

Abstract
Variations in hydrogen-bond strengths are investigated for complexes of nine para-substituted phenols (XPhOH) with a water molecule and chloride ion. Results from ab initio HF/6-311+G(d, p) and MP2/6-311+G(d, p)//HF/6-311+G(d, p) calculations are compared with those from the OPLS-AA and OPLS/CM1A force fields. In the OPLS-AA model, the partial charges on the hydroxyl group of phenol are not affected by the choice of para substituent, while the use of CM1A charges in the OPLS/CM1A approach does provide charge redistribution. The ab initio calculations reveal a 2.0-kcal/mol range in hydrogen-bond strengths for the XPhOH⋯OH2 complexes in the order X = NO2 > CN > CF3 > Cl > F > H >OH >CH3 > NH2. The pattern is not well-reproduced with OPLS-AA, which also compresses the variation to 0.7 kcal/mol. However, the OPLS/CM1A results are in good accord with the ab initio findings for both the ordering and range, 2.3 kcal/mol. The hydrogen bonding is, of course, weaker with XPhOH as acceptor, the order for X is largely inverted, and the range is reduced to ca. 1.0 kcal/mol. The substituent effects are found to be much greater for the chloride ion complexes with a range of 11 kcal/mol. For quantitative treatment of such strong ion-molecule interactions the need for fully polarizable force fields is demonstrated.
Introduction
In our development of non-nucleoside inhibitors of HIV-1 reverse transcriptase (NNRTIs), high sensitivity to substitution at the 4-position in the phenyl ring has been found for the thiazole series 1 and the pyrimidines 2, as summarized in Table 1.1,2 Specifically, for the thiazoles, there is a 50-fold enhancement in activity as the substituent X is made more electronegative in going from X = H to CN, while a 1500-fold enhancement is obtained in the pyrimidine series. The structures of the complexes of such NNRTIs with HIV-RT have been well established through X-ray crystallography and computation.3-5 A key feature is a short hydrogen bond between the amino group of the NNRTI and the carbonyl oxygen of Lys101 of HIV-RT (Figure 1). The β-nitrogen in the heterocycle is also in a longer hydrogen bond with the backbone NH of Lys101.
Table 1.
Anti-HIV Activity (EC50 in μM for Protection of MT-2 Cells) for Compounds 1 and 2a
| X | 1 | 2 |
|---|---|---|
| H | 10.0 | 30.0 |
| CH3 | 3.0 | 2.8 |
| Cl | 0.30 | 0.20 |
| CN | 0.21 | 0.02 |
Figure 1.

Partial computed structure for 2 (X = Cl) bound to HIV-1 reverse transcriptase highlighting the hydrogen bonds with the backbone of Lysine101. Carbon atoms of the ligand are colored gold for clarity.
Questions that then arise are (a) how sensitive are such hydrogen-bond strengths to substitution in the phenyl rings, and (b) are such effects adequately reflected in the force-field calculations that are often used to examine the energetics of protein-ligand binding?6-8 For example, successful guidance of lead-optimization by performing free-energy perturbation calculations to predict the effects of changes in substituents on rings and of choices of heterocycles on binding affinities is expected to require proper representation of such effects.1,9,10 Electronic polarization is a central issue here since change in X alters the charge distribution including for the key hydrogen-bond donating hydrogen of the ligand.7,11 Related effects on acidities of substituted benzoic acids and phenols led to the development of the Hammett equation and the σ and σ− substituent constants.12 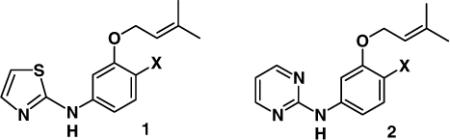
As summarized below, these ideas were pursued by performing ab initio calculations on prototypical hydrogen-bonded systems and comparing the results to those obtained from the non-polarizable OPLS-AA force field13 and its OPLS/CM1A variant,8 which incorporates polarized partial atomic charges that are obtained from the quantum mechanical CM1A procedure.14 The CM1A method, which is based on AM1 wavefunctions, was derived to reproduce dipole moments for organic molecules in the gas phase.14 The CM1A charges when enhanced by 14% (1.14*CM1A) were also found to perform well for computing free energies of hydration of 25 diverse organic molecules15 in explicit TIP4P water16 with all other force-field parameters taken from OPLS-AA. The systems chosen for initial study of substituent effects on hydrogen bonding are the phenol-water and phenol-chloride ion complexes, 3 – 5. 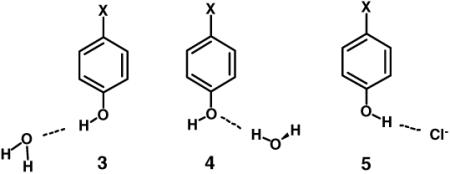
Computational Details
The principal interest here is comparison of ab initio and force field predictions for the effects of the substituents X on the hydrogen-bond strengths. Ab initio and density functional theory calculations were carried out with Gaussian 03 and all geometrical degrees of freedom were optimized for the complexes and separated components.17 In Table 2, results for the PhOH⋯OH2 (3) complex and water dimer are compared at the HF/6-31G(d), B3LYP/6-31G(d), HF/6-311+G(d, p), and MP2/6-311+G(d, p)// HF/6-311+G(d, p) levels. The interaction energies at the latter two levels bracket what is accepted as the true value for the water dimer, −5.4 ± 0.7 kcal/mol from experiment18 and −5.1 ± 0.2 kcal/mol from theory.19 The ΔE results for the phenol-water complex are also similar to those from the highest-level calculations in a prior study, i.e. −7 to −8 kcal/mol at the MP2/aug-cc-pVDZ level.20 Thus, the substituent effects were explored with the HF/6-311+G(d, p) and MP2/6-311+G(d, p) calculations. Counterpoise corrections have not been made since they are expected to show little variation with the choice of substituent X.
Table 2.
Computed Interaction Energies (kcal/mol) and OO Distances (Å)a
| PhOH---OH2 | (H2O)2 | |||
|---|---|---|---|---|
| Method | − Δ E | r(OO) | − Δ E | r(OO) |
| HF/6-31G(d) | 7.35 | 2.901 | 5.62 | 2.971 |
| B3LYP/6-31G(d) | 9.70 | 2.808 | 7.68 | 2.861 |
| HF/6-311+G(d, p) | 6.24 | 2.940 | 4.83 | 3.000 |
| MP2/6-311+G(d, p)b | 8.13 | (2.940) | 5.91 | (3.000) |
| MP2/6-311++G(2d, 2p)c | 5.44 | 2.911 | ||
For A + H2O → A---H2O, ΔE = E(A---H2O) − E(A) − E(H2O)
Using HF/6-311+G(d, p) optimized structures.
Reference 19a.
The corresponding force field calculations were carried out first with the substituted benzenes described with the OPLS-AA force field21 and with the water molecule represented by the TIP4P model.16 Complete energy minimizations were carried out with the BOSS program22 except that the internal geometry of the water molecule is fixed in the TIP4P model, r(OH) = 0.9572 Å and ∠HOH = 104.52°. Notably, in the reported OPLS-AA model for para-substituted benzenes, the net charge on the substituent plus attached benzene carbon atom is zero.21 This permits transferability that simplifies the modeling of arbitrary substituted benzenes, but it ignores associated polarization effects. Thus, the partial atomic charges on the COH group of all para-substituted phenols are the same (Figure 2). Though testing for numerous mono- and di-substituted benzenes has revealed modest average errors for computed free energies of hydration (0.5 kcal/mol) and pure liquid heats of vaporization (1.0 kcal/mol) and densities (0.02 g/mL),21,23 differential polarization effects are expected to be more apparent upon examination of specific hydrogen-bond strengths as in protein-ligand binding.
Figure 2.

Examples of OPLS-AA (top) and OPLS/CM1A (bottom) atomic charges for substituted phenols. All unsubstituted phenyl C and H atoms have charges of −0.115 e and +0.115 e in the OPLS-AA model. The CM1A charges for neutral molecules are scaled by a factor of 1.14 for the OPLS/CM1A force field.
For the OPLS/CM1A approach,8 OPLS-AA parameters are used except for the partial atomic charges, which are obtained from the CM1A method.14 A sequence of geometry optimizations and CM1A calculations is performed with a BOSS script until the charges are converged. For neutral molecules, it is noted again that the CM1A charges are scaled by a factor of 1.14 for use in the OPLS/CM1A force field.15 For ions, the CM1A charges are not scaled to avoid non-physical net charges.24 The program also symmetrizes the charges for equivalent atoms, e.g., the charges are averaged for equivalent methyl hydrogens or the ortho carbons and hydrogens in Figure 2. Without the symmetrization, artifacts arise for general molecular modeling such as introduction of spurious minima in conformational searching. Optimization of the complexes is then performed with the converged charges and with the internally rigid TIP4P water molecule. Further polarization of the charge distribution for the substituted benzenes upon complex formation with water is not carried out. For the much stronger phenolchloride ion interactions, the importance of a full treatment of polarization effects is considered below.
Results and Discussion
XPhOH-Water Complexes 3
The computed interaction energies ΔE for the complexes 3 with the phenol as the hydrogen-bond donor are summarized in Table 3 and the optimized OO distances are in Table 4. The trend in the ab initio results is as expected with electron-withdrawing substituents acidifying the hydroxyl group, increasing the hydrogen-bond strengths (Table 3), and decreasing the hydrogen-bond lengths (Table 4). However, there are fine points. For example, the π-donating character of the amino substituent outweighs its σ-withdrawing character to yield the weakest hydrogen bond. For fluorine and chlorine, the opposite pattern seems to be operative as the hydrogen bonds are stronger than for phenol (X = H) in those cases. As shown in Figure 3, the ab initio hydrogen-bond strengths roughly follow the trend of Hammett σ constants, though this is not fully expected in view of the differences in the processes, i.e., substituent effects on phenol-water hydrogen-bond strengths and on acidities of substituted benzoic acids in aqueous solution.
Table 3.
Computed Interaction Energies (−ΔE, kcal/mol) for Complexes 3
| X | σ a | HFb | MP2c | OPLS-AA | OPLS/CM1A |
|---|---|---|---|---|---|
| NH2 | −0.57 | 5.90 | 7.79 | 7.19 | 7.03 |
| CH3 | −0.14 | 6.05 | 7.98 | 7.00 | 7.23 |
| OH | −0.38 | 6.18 | 8.10 | 7.22 | 7.40 |
| H | 0.0 | 6.24 | 8.13 | 7.09 | 7.28 |
| F | 0.15 | 6.65 | 8.57 | 7.51 | 7.88 |
| Cl | 0.24 | 6.87 | 8.74 | 7.50 | 7.95 |
| CF3 | 0.53 | 7.24 | 9.20 | 7.67 | 8.34 |
| CN | 0.70 | 7.64 | 9.54 | 7.59 | 8.64 |
| NO2 | 0.81 | 7.94 | 9.72 | 7.66 | 9.33 |
| mue | 1.90 | (0) | 1.26 | 0.74 |
Table 4.
Computed Oxygen-Oxygen Distances (Å) for Complexes 3 and 4a
| X | HFb | OPLS-AA | OPLS/CM1A |
|---|---|---|---|
| NH2 | 2.955, 3.029 | 2.734, 2.767 | 2.759, 2.844 |
| CH3 | 2.945, 3.044 | 2.734, 2.766 | 2.757, 2.851 |
| OH | 2.947, 3.028 | 2.733, 2.769 | 2.760, 2.837 |
| H | 2.940, 3.050 | 2.728, 2.767 | 2.754, 2.850 |
| F | 2.936, 3.036 | 2.731, 2.770 | 2.751, 2.843 |
| Cl | 2.926, 3.048 | 2.729, 2.770 | 2.749, 2.846 |
| CF3 | 2.913, 3.056 | 2.731, 2.772 | 2.746, 2.853 |
| CN | 2.904, 3.061 | 2.730, 2.759 | 2.739, 2.845 |
| NO2 | 2.895, 3.068 | 2.731, 2.772 | 2.735, 2.867 |
Values x, y are for 3 and 4.
HF/6-311+G(d, p).
Figure 3.

Computed interaction energies for the p-XPhOH⋯OH2 complexes 3 vs. σ(X).
The substituent effects on the hydrogen-bond strengths are substantial with a 2 kcal/mol range from both the HF and MP2 calculations for the complexes 3. In view of the constancy of the OPLS-AA partial charges for the phenolic hydroxyl group, it is not surprising that the range for ΔE is compressed to 0.7 kcal/mol and the ordering of the values is poor. There is also negligible variation in the OO distances in Table 4 with OPLS-AA, while the HF results show a reduction of the hydrogen-bond lengths by 0.06 Å in going from X = NH2 to NO2 for the complexes 3. In contrast, use of the 1.14*CM1A charges nicely corrects the problems with the interaction energies and yields absolute values roughly midway between the HF and MP2 results (Table 3 and Figure 3). The level of accord was not anticipated, but it suggests that the polarization of the charge distributions by the CM1A method is remarkably accurate within the context of the simple point charge model for the force fields (single atom-centered partial charges). The hydrogen-bond lengths with OPLS/CM1A also decrease with increasing strength, though the range is less than from the HF optimizations (Table 4). The absolute hydrogen-bond lengths are 0.15 – 0.20 Å shorter from the force fields than from the ab initio calculations, which is normal for fixed-charge force fields that are intended for condensed-phase simulations.7,8,13,16
XPhOH-Water Complexes 4
Turning to the complexes 4 with the phenol as the hydrogen-bond acceptor, the trends for hydrogen-bond strengths and lengths from the ab initio calculations are now opposite with electron-withdrawing substituents weakening the basicity of the phenolic oxygen. Thus the MP2 results for 3 range from −7.8 to −9.7 kcal/mol in going from X = NH2 to NO2, while the corresponding values for 4 are −5.4 to −4.9 kcal/mol (Table 5), i.e., opposite in trend, much weaker, and in a narrower range. Qualitatively similar results are obtained from the HF calculations, though the range for the complexes 4 is 1.0 kcal/mol. As before, the OPLS-AA results are too invariant, while the OPLS/CM1A model is successful in showing the weakening of the hydrogen bond for 4 with increasing electron-withdrawing character for the substituent X.
Table 5.
Computed Interaction Energies (−ΔE, kcal/mol) for Complexes 4
| X | HFa | MP2a | OPLS-AA | OPLS/CM1A |
|---|---|---|---|---|
| NH2 | 3.99 | 5.42 | 5.95 | 5.51 |
| CH3 | 3.74 | 5.19 | 6.05 | 5.23 |
| OH | 3.88 | 5.48 | 5.93 | 4.92 |
| H | 3.60 | 5.05 | 5.98 | 5.16 |
| F | 3.55 | 5.20 | 5.72 | 4.80 |
| Cl | 3.39 | 5.13 | 5.73 | 4.73 |
| CF3 | 3.22 | 5.11 | 5.65 | 4.44 |
| CN | 3.11 | 4.94 | 5.85 | 4.35 |
| NO2 | 3.00 | 4.90 | 5.65 | 3.87 |
| mue | 1.66 | (0) | 0.68 | 0.49 |
As in Table 3.
A key point from Tables 3 and 5 is the increasing gap between the substituted phenol's ability to act as a hydrogen bond donor and acceptor with increasing electron-withdrawing character for X. E.g., for p-cyanophenol as donor and acceptor the difference in hydrogen-bond strengths is 4.6 kcal/mol from the MP2 results and 4.3 kcal/mol with OPLS/CM1A, while the difference is only 1.7 kcal/mol from the OPLS-AA calculations. It is clear that (a) such modulation of hydrogen-bonding ability is important for proper description of intermolecular interactions, and (b) its accurate description requires methodology that allows polarization of the charge distributions. It is also apparent from Figure 2 and Tables 3 and 5 that the hydrogen-bond strengths are very sensitive to small changes in the atomic charges. The variations for the hydroxyl oxygen and hydrogen are only ca. 0.01 e with OPLS/CM1A; the variation for the ipso carbon is actually much greater, 0.1 e. For phenol itself, if the OPLS-AA charges for the hydroxyl oxygen and hydrogen are changed by 0.01 e, the strength of the hydrogen-bond for the complex 3 changes by ca. ±0.3 kal/mol in the expected manner. This sensitivity is well known and has always been a challenge in the development of force fields.7,8
XPhOH-Cl− Complexes 5
Naturally, the substituent effects are much enhanced for the complexes with chloride ion, 5 (Table 6). It is noted that the OPLS chloride ion parameters (q = −1.0 e, σ = 4.02 Å, ε = 0.71 kcal/mol) that were used here are from a recent, comprehensive study of the hydration of halide and alkali ions.24 For the complexes with the substituted phenols, the ranges for the interaction energies are −17 to −28 (HF), −23 to −35 (MP2), −14 to −19 (OPLS-AA), and −15 to −27 kcal/mol (OPLS/CM1A). Some experimental data from high-pressure mass spectrometry are also available for complexes 5, as listed in Table 6.25 For anion-molecule complexes like these, conversion of the computed electronic energy change ΔE to ΔH at 298 K involves a correction of ca. +0.9 kcal/mol.26 It is apparent that the MP2 results are in close accord with the experimental data, while the HF and force-field results significantly underestimate the hydrogen-bond strengths. However, the OPLS/CM1A approach again does much better than OPLS-AA for the magnitude, pattern, and range for the interaction energies. In this case, polarization of the substituted phenol by the chloride ion can be expected to be significant and the fixed-charge OPLS-AA and OPLS/CM1A models are both inadequate. This is the case in spite of the fact that the optimal interaction energy for Cl− with a TIP4P water molecule of −13.0 kcal/mol is in good agreement with the best available estimates.24 The larger phenols are more polarizable than a water molecule.
Table 6.
Interaction Energies (−ΔE) and Enthalpies (−ΔH) for Complexes 5 (kcal/mol)
| X | HFa | MP2a | OPLS-AA | OPLS/CM1A | −ΔH, exptl.b |
|---|---|---|---|---|---|
| NH2 | 17.01 | 23.49 | 15.39 | 14.98 | |
| CH3 | 17.60 | 24.38 | 14.25 | 15.69 | 24.1 |
| OH | 18.11 | 24.70 | 15.39 | 16.25 | |
| H | 18.34 | 24.95 | 14.90 | 16.06 | 24.5 |
| F | 20.94 | 27.62 | 17.64 | 19.13 | 26.4 |
| Cl | 22.17 | 28.61 | 17.66 | 19.46 | 28.1 |
| CF3 | 24.24 | 31.79 | 18.87 | 21.34 | |
| CN | 26.80 | 33.84 | 18.44 | 23.38 | 33.6 |
| NO2 | 28.19 | 34.93 | 18.93 | 26.77 | |
| mue | 6.77 | (0) | 11.43 | 9.03 |
For proper treatment of such strong ion-molecule interactions, it is accepted that a fully polarizable force field is required.7,11 Thus, we have been exploring the addition of inducible dipoles to the OPLS models. A simple approach has been taken by which an inducible dipole can be added to non-hydrogen atoms. Furthermore, the electric field that determines the inducible dipoles is only computed from the permanent charges (eq 1) and the total polarization energy is given by the usual formula, eq 2. The key approximation
| (1) |
| (2) |
is that the induced dipoles do not contribute to the electric field, which simplifies the computations since iterative solution for the dipoles is not required. Addition of the induced dipoles to OPLS-AA and OPLS/CM1A yields OPLS-AAP and OPLS/CM1AP. The implementations are residue-based in that the electric field at an atom is determined by the charges on all other atoms not in the same residue. For the complex 5, the substituted phenol and chloride ion are treated as separate residues, so the induced dipoles for the phenol are only determined by the field from the chloride ion. The same polarization model has been used by others27,28 and it performed well in a previous study of ours for reproducing solvent effects for the gauche/anti equilibrium for 1,2-dichloroethane in multiple solvents and the free energy of solvation of water in cyclohexane.29
Modest parameter optimization has been carried out for the polarizabilities α to reproduce gas-phase complexation energies (MP2/6-311G(d, p)) for ca. 30 ion-molecule complexes focusing on cation-π interactions. This led to setting αi = 1.0 Å3 for carbon and αi = 1.5 Å3 for heteroatoms. With these choices, the phenol-chloride ion complexes were optimized yielding the results in Table 7. Inclusion of the induced dipoles is found to enhance the interaction energies by 5-6 kcal/mol. The hydrogen-bond lengths are also shortened by ca. 0.1 Å to yield the values that are listed in Table 7. The agreement between the ab initio and OPLS/CM1AP results is certainly respectable, while the OPLS-AAP results still suffer from the underlying problems with the invariant partial charges in the OPLS-AA model. For the phenol-water complexes, addition of the inducible dipoles to the force fields strengthens the hydrogen bonds uniformly by 0.4 - 0.5 kcal/mol and shortens them by ca. 0.02 Å.
Table 7.
Computed Interaction Energies (−ΔE, kcal/mol) for Complexes 5 using Polarizable Force Fields, and Optimized O-Cl Distances (Å)
| − Δ E | − Δ E | r(O-Cl) | r(O-Cl) | r(O-Cl) | |
|---|---|---|---|---|---|
| X | OPLS-AAP | OPLS/CM1AP | HFa | OPLS-AAP | OPLS/CM1AP |
| NH2 | 20.48 | 19.93 | 3.181 | 3.119 | 3.146 |
| CH3 | 19.32 | 20.74 | 3.164 | 3.124 | 3.136 |
| OH | 20.59 | 21.40 | 3.164 | 3.111 | 3.129 |
| H | 19.95 | 21.08 | 3.154 | 3.121 | 3.133 |
| F | 22.83 | 24.35 | 3.145 | 3.111 | 3.120 |
| Cl | 22.91 | 24.76 | 3.125 | 3.113 | 3.118 |
| CF3 | 24.16 | 26.76 | 3.098 | 3.109 | 3.106 |
| CN | 23.69 | 28.89 | 3.080 | 3.112 | 3.101 |
| NO2 | 24.17 | 32.47 | 3.059 | 3.109 | 3.082 |
| mueb | 6.25 | 3.77 |
HF/6-311+G(d, p).
Mean unsigned error to MP2 ΔE in Table 6.
Conclusion
Substituent effects on the interaction energies for complexes of phenols with water and chloride ion have been investigated. For the complexes with water, the OPLS/CM1A model was found to yield good reproduction of ab initio results, while the OPLS-AA force field with invariant partial charges for the hydroxyl group compresses the substituent effects. For the complexes with chloride ion, the interaction energies and substituent effects are much magnified, and the need for explicit treatment of the intermolecular polarization energy is apparent. Addition of inducible dipoles on non-hydrogen atoms was found to enhance the interaction energies by 5-6 kcal/mol. The resultant OPLS/CM1AP model performed well and warrants further investigation. It is emphasized that the ability to predict accurately substituent effects on intermolecular interactions is central to key applications of molecular modeling, for example, in the design of drugs, materials, and catalysts.
Supplementary Material
Acknowledgments
Gratitude is expressed to the National Science Foundation (CHE-0446920) and National Institutes of Health (GM32136) for support.
Footnotes
Supporting Information Available: An Excel file is provided with the ab initio energies for the substituted phenols and complexes 3 - 5. These materials are available free of charge via the Internet at (http://pubs.acs.org/instruct/jacsat.pdf).
References
- 1.Jorgensen WL, Ruiz-Caro J, Tirado-Rives J, Basavapathruni A, Anderson KS, Hamilton AD. Bioorg. Med. Chem. Lett. 2006;16:663–667. doi: 10.1016/j.bmcl.2005.10.038. [DOI] [PubMed] [Google Scholar]
- 2.Ruiz-Caro J, Basavapathruni A, Kim JT, Wang L, Bailey CM, Anderson KS, Hamilton AD, Jorgensen WL. Bioorg. Med. Chem. Lett. 2006;16:668–671. doi: 10.1016/j.bmcl.2005.10.037. [DOI] [PubMed] [Google Scholar]
- 3.Ren J, Esnouf RM, Hopkins AL, Warren J, Balzarini J, Stuart DI, Stammers DK. Biochemistry. 1998;37:14394–14403. doi: 10.1021/bi981309m. [DOI] [PubMed] [Google Scholar]
- 4.Blagović MU, Tirado-Rives J, Jorgensen WL. J. Am. Chem. Soc. 2003;125:6016–6017. doi: 10.1021/ja034308c. [DOI] [PubMed] [Google Scholar]
- 5.Das K, Clark AD, Jr., Lewi PJ, Heeres J, de Jonge MR, Koymans LMH, Vinkers HM, Daeyaert F, Ludovici DW, Kukla MJ, De Corte B, Kavash RW, Ho CY, Ye H, Lichtenstein MA, Andries K, Pauwels R, de Béthune M-P, Boyer PL, Clark P, Hughes SH, Janssen PAJ, Arnold E. J. Med. Chem. 2004;47:2550–2560. doi: 10.1021/jm030558s. [DOI] [PubMed] [Google Scholar]
- 6.Gohlke H, Klebe G. Angew. Chem. Int. Ed. 2002;41:2644–2676. doi: 10.1002/1521-3773(20020802)41:15<2644::AID-ANIE2644>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 7.Ponder JW, Case DA. Adv. Protein Chem. 2003;66:27–85. doi: 10.1016/s0065-3233(03)66002-x. [DOI] [PubMed] [Google Scholar]
- 8.Jorgensen WL, Tirado-Rives J. Proc. Nat. Acad. Sci USA. 2005;102:6665–6670. doi: 10.1073/pnas.0408037102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thakur VV, Kim JT, Hamilton AD, Bailey CM, Domaoal RA, Wang L, Anderson KS, Jorgensen WL. Bioorg. Med. Chem. Lett. 2006;16:5664–5667. doi: 10.1016/j.bmcl.2006.08.037. [DOI] [PubMed] [Google Scholar]
- 10.Kim JT, Hamilton AD, Bailey CM, Domaoal RA, Wang L, Anderson KS, Jorgensen WL. J. Am. Chem. Soc. 2006;128:15372–15373. doi: 10.1021/ja066472g. [DOI] [PubMed] [Google Scholar]
- 11.Rick SW, Stuart SJ. Rev. Comput. Chem. 2002;18:89–146. [Google Scholar]
- 12.Smith MB, March J. March's Advanced Organic Chemistry. 5th Edit. Chapt. 9. Wiley; New York: 2001. [Google Scholar]
- 13.Jorgensen WL, Maxwell DS, Tirado-Rives J. J. Am. Chem. Soc. 1996;118:11225–11236. [Google Scholar]
- 14.Storer JW, Giesen DJ, Cramer CJ, Truhlar DG. J. Comput.-Aided Mol. Des. 1995;9:87–110. doi: 10.1007/BF00117280. [DOI] [PubMed] [Google Scholar]
- 15.Blagović MU, Morales de Tirado P, Pearlman SA, Jorgensen WL. J. Comput. Chem. 2004;25:1322–1332. doi: 10.1002/jcc.20059. [DOI] [PubMed] [Google Scholar]
- 16.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. J. Phys. Chem. 1983;79:926–935. [Google Scholar]
- 17.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr., Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03. Gaussian, Inc.; Wallingford CT: 2004. Revision C.02. [Google Scholar]
- 18.Curtiss LA, Frurip DJ, Blander M. J. Chem. Phys. 1979;71:2703–2711. [Google Scholar]
- 19.a Frisch MJ, Del Bene JE, Binkley JS, Schaefer HF., III J. Chem. Phys. 1986;84:2279–2289. [Google Scholar]; b Halkier A, Koch H, Jorgensen P, Christiansen O, Beck Nielsen IM, Helgaker T. Theor. Chem. Acc. 1997;97:150–157. [Google Scholar]
- 20.Feller D, Feyereisen MW. J. Comput. Chem. 1993;14:1027–1035. [Google Scholar]
- 21.a Jorgensen WL, Nguyen TB. J. Comput. Chem. 1993;13:195–205. [Google Scholar]; b Jorgensen WL, Laird ER, Nguyen TB, Tirado-Rives J. J. Comput. Chem. 1993;13:206–215. [Google Scholar]
- 22.Jorgensen WL, Tirado-Rives J. J. Comput. Chem. 2005;26:1689–1700. doi: 10.1002/jcc.20297. [DOI] [PubMed] [Google Scholar]
- 23.Price DJ, Brooks CL., III J. Comput. Chem. 2005;26:1529–1541. doi: 10.1002/jcc.20284. [DOI] [PubMed] [Google Scholar]
- 24.Jensen KP, Jorgensen WL. J. Chem. Theory Comput. 2006;2:1499–1509. doi: 10.1021/ct600252r. [DOI] [PubMed] [Google Scholar]
- 25.a Cumming JB, French MA, Kebarle P. J. Am. Chem. Soc. 1977;99:6999–7003. [Google Scholar]; b Paul GJC, Kebarle P. Can. J. Chem. 1990;68:2070–2077. [Google Scholar]
- 26.Gao J, Garner DS, Jorgensen WL. J. Am. Chem. Soc. 1986;108:4784–4790. [Google Scholar]
- 27.King G, Warshel A. J. Chem. Phys. 1990;93:8682–8692. [Google Scholar]
- 28.Straatsma TP, McCammon JA. Chem. Phys. Lett. 1991;177:433–440. [Google Scholar]
- 29.Jorgensen WL, McDonald NA, Selmi M, Rablen PR. J. Am. Chem. Soc. 1995;117:11809–11810. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.