone-receptor interface. Science 1995;267:383-6 [DOI] [PubMed] [Google Scholar]
- 42. Oltersdorf T, Elmore SW, Shoemaker AR, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005;435:677-81 [DOI] [PubMed] [Google Scholar]
- 43. Sasi N, Hwang M, Jaboin J, Csiki I, Lu B. Regulated cell death pathways: new twists in modulation of BCL2 family function. Mol Cancer Ther 2009;8:1421-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Berg T, Cohen SB, Desharnais J, et al. Small-molecule antagonists of Myc/Max dimerization inhibit Myc-induced transformation of chicken embryo fibroblasts. Proc Natl Acad Sci U S A 2002;99:3830-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kiessling A, Wiesinger R, Sperl B, Berg T. Selective inhibition of c-Myc/Max dimerization by a pyrazolo[1,5-a]pyrimidine. ChemMedChem 2007;2:627-30 [DOI] [PubMed] [Google Scholar]
- 46. Kiessling A, Sperl B, Hollis A, Eick D, Berg T. Selective inhibition of c-Myc/Max dimerization and DNA binding by small molecules. Chem Biol 2006;13:745-51 [DOI] [PubMed] [Google Scholar]
- 47. Galarneau A, Primeau M, Trudeau LE, Michnick SW. Beta-lactamase protein fragment complementation assays as in vivo and in vitro sensors of protein protein interactions. Nat Biotechnol 2002;20:619-22 [DOI] [PubMed] [Google Scholar]
- 48. Wehrman T, Kleaveland B, Her JH, Balint RF, Blau HM. Protein-protein interactions monitored in mammalian cells via complementation of beta-lactamase enzyme fragments. Proc Natl Acad Sci U S A 2002;99:3469-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lee HK, Brown SJ, Rosen H, Tobias PS. Application of beta-lactamase enzyme complementation to the high-throughput screening of toll-like receptor signaling inhibitors. Mol Pharmacol 2007;72:868-75 [DOI] [PubMed] [Google Scholar]
- 50. Boger DL, Lee JK, Goldberg J, Jin Q. Two comparisons of the performance of positional scanning and deletion synthesis for the identification of active constituents in mixture combinatorial libraries. J Org Chem 2000;65:1467-74 [DOI] [PubMed] [Google Scholar]
- 51. Xu Y, Shi J, Yamamoto N, Moss JA, Vogt PK, Janda KD. A credit-card library approach for disrupting protein-protein interactions. Bioorg Med Chem 2006;14:2660-73 [DOI] [PubMed] [Google Scholar]
- 52. Lu X, Vogt PK, Boger DL, Lunec J. Disruption of the MYC transcriptional function by a small-molecule antagonist of MYC/MAX dimerization. Oncol Rep 2008;19:825-30 [PubMed] [Google Scholar]
- 53. Bowman T, Broome MA, Sinibaldi D, et al. Stat3-mediated Myc expression is required for Src transformation and PDGF-induced mitogenesis. Proc Natl Acad Sci U S A 2001;98:7319-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mateyak MK, Obaya AJ, Adachi S, Sedivy JM. Phenotypes of c-Myc-deficient rat fibroblasts isolated by targeted homologous recombination. Cell Growth Differ 1997;8:1039-48 [PubMed] [Google Scholar]
- 55. Follis AV, Hammoudeh DI, Wang H, Prochownik EV, Metallo SJ. Structural rationale for the coupled binding and unfolding of the c-Myc oncoprotein by small molecules. Chem Biol 2008;15:1149-55 [DOI] [PubMed] [Google Scholar]
- 56. Hammoudeh DI, Follis AV, Prochownik EV, Metallo SJ. Multiple independent binding sites for small-molecule inhibitors on the oncoprotein c-Myc. J Am Chem Soc 2009;131:7390-401 [DOI] [PubMed] [Google Scholar]
- 57. Richmond NJ, Abrams CA, Wolohan PR, Abrahamian E, Willett P, Clark RD. GALAHAD: 1. Pharmacophore identification by hypermolecular alignment of ligands in 3D. J Comput Aided Mol Des 2006;20:567-87 [DOI] [PubMed] [Google Scholar]
- 58. Shepphird JK, Clark RD. A marriage made in torsional space: using GALAHAD models to drive pharmacophore multiplet searches. J Comput Aided Mol Des 2006;20:763-71 [DOI] [PubMed] [Google Scholar]
- 59. Irwin JJ, Shoichet BK. ZINC: a free database of commercially available compounds for virtual screening. J Chem Inf Model 2005;45:177-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wrighton SA, Schuetz EG, Thummel KE, Shen DD, Korzekwa KR, Watkins PB. The human CYP3A subfamily: practical considerations. Drug Metab Rev 2000;32:339-61 [DOI] [PubMed] [Google Scholar]
- 61. Erlanson DA, Wells JA, Braisted AC. Tethering: fragment-based drug discovery. Annu Rev Biophys Biomol Struct 2004;33:199-223 [DOI] [PubMed] [Google Scholar]
- 62. Liu J, Begley D, Mitchell DD, Verlinde CL, Varani G, Fan E. Multivalent drug design and inhibition of cholera toxin by specific and transient protein-ligand interactions. Chem Biol Drug Des 2008;71:408-19 [DOI] [PubMed] [Google Scholar]
- 63. Fieber W, Schneider ML, Matt T, Krautler B, Konrat R, Bister K. Structure, function, and dynamics of the dimerization and DNA-binding domain of oncogenic transcription factor v-Myc. J Mol Biol 2001;307:1395-410 [DOI] [PubMed] [Google Scholar]
- 64. Nair SK, Burley SK. X-ray structures of Myc–Max and Mad-Max recognizing DNA: molecular bases of regulation by proto-oncogenic transcription factors. Cell 2003;112:193-205 [DOI] [PubMed] [Google Scholar]
- 65. Uversky VN, Oldfield CJ, Dunker AK. Showing your ID: intrinsic disorder as an ID for recognition, regulation and cell signaling. J Mol Recognit 2005;18:343-84 [DOI] [PubMed] [Google Scholar]
- 66. Miyamoto-Sato E, Fujimori S, Ishizaka M, et al. A comprehensive resource of interacting protein regions for refining human transcription factor networks. PLoS One 2010;5:e9289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sigalov AB. Protein intrinsic disorder and oligomericity in cell signaling. Mol Biosyst 2010;6:451-61 [DOI] [PubMed] [Google Scholar]
- 68. Guo J, Parise RA, Joseph E, et al. Efficacy, pharmacokinetics, tisssue distribution, and metabolism of the Myc–Max disruptor, 10058-F4 [Z,E]-5-4-ethylbenzylidine]-2-thioxothiazolidin-4-one, in mice. Cancer Chemother Pharmacol 2009;63:615-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Giordano A. Molecular basis of different outcomes for drug-eluting stents that release sirolimus or tacrolimus. Curr Opin Drug Discov Devel 2010;13:159-68 [PubMed] [Google Scholar]
- 70. Zhang F, Dong L, Ge J. Meta-analysis of five randomized clinical trials comparing sirolimusversus paclitaxel-eluting stents in patients with diabetes mellitus. Am J Cardiol 2010;105:64-8 [DOI] [PubMed] [Google Scholar]
- 71. De Feo M, Forte A, Onorati F, et al. Rat carotid arteriotomy: c-myc is involved in negative remodelling and apoptosis. J Cardiovasc Med (Hagerstown) 2006;7:61-7 [DOI] [PubMed] [Google Scholar]
- 72. Forte A, Galderisi U, De Feo M, et al. c-Myc antisense oligonucleotides preserve smooth muscle differentiation and reduce negative remodelling following rat carotid arteriotomy. J Vasc Res 2005;42:214-25 [DOI] [PubMed] [Google Scholar]
- 73. Kipshidze N, Tsapenko M, Iversen P, Burger D. Antisense therapy for restenosis following percutaneous coronary intervention. Expert Opin Biol Ther 2005;5:79-89 [DOI] [PubMed] [Google Scholar]
- 74. Andreoli CM, Miller JW. Anti-vascular endothelial growth factor therapy for ocular neovascular disease. Curr Opin Ophthalmol 2007;18:502-8 [DOI] [PubMed] [Google Scholar]
- 75. Emerson MV, Lauer AK. Emerging therapies for the treatment of neovascular age-related macular degeneration and diabetic macular edema. BioDrugs 2007;21:245-57 [DOI] [PubMed] [Google Scholar]
- 76. Figueroa MS, Contreras I, Noval S. Anti-angiogenic drugs as an adjunctive therapy in the surgical treatment of diabetic retinopathy. Curr Diabetes Rev 2009;5:52-6 [DOI] [PubMed] [Google Scholar]
- 77. Blackwood EM, Eisenman RN. Max: a helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science 1991;251:1211-7 [DOI] [PubMed] [Google Scholar]
- 78. Blackwood EM, Luscher B, Eisenman RN. Myc and Max associate in vivo. Genes Dev 1992;6:71-80 [DOI] [PubMed] [Google Scholar]
- 79. Yin X, Grove L, Prochownik EV. Lack of transcriptional repression by max homodimers. Oncogene 1998;16:2629-37 [DOI] [PubMed] [Google Scholar]
- 80. Lindeman GJ, Harris AW, Bath ML, Eisenman RN, Adams JM. Overexpressed max is not oncogenic and attenuates myc-induced lymphoproliferation and lymphomagenesis in transgenic mice. Oncogene 1995;10:1013-7 [PubMed] [Google Scholar]
- 81. Cogliati T, Dunn BK, Bar-Ner M, Cultraro CM, Segal S. Transfected wild-type and mutant max regulate cell growth and differentiation of murine erythroleukemia cells. Oncogene 1993;8:1263-8 [PubMed] [Google Scholar]
- 82. Kretzner L, Blackwood EM, Eisenman RN. Transcriptional activities of the Myc and Max proteins in mammalian cells. Curr Top Microbiol Immunol 1992;182:435-43 [DOI] [PubMed] [Google Scholar]
- 83. Morris GM, Goodsell DS, Halliday RS, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem 1998;19:1639-62 [Google Scholar]
- 84. Brooijmans N, Kuntz ID. Molecular recognition and docking algorithms. Annu Rev Biophys Biomol Struct 2003;32:335-73 [DOI] [PubMed] [Google Scholar]
- 85. Li C, Xu L, Wolan DW, Wilson IA, Olson AJ. Virtual screening of human 5-aminoimidazole-4-carboxamide ribonucleotide transformylase against the NCI diversity set by use of AutoDock to identify novel nonfolate inhibitors. J Med Chem 2004;47:6681-90 [DOI] [PubMed] [Google Scholar]
- 86. Dickerson TJ, Beuscher AET, Rogers CJ, et al. Discovery of acetylcholinesterase peripheral anionic site ligands through computational refinement of a directed library. Biochemistry 2005;44:14845-53 [DOI] [PubMed] [Google Scholar]
- 87. Rogers JP, Beuscher AET, Flajolet M, et al. Discovery of protein phosphatase 2C inhibitors by virtual screening. J Med Chem 2006;49:1658-67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Mustata G, Follis AV, Hammoudeh DI, et al. Discovery of novel myc-max heterodimer disruptors with a three-dimensional pharmacophore model. J Med Chem 2009;52:1247-50 [DOI] [PMC free article] [PubMed] [Google Scholar]