

Abstract
Proneurotrophins and mature neurotrophins activate different signaling pathways with distinct effects on their target cells: proneurotrophins can induce apoptotic signaling via p75NTR, whereas mature neurotrophins activate Trk receptors to influence survival and differentiation. Here, we demonstrate that the PTEN (phosphatase and tensin homolog deleted on chromosome 10) phosphatase represents a novel switch between the survival and apoptotic signaling pathways in rat CNS neurons. Simultaneous activation of p75NTR by proNGF and TrkB signaling by BDNF elicited apoptosis despite TrkB phosphorylation. Apoptosis induced by p75NTR required suppression of TrkB-induced phosphoinositide-3 kinase signaling, mediated by induction of PTEN, for apoptosis to proceed. Inhibition of PTEN restored the ability of BDNF to phosphorylate Akt and protect cultured basal forebrain neurons from proNGF-induced death. In vivo, inhibition or knockdown of PTEN after pilocarpine-induced seizures protected CNS neurons from p75NTR-mediated death, demonstrating that PTEN is a crucial factor mediating the balance between p75NTR-induced apoptotic signaling and Trk-mediated survival signaling in brain neurons.
Introduction
The neurotrophin growth factor family is critical for the development and maintenance of the nervous system, affecting multiple aspects of neuronal function (Lewin and Barde, 1996; Bibel and Barde, 2000; Huang and Reichardt, 2001). The diversity of neurotrophin functions depends on two different receptors: Trk receptors, which are necessary for neuronal survival, differentiation, and synaptic formation (Friedman and Greene, 1999; Reichardt, 2006); and p75NTR, which is important for cell death during developmental and in pathological conditions (Roux and Barker, 2002). Proneurotrophins, the uncleaved neurotrophin precursors, can be secreted and can bind to a p75NTR–sortilin receptor complex with high affinity to induce neuronal apoptosis (Lee et al., 2001; Beattie et al., 2002; Teng et al., 2005). Since neurons are likely to be exposed to multiple factors in their environment, including both mature neurotrophins and proneurotrophins, in basal and pathophysiological situations, we have investigated the consequences of exposing neurons to both mature neurotrophins and proneurotrophins simultaneously. Basal forebrain (BF) neurons express all three Trk receptors, and BDNF is an established survival factor for these neurons (Alderson et al., 1990; Friedman et al., 1993). BF neurons also express p75NTR and are one of the few CNS populations to express this receptor throughout life. Our previous studies demonstrated that proneurotrophins could induce death of BF neurons via the p75NTR–sortilin receptor complex, even when Trk receptors were activated on the same neurons. Although proNGF did not prevent receptor phosphorylation, it inhibited the downstream activation of Akt normally induced by BDNF (Volosin et al., 2006). We have now investigated the mechanisms of interaction between p75NTR and TrkB signaling and demonstrate that while proNGF activates apoptotic signaling by p75NTR via the intrinsic caspase pathway (Wang et al., 2001; Troy et al., 2002), it must simultaneously suppress activation of the phosphoinositide-3 kinase (PI3K) pathway by Trk receptors for apoptosis to ensue. This suppression is mediated by PTEN (phosphatase and tensin homolog deleted on chromosome 10), a dual lipid/protein phosphatase that can dephosphorylate phosphatidylinositol 3,4,5-trisphosphate (PIP3) and convert it to PIP2, thereby antagonizing the activation of Akt by PI3K (Maehama and Dixon, 1998).
In this study, we demonstrate that proNGF induces PTEN in BF neurons, even in the presence of BDNF. Regulation of PTEN by proNGF is independent of transcription but requires new protein synthesis. The induction of PTEN prevents activation of Akt by BDNF and leads to cell death. When PTEN is inhibited, BDNF treatment can reverse the inhibition of Akt by proNGF and protect BF neurons from apoptosis. Thus, proNGF induction of neuronal cell death requires both apoptotic signaling via the intrinsic caspase pathway and simultaneous suppression of Trk-mediated survival signaling via Akt. To determine whether this dual mechanism is also required in vivo, we investigated whether inhibition of PTEN can prevent neuronal loss in the hippocampus after seizure, a model of p75NTR-mediated neuronal death that we have previously established. Infusion of a PTEN inhibitor or small interfering RNA (siRNA) into the hippocampus provided significant neuroprotection, indicating that the balance of these signaling pathways are critical determinants of neuronal survival or death after seizures.
Materials and Methods
Reagents.
BDNF was a gift from C. F. Ibanez (Karolinska Institute, Stockholm, Sweden). Anti-p75 antibodies were purchased from Millipore Bioscience Research Reagents or Millipore; other anti-p75 antisera were generously provided by M. V. Chao (Skirball Institute, New York University, New York, NY). Antibodies to p-Akt, Akt, p-Erk, Erk, cleaved caspase-3, and PTEN (catalog #9559), p-PTEN (catalog #9554 recognizing phosphorylation sites Ser380/Thr382/383) and the inhibitors LY294002 and PD98059 were purchased from Cell Signaling Technologies. The inhibitor to phospholipase C (PLC), U-73122, was purchased from Cayman Chemical. PTEN inhibitor [bpV(pic)] was from Calbiochem. Alexa 488 and Alexa 594 anti-rabbit and anti-mouse secondary antibodies were purchased from Invitrogen. Poly-d-lysine, glucose, putrescine, progesterone, transferrin, selenium, and insulin were purchased from Sigma.
Neuronal cultures.
Pregnant rats were killed by exposure to CO2 and soaked in 80% ethanol for 10 min. Embryonic day 16 (E16) rat fetuses were removed under sterile conditions and kept in PBS on ice. BFs were dissected and dissociated in serum-free medium composed of a 1:1 mixture of Eagle's MEM and Ham's F-12 supplemented with glucose (6 mg/ml), putrescine (60 μm), progesterone (20 nm), transferrin (100 μg/ml), selenium (30 nm), penicillin (0.5 U/ml), and streptomycin (0.5 μg/ml). For hippocampal cultures, fetuses were dissected at E18. The cells were then plated on tissue culture dishes that were precoated overnight with poly-d-lysine (0.1 mg/ml). The cells were maintained in serum-free medium for 5 d at 37°C (Farinelli et al., 1998; Friedman, 2000).
Survival assay.
Survival of cultured BF neurons was assayed by a method we have described previously (Farinelli et al., 1998; Maroney et al., 1999; Friedman, 2000). After removal of the medium, cultured cells were lysed, and intact nuclei were counted using a hemacytometer. Nuclei of dead cells either disintegrate or, if in the process of dying, appear pyknotic and irregularly shaped. In contrast, nuclei of healthy cells are phase bright and have clearly defined limiting membranes. Cell counts were performed in triplicate wells.
Real-time PCR.
BF neurons were cultured for 5 d before RNA was isolated using TRIZOL reagent (Invitrogen). cDNA was synthesized using superscript reverse transcriptase (Invitrogen). Real-time PCR (qPCR) was performed with the LightCycler 480 (Roche) using Sybr green master mix. The primers were synthesized by Invitrogen. The sequences were ACACCGCCAAATTTAACTGC (forward) and TACACCAGTCCGTCCTTTCC (reverse). Quantification of PTEN mRNA was assessed relative to glyceraldehyde 3-phosphate dehydrogenase (GAPDH).
siRNA.
Two distinct PTEN siRNAs with 5′ thiol modifications on the sense strands were synthesized by Dharmocon RNAi Technologies. The sense strand sequences were GATGGCTGTCATGTCTGGGAG (targeting the 5′ region) and GUAUAGAGCGUGCGGAUAA (targeting the coding domain). The siRNAs were mixed with Penetratin1 (Qbiogene), heated for 15 min at 65°C, and incubated for 1 h at 37°C. Before treating cells, the siRNAs were denatured at 65°C for 15 min and used at a concentration of 80 nm. A nonspecific penetratin-linked siRNA was used as the control (Ho et al., 2009).
Western blot analysis.
After 5 d in culture, the neurons were treated for the indicated times with vehicle, mature neurotrophins, and/or proneurotrophins. Cells were lysed in buffer containing 120 mm Tris, 2% SDS, 10% glycerol, and protease inhibitors. Equal amounts of protein were run on a 10 or 12% polyacrylamide gel and transferred to nitrocellulose membrane. The blots were blocked with 5% nonfat milk and incubated in a primary antibody overnight. After washing three times with TBS with 0.05% Tween 20 (TBST) for 15 min each, the blots were incubated with appropriate secondary antibodies for 1 h at room temperature. The membrane was washed three times with TBST before being visualized either using an ECL kit (Pierce) or being scanned with the Odyssey infrared imaging system (LI-COR Bioscience). All blots shown are representative of at least three independent experiments.
Pilocarpine-induced seizures.
Male Sprague Dawley rats (250–275 g) were cannulated 1 week before the induction of seizures. Animals were anesthetized with ketamine/xylazine and placed in a stereotaxic frame. Two cannulas were implanted bilaterally into the dorsal hippocampus. After 1 week of recovery, animals were pretreated with methyl-scopolamine (1 mg/kg, s.c.; Sigma-Aldrich) 30 min before being treated with pilocarpine hydrochloride (350 mg/kg, i.p.; Sigma-Aldrich). After 1 h of status epilepticus, diazepam (10 mg/kg; Abbott Laboratories) and phenytoin (50 mg/kg, i.p.) were injected to stop the seizure. Immediately after the seizure and twice per day for 3 d, the PTEN inhibitor bpv(pic) (5 μm in 0.5 μl) was injected through the cannula into the hippocampus on one side of the brain, and saline was injected into the contralateral side. Alternatively, PTEN siRNA (80 nm in 0.5 μl) was infused on one side of the brain. Three days after seizure, rats were anesthetized with ketamine/xylazine and perfused transcardially with saline followed by 4% paraformaldehyde. The brains were removed and fixed in 4% paraformaldehyde for 2 h before being cryoprotected in 30% sucrose for 2 d. Brains were then sectioned on a cryostat (Leica), mounted onto charged slides for immunostaining.
Immunocytochemistry.
Frozen brain sections (12 μm) were warmed at 37°C for 2 min and washed with PBS for 10 min. Before primary antibody incubation, the tissue was permeabilized and blocked with PBS/0.3% Triton X-100/10%goat serum for 30 min at room temperature. Primary antibodies were applied to the sections overnight at 4°C. The sections were then washed three times with PBS for 15 min. The secondary antibodies anti-rabbit Alexa 488 (green) and anti-mouse 594 (red) (1:500) were applied to sections in the dark for 1 h, washed three times with PBS for 15 min, and mounted. Hoechst 33342 dye (1 μg/ml; Sigma) was added into PBS during the last wash to label nuclei, and the sections were examined by fluorescence microscopy (Troy et al., 2002). In both anterior and posterior directions to the cannula tip (anteroposterior, −0.31 cm; lateral, ±0.2 cm; vertical, −0.3 cm from bregma), every 15th section was examined. In each section, all the cells labeled with p75NTR and cleaved caspase 3 in the hilus were counted. The number of labeled cells on each side of the hippocampus from three different rats was obtained, and the average was calculated for comparison. For infusion of the PTEN inhibitor bpv(pic), sections within 900 μm from the cannula showed significant differences between the two sides of the brain. Infusion of the PTEN siRNA showed differences between the two hemispheres within 600 μm of the cannula, indicating the diffusion range of the inhibitor and the siRNA, respectively.
All animal studies were conducted using the National Institutes of Health guidelines for the ethical treatment of animals with approval of the Rutgers Animal Care and Facilities Committee.
Statistical analysis.
Statistical significance on the multipart in vitro survival assays was determined by ANOVA with Bonferroni or Newman–Keuls post hoc analysis. For in vivo analysis of control versus PTEN inhibition, statistical significance was determined by Student's t test.
Results
ProNGF increases PTEN in BF neurons
Our previous studies had shown that treatment of BF neurons with proNGF induced neuronal apoptosis even when BDNF was present and TrkB was phosphorylated. However, once Akt was phosphorylated, no apoptosis was observed. Moreover, treatment with proNGF prevented the phosphorylation of Akt by BDNF but did not prevent phosphorylation of TrkB, suggesting that a critical interaction between p75-mediated apoptosis and TrkB-mediated survival signaling might occur downstream of the receptor, but upstream of Akt activation (Volosin et al., 2006). Since the PTEN phosphatase is known to antagonize activation of Akt by PI3K, levels of PTEN in these BF neurons were determined with and without proNGF treatment. BF neurons treated with proNGF showed a rapid and strong increase in PTEN levels that was maintained for at least 2 h (Fig. 1 A). The level of phosphorylated PTEN (p-PTEN) did not change significantly, thus the increase of total PTEN relative to p-PTEN indicates an increase in the nonphosphorylated form of PTEN, which is the active form of the phosphatase (Vazquez et al., 2000) (Fig. 1 B). This rapid increase in PTEN protein did not require new mRNA synthesis, as proNGF did not alter levels of PTEN mRNA (Fig. 1 E) and treatment of the neurons with actinomycin D did not prevent the proNGF-induced increase in the protein (Fig. 1 F). However, cycloheximide prevented the increase in PTEN, indicating that new protein synthesis was required (Fig. 1 F). The presence of BDNF did not prevent the proNGF-induced upregulation of PTEN (Fig. 1 G,H), consistent with the ability of proNGF to induce apoptosis in the presence of BDNF.
Figure 1.

ProNGF increases PTEN in BF neurons. A, E16 BF neurons were cultured for 5 d and treated with proNGF (1 ng/ml) at different time points, as indicated. Cells were lysed for Western blot analysis and probed for p-PTEN and PTEN. Blots were stripped and reprobed for actin. The blot shown is representative of five independent experiments. B, Quantification of PTEN relative to p-PTEN. Data are from five independent experiments and shown as mean band density ± SE. The asterisk indicates values different from control by ANOVA with Bonferroni post hoc analysis (p < 0.05). C, Induction of PTEN by proNGF requires both p75 and sortilin receptors. BF neurons were pretreated with anti-sortilin or anti-p75NTR antibodies for 30 min and then treated with proNGF (10 ng/ml, 30 min) alone or with the antibodies. The level of PTEN protein was measured by Western blot analysis. The blots were stripped and reprobed for tubulin. The blots shown are representative of three independent experiments. D, Quantification of three independent experiments exemplified in C and represented as mean band density ± SE. Asterisks indicates values different from control by ANOVA with Bonferroni post hoc analysis (p < 0.05). E, ProNGF does not regulate PTEN mRNA in BF neurons. BF neurons were treated with proNGF for the indicated times. Total RNA was extracted, and qPCR was performed to detect the level of PTEN mRNA relative to GAPDH mRNA. Data are from three independent experiments. F, New protein synthesis, but not mRNA synthesis, is required for PTEN induction. Cultured BF neurons were treated with proNGF (1 ng/ml, 30 min), cycloheximide (CHX; 1 μg/ml, 60 min), actinomycin-D (actinoD; 1.5 μm, 60 min), or both. Western blot analysis was performed to detect the level of PTEN. Blots were then stripped and reprobed for tubulin. The blot is representative of three independent experiments. G, ProNGF increases PTEN in the presence of BDNF. Cultured BF neurons were treated with BDNF (10 ng/ml), proNGF(1 ng/ml), or both for 30 min. Western blot was performed to probe for PTEN and stripped and reprobed for Erk to demonstrate equal protein loading. The blot is representative of three independent experiments. H, Quantification of PTEN relative to Erk from three independent experiments. Bars represent mean band density ± SE. The asterisk indicates values different from control by ANOVA with Bonferroni post hoc analysis, p < 0.05. con, Control.
Both p75NTR and sortilin receptors are required for proNGF-induced PTEN upregulation
Since proNGF binds to a receptor complex comprising p75NTR and sortilin to induce death of BF neurons (Volosin et al., 2006), we determined whether this receptor complex mediated the upregulation of PTEN. Blocking antibodies to either p75NTR or sortilin prevented the increase in PTEN, indicating that PTEN induction occurs through p75NTR–sortilin receptor binding (Fig. 1 C,D).
BDNF increases phospho-PTEN via the PI3K pathway, which can be blocked by proNGF
To determine whether there might be reciprocal regulation of PTEN by mature neurotrophins, BF neurons were treated with BDNF at different time points. Interestingly, levels of PTEN protein were unaffected by BDNF; however, phosphorylation was increased after treatment (Fig. 2 A). Since phosphorylation of PTEN suppresses its phosphatase activity (Vazquez et al., 2000), the increase in p-PTEN induced by BDNF would be likely to facilitate Akt activation and survival signaling. However, simultaneous treatment with proNGF prevented BDNF-induced PTEN phosphorylation (Fig. 2 B). Moreover, BDNF-induced phosphorylation of PTEN required activation of the PI3K pathway (Fig. 2 C). Thus, although BDNF can phosphorylate PTEN, this is prevented by proNGF since the induction of PTEN blocks PI3K signaling, thereby inhibiting phosphorylation of PTEN. These results suggest that proneurotrophins and mature neurotrophins have opposing effects on PTEN activity, with the consequences for neuronal survival depending on the balance between these signaling pathways.
Figure 2.

BDNF induces phosphorylation of PTEN via the PI3K pathway, which can be blocked by proNGF. A, BDNF increases phosphorylated PTEN protein in BF neurons. E16 neurons were cultured for 5 d before being treated with BDNF (10 ng/ml) for the indicated time points. Cells were lysed for Western blot to probe for p-PTEN and PTEN. Blots were stripped and reprobed for actin. The blot is representative of three independent experiments. B, ProNGF inhibits BDNF-induced phosphorylation of PTEN. BF neurons were treated with BDNF for 10 and 30 min in the presence or absence of proNGF. The levels of p-PTEN and PTEN were measured by Western blot. The blot was stripped and reprobed for tubulin. The blot is representative of three independent experiments. C, The PI3K pathway inhibitor blocks induction of p-PTEN by BDNF. BF neurons were treated with PI3K pathway inhibitor LY294002 (50 μm), MAPK pathway inhibitor PD98059 (10 μm), or PLCγ inhibitor U73122 (1 μm) before being treated with BDNF for 30 min. Lysates were analyzed by Western blot for p-PTEN and PTEN. The blot was stripped and reprobed for tubulin. The blot is representative of three independent experiments. con, Control.
PTEN inhibition allows BDNF-induced Akt phosphorylation and neuronal survival in the presence of proNGF
To understand the potential role of PTEN in proNGF-induced apoptosis, we treated cultured BF neurons with a potent and selective PTEN inhibitor, bpv(pic) (5 μm) (Schmid et al., 2004), to block PTEN activity. After bpv(pic) treatment, proNGF no longer inhibited BDNF-induced Akt phosphorylation, confirming that PTEN blocked the PI3K pathway (Fig. 3 A).
Figure 3.

PTEN inhibitor reverses the inhibition by proNGF of BDNF-induced Akt phosphorylation and survival of BF neurons. A, PTEN inhibitor reverses the inhibition of Akt activation by proNGF. E16 BF neurons were cultured in the presence of BDNF for 5 d. The cells were then treated with BDNF (10 ng/ml, re-added to ensure that TrkB receptors were activated), proNGF (1 ng/ml), or BDNF plus proNGF in the presence or absence of the PTEN inhibitor bpv(pic) (5 μm). Western blot analysis was performed to probe for p-Akt. Blots were then stripped and reprobed for total Akt. The blot is representative of four independent experiments. CON, Control. B, PTEN inhibitor rescues BF neurons from proNGF-induced death only in the presence of BDNF. Cultured E16 BF neurons were treated overnight with proNGF (1 ng/ml) alone or proNGF plus bpv(pic) (5 μm) in the presence or absence of BDNF (10 ng/ml). A survival assay was performed to count the number of healthy neurons in the culture and indicated as the percentage of total cells. Data are from three independent experiments and expressed as mean percent survival ± SE. The asterisk indicates values different from control by ANOVA with Bonferroni post hoc analysis at p < 0.05.
Since inhibiting PTEN restored the ability of BDNF to activate Akt, we tested whether inhibiting PTEN could also restore the ability of BDNF to protect neurons from proNGF-induced apoptosis. Cultured BF neurons were treated with bpv(pic), in the absence or presence of BDNF, and assayed for effects of proNGF on survival. The presence of the PTEN inhibitor provided significant protection from cell death induced by proNGF, but only in the presence of BDNF (Fig. 3 B). This effect was not observed in the absence of BDNF, suggesting that TrkB-mediated signaling must be activated for the inhibition of PTEN to protect neurons from proNGF-induced apoptosis.
To confirm the effects of the pharmacologic PTEN inhibitor, we used siRNAs to knock down PTEN in the BF neurons. The siRNA was linked to penetratin to facilitate entry into neurons (Davidson et al., 2004). Long-term treatment of BF neurons with the penetratin-linked PTEN siRNA decreased basal PTEN levels (Fig. 4 A), but more important, pretreatment of BF neurons with the PTEN siRNA for 30 min prevented the rapid proNGF-induced PTEN upregulation (Fig. 4 B). When PTEN induction was blocked by the siRNA, treatment with BDNF was able to rescue BF neurons from proNGF-induced death (Fig. 4 C,D). Two distinct PTEN siRNAs gave the same result. No rescue occurred in the absence of BDNF treatment, confirming previous results with the pharmacological inhibitor and indicating that PTEN does not directly mediate apoptotic signaling by proNGF but, rather, is necessary to suppress survival signaling mediated by BDNF activation of Akt via TrkB.
Figure 4.

PTEN siRNA rescues BF neurons from proNGF-induced apoptosis in the presence of BDNF. A, A penetratin-linked PTEN siRNA was used to knock down PTEN protein levels. BF neurons were treated with PTEN siRNA for the indicated times and analyzed by Western blot to detect PTEN protein. The blot was stripped and reprobed for actin. B, PTEN siRNA inhibited the increase of PTEN induced by proNGF. BF neurons were pretreated with control or PTEN siRNA for 30 min before being treated with proNGF (1 ng/ml) for 30 min. Western blot analysis was performed to detect PTEN protein. The blot is representative of three independent experiments. C, D, PTEN siRNA blocks proNGF-induced death only in the presence of BDNF. Cultured BF neurons were treated overnight with vehicle (control), proNGF alone (1 ng/ml), control siRNA or PTEN siRNA alone, or siRNA combined with proNGF in the presence (C) or absence (D) of BDNF (10 ng/ml). The number of healthy neurons was counted and indicated as the percentage of total cells ± SE. Data are from three independent experiments. The asterisk indicates values different from control by ANOVA with Bonferroni post hoc analysis at p < 0.05. con, Control; con si, control siRNA; si PTEN, PTEN siRNA.
PTEN is required for p75NTR-mediated death in the hippocampus in vivo
Since neurons in vivo are exposed to multiple factors in their environment, including both mature neurotrophins and proneurotrophins, we investigated the potential function of PTEN in regulating the balance between p75NTR-mediated apoptotic signaling and TrkB-mediated survival signaling after pilocarpine-induced seizures in vivo.
We have previously demonstrated that infusion of an anti-proNGF antibody into the hippocampus provided significant protection from neuronal death after seizures (Volosin et al., 2006, 2008). To use this model to assess whether PTEN activity was necessary to facilitate neuronal apoptosis, we first examined whether proNGF could regulate PTEN in cultured hippocampal neurons as in BF neurons. ProNGF treatment of hippocampal neurons increased PTEN protein and blocked BDNF-induced Akt activation in hippocampal neurons as in BF neurons (supplemental Fig. S2, available at www.jneurosci.org as supplemental material).
The PTEN inhibitor, bpv(pic) (5 μm), was infused through an implanted cannula into the dorsal hippocampus on one side of the brain while saline was injected into the contralateral side after pilocarpine-induced seizure. Three days after the seizures, the time of maximal neuronal death (Volosin et al., 2008), double immunostaining for p75NTR and cleaved caspase 3 revealed fewer apoptotic neurons in the hilus on the side of bpv(pic) infusion compared with the control side in the same brain, indicating that inhibiting PTEN can rescue hippocampal neurons from proNGF-induced death (Fig. 5 A,B). Labeling degenerating neurons with Fluoro-Jade B confirmed the decrease in dying neurons on the side of the brain receiving the PTEN inhibitor compared with the contralateral side that received saline (Fig. 5 C). Adjacent sections labeled for P-Trk and P-Akt demonstrated numerous double-labeled neurons on the saline-injected side and an increase in P-Akt-labeled neurons on the side that received the PTEN inhibitor (Fig. 5 D), indicating that antagonizing the PI3 kinase pathway is necessary for neuronal death.
Figure 5.
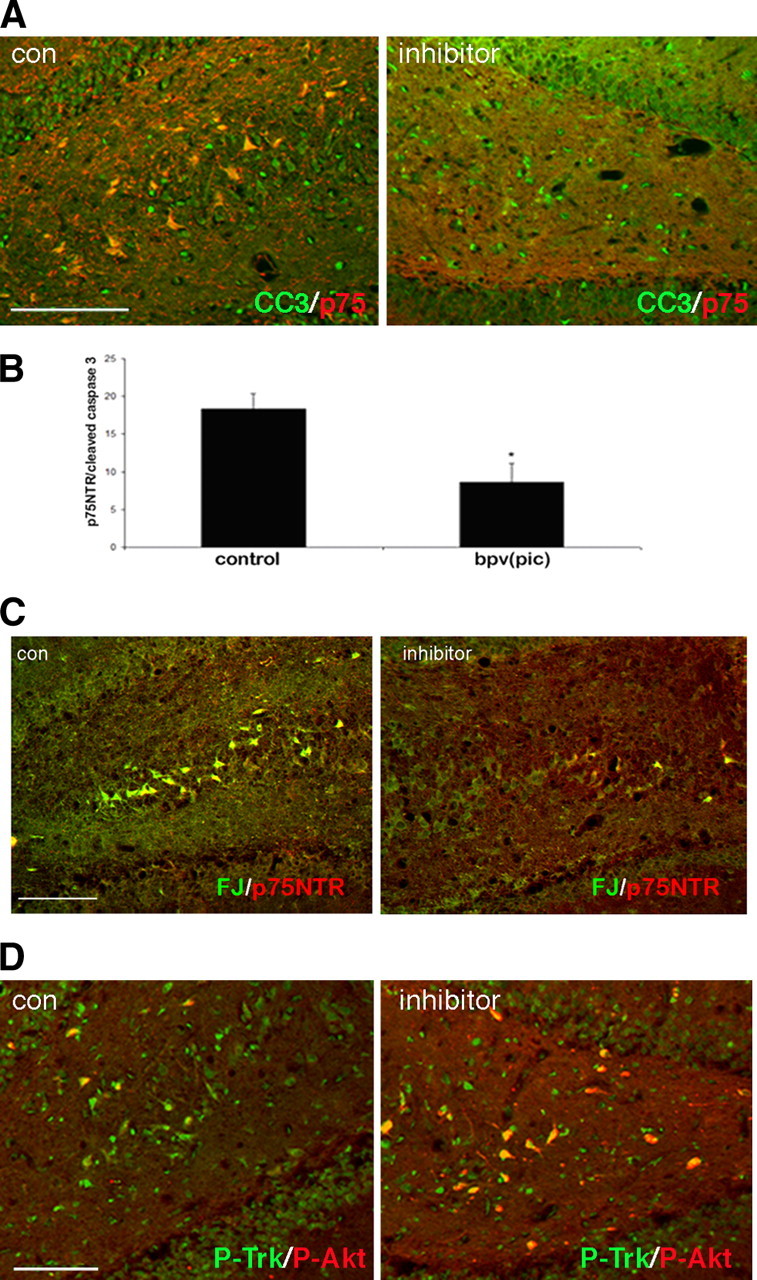
PTEN inhibitor rescues neuronal loss after seizure. Adult rats were cannulated bilaterally in the dorsal hippocampus 1 week before pilocarpine-induced seizure. After seizures, PTEN inhibitor [bpv(pic), 5 μm] was infused into one side and saline was infused into the contralateral side twice per day for 3 d. A, Double immunostaining for p75NTR and cleaved caspase 3 was performed on hippocampal brain sections. There is a decrease in the number of cells expressing both p75NTR and cleaved caspase 3 on the side with the inhibitor relative to the control side in the same brain sections. B, The number of cells expressing both p75NTR and cleaved caspase 3 was counted in both hippocampal hemispheres. The average number of cells coexpressing p75NTR and cleaved caspase 3 from three independent experiments was calculated and shown as mean ± SE. The asterisk indicates the value significantly different from control at p < 0.05 by Student's t test. C, Double staining for Fluoro-Jade B (FJ) and p75NTR demonstrated fewer dying neurons on the side receiving inhibitor relative to the control side. D, Double immunostaining for phospho-Trk and phospho-Akt on hippocampal sections. More cells expressing p-Akt were observed on the side with the PTEN inhibitor relative to the side receiving saline in the same brain. Scale bars, 50 μm. con, Control.
To confirm the effects of the PTEN inhibitor, penetratin-linked PTEN siRNA was infused on one side of the brain for 3 d after seizures and showed a similar decrease in apoptotic hippocampal neurons compared with the contralateral side that received a control penetratin-linked siRNA (Fig. 6). Thus, inhibition of PTEN either pharmacologically or by knockdown after seizures attenuated neuronal apoptosis.
Figure 6.

PTEN siRNA rescues neuronal loss after seizure. Adult rats were cannulated bilaterally in the dorsal hippocampus 1 week before pilocarpine-induced seizure. After seizures, penetratin-linked PTEN siRNA was infused into one side and a control penetratin-linked siRNA was infused into the contralateral side twice per day for 3 d. A, Double immunostaining for p75NTR and cleaved caspase 3 was performed on hippocampal brain sections. Scale bar, 50 μm. B, The average number of cells coexpressing p75NTR and cleaved caspase 3 from three independent experiments was calculated and is shown as mean ± SE. The asterisk indicates the value significantly different from control at p < 0.05 by Student's t test.
Discussion
In this study, we have investigated the interaction of survival and apoptotic signaling pathways that ultimately determine the fate of CNS neurons. Neurotrophins regulate multiple cellular functions by binding to either Trk receptors or p75NTR (Arevalo and Wu, 2006; Reichardt, 2006). Mature neurotrophins bind preferentially to Trk receptors, which have been associated with survival and differentiation of cells via both the PI3K and mitogen-activated protein kinase (MAPK) pathways (Patapoutian and Reichardt, 2001). In contrast, proneurotrophins bind with high affinity to a p75NTR/sortilin receptor complex (Nykjaer et al., 2004), which activates signaling pathways important in regulating apoptosis (Roux and Barker, 2002; Volosin et al., 2006). ProNGF and proBDNF induce cell death via p75NTR in neuronal cultures (Lee et al., 2001; Beattie et al., 2002; Teng et al., 2005; Volosin et al., 2006). In vivo, these proneurotrophins are increased in neurons and astrocytes after seizures and are secreted into the CSF (Volosin et al., 2006; 2008). Proneurotrophins induce neuronal apoptosis in several pathophysiological conditions, further implicating their involvement in neuronal loss after injury, in seizures, and in age-related disease (Harrington et al., 2004; Peng et al., 2004; Pedraza et al., 2005; Volosin et al., 2006; Domeniconi et al., 2007).
As cells are normally exposed to multiple factors in their environment, what happens when neurons are exposed to both mature neurotrophins and proneurotrophins, particularly after injury when neurotrophin mRNAs are upregulated and release of both mature neurotrophins and proneurotrophins can occur? Since activation of Trk receptors supports neuronal survival and activation of p75NTR leads to apoptosis, how do the signaling pathways activated by the different receptors interact to determine the outcome for the cell?
BF cholinergic neurons are among the few CNS neuronal populations that express p75NTR throughout life. These cells also express all three Trk receptors, and mature neurotrophins promote survival of BF neurons (Alderson et al., 1990; Friedman et al., 1993). These neurons are important for memory and learning (Conner et al., 2003), and loss of BF neurons is one of the early features of Alzheimer's disease (Whitehouse et al., 1982), although the mechanism of this loss remains unknown.
Under both physiological and pathological conditions, BF neurons may be exposed to both mature neurotrophins and proneurotrophins from neurons in the target hippocampus and cortex, or from glia in the local BF environment (Seiler and Schwab, 1984; Wu et al., 2004). Our previous studies have demonstrated increased expression of proNGF and proBDNF in both BF and hippocampal astrocytes, as well as hippocampal neurons, after seizures (Volosin et al., 2008). In Alzheimer's disease, there is increased proNGF in the brain (Fahnestock et al., 2001; Peng et al., 2004), which is capable of inducing neuronal apoptosis (Pedraza et al., 2005; Podlesniy et al., 2006). The increase in proNGF that occurs after injury, after seizures, or in disease may shift the balance of proneurotrophins relative to mature neurotrophins. Our current study suggests that suppression of the PI3K–Akt pathway by PTEN is an important mechanism in the induction of apoptosis by proneurotrophins.
The role of PTEN in proNGF-induced death of CNS neurons
We initially investigated the interaction of Trk and p75 signaling in BF neurons since all these neurons express p75NTR together with a Trk receptor, primarily TrkB. We previously showed that proNGF elicited death of BF neurons through p75NTR-mediated signaling, even in the presence of activated TrkB receptors, and that proNGF can inhibit BDNF-induced Akt phosphorylation (Volosin et al., 2006). Because PTEN can block PI3K-mediated signaling, we evaluated whether PTEN might play a role in the interaction between p75NTR and TrkB-mediated signaling in BF neurons. We found an increased level of PTEN after proNGF treatment, which was not blocked by BDNF, consistent with our previous observation that proNGF prevented Akt phosphorylation and induced neuronal apoptosis in the presence of BDNF. Moreover, blocking PTEN with either a pharmacological inhibitor or siRNA-mediated knockdown reversed the effects on both Akt phosphorylation and neuronal survival. Thus, when PTEN activity was prevented, BDNF was able to induce Akt phosphorylation and neuronal survival even in the presence of proNGF, demonstrating that PTEN plays a critical role in proNGF apoptotic signaling by suppressing activation of Akt (Fig. 7).
Figure 7.

Schematic diagram of the interaction of p75NTR and TrkB signaling. Apoptosis induced by proNGF via p75NTR requires suppression of the TrkB-induced PI3K pathway by PTEN. When PTEN is inhibited, BDNF-induced activation of Akt can inhibit apoptosis.
To investigate the role of PTEN in vivo, we used a model of pilocarpine-induced seizures that elicits p75NTR-mediated neuronal death in the hippocampus. After confirming that proNGF also induced PTEN in cultured hippocampal neurons, we showed that blocking PTEN, either pharmacologically or by siRNA-mediated knockdown, provided significant neuroprotection after seizures.
The balance between survival and death signaling
Because of the expression of both p75NTR and Trk receptors in BF neurons, appropriate ligands for each of these receptors can activate their respective signaling pathways (Volosin et al., 2006), but how these signaling pathways interact to determine whether the cell dies or survives remained to be elucidated. In this study, we have demonstrated that one interaction point is at the level of Akt activation; BDNF signaling via TrkB activates Akt via the PI3K pathway, whereas proNGF signaling via p75NTR inhibits Akt. PTEN is regulated by proNGF through p75NTR/sortilin signaling and antagonizes PI3K activation, thereby inhibiting BDNF-induced survival signaling. The activity of PTEN can be regulated in several ways including phosphorylation, acetylation, and oxidation (Lee et al., 2002; Okumura et al., 2006; Maccario et al., 2007). Phosphorylation of PTEN at specific residues renders it unstable and less active. Interestingly, in contrast to the effect of proNGF, BDNF increased PTEN phosphorylation in BF neurons, although the level of PTEN protein remained unaltered. However, the effect of BDNF on PTEN phosphorylation required signaling via the PI3K pathway and was prevented in the presence of proNGF. Thus, the presence of proNGF shifted the balance of these signaling pathways toward apoptosis.
The phosphorylation of serine/threonine residues on the C terminal of PTEN decreases its phosphatase activity (Vazquez et al., 2000). Casein kinase II (CK2) can phosphorylate some of these sites both in vitro and in intact cells, and high levels of CK2 expression are present in human solid tumors, supporting the notion that CK2 negatively regulates PTEN activity as a tumor suppressor (Daya-Makin et al., 1994; Vazquez et al., 2001). Studies investigating signaling pathways regulating PTEN phosphorylation and CK2 activity suggest that PI3K facilitates phosphorylation of PTEN on its C terminal (Birle et al., 2002). Additionally, epidermal growth factor receptor-activated ERK2 can bind and activate CK2α (Ji et al., 2009). We have observed that BDNF increased PTEN phosphorylation in BF neurons, suggesting that TrkB activation can phosphorylate and negatively regulate PTEN activity. Since both the PI3K and MAPK pathways can be activated by TrkB, PTEN phosphorylation may be regulated by either or both of the pathways. However, in BF neurons we showed that the PI3K inhibitor, but not the MEK inhibitor, prevented BDNF-induced PTEN phosphorylation, indicating that in these neurons the PI3K pathway is more likely to mediate PTEN phosphorylation, possibly via activation of CK2.
BDNF can induce phosphorylation of PTEN, suggesting that TrkB signaling might inhibit apoptosis by converting PTEN to its unstable form. Collectively, the fate of these CNS neurons is determined by competition between Trk and p75NTR signaling. When both receptors are activated, the cross talk between these two pathways contributes to the fate of these cells. Here, we demonstrate that induction of the PTEN phosphatase is an important mechanism involved in this cross talk by regulating Akt activation. For apoptotic signaling to proceed via the intrinsic caspase pathway previously identified, there must be simultaneous suppression of survival signaling. That BDNF can protect these neurons from proNGF-induced death when PTEN is inhibited demonstrates the critical role of this protein in mediating the balance between survival and death of CNS neurons, especially in pathological states.
PTEN, p75NTR, and neuronal disorders
PTEN is widely expressed in the brain (Lachyankar et al., 2000; Perandones et al., 2004), and multiple studies have described important roles for PTEN in neuronal death (Gary and Mattson, 2002; Li et al., 2002; Xu et al., 2003). Mutations in PTEN have been linked to deficient cell death, thus a high frequency of PTEN mutations occur in glioblastomas (Knobbe et al., 2008). Conditionally deleting PTEN resulted in increased brain size over time (Backman et al., 2001; Kwon et al., 2001), and several illnesses can be linked to a mutation of the PTEN gene, including autism, Cowden's syndrome, Bannayan-Riley-Ruvalcada syndrome, and Lhermitte-Duclos disease (Butler et al., 2005; Kwon et al., 2006; Herman et al., 2007).
ProNGF/p75NTR interactions have also been linked to neuronal pathologies, and changes in p75NTR expression have been directly related to glioblastoma invasion and neurodegeneration in Alzheimer's disease (Johnston et al., 2007; Fombonne et al., 2009). Here, we describe for the first time that PTEN plays an important role in proNGF/p75NTR apoptotic signaling. The p75NTR receptor is induced in many cell types in a variety of injury situations. The ability of p75NTR signaling to induce PTEN may regulate Akt activation by growth factors other than neurotrophins as well, therefore these observations may have broader implications. The interactions between proNGF, p75NTR, and PTEN may provide a new target for neuroprotection and therapeutic treatment of neurodegenerative diseases.
Footnotes
This work was supported by National Institutes of Health Grants NS045556 (W.J.F.) and NS30687 (B.L.H.). We thank Carol Troy for advice on the penetratin-linked siRNA and for providing the control siRNA. We thank Richard Farias and Matthew Wilkins for technical assistance.
References
- Alderson RF, Alterman AL, Barde Y-A, Lindsay RM. Brain-derived neurotrophic factor increases survival and differentiated functions of rat septal cholinergic neurons in culture. Neuron. 1990;5:297–306. doi: 10.1016/0896-6273(90)90166-d. [DOI] [PubMed] [Google Scholar]
- Arevalo JC, Wu SH. Neurotrophin signaling: many exciting surprises! Cell Mol Life Sci. 2006;63:1523–1537. doi: 10.1007/s00018-006-6010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backman SA, Stambolic V, Suzuki A, Haight J, Elia A, Pretorius J, Tsao MS, Shannon P, Bolon B, Ivy GO, Mak TW. Deletion of Pten in mouse brain causes seizures, ataxia and defects in soma size resembling Lhermitte-Duclos disease. Nat Genet. 2001;29:396–403. doi: 10.1038/ng782. [DOI] [PubMed] [Google Scholar]
- Beattie MS, Harrington AW, Lee R, Kim JY, Boyce SL, Longo FM, Bresnahan JC, Hempstead BL, Yoon SO. ProNGF induces p75-mediated death of oligodendrocytes following spinal cord injury. Neuron. 2002;36:375–386. doi: 10.1016/s0896-6273(02)01005-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibel M, Barde YA. Neurotrophins: key regulators of cell fate and cell shape in the vertebrate nervous system. Genes Dev. 2000;14:2919–2937. doi: 10.1101/gad.841400. [DOI] [PubMed] [Google Scholar]
- Birle D, Bottini N, Williams S, Huynh H, deBelle I, Adamson E, Mustelin T. Negative feedback regulation of the tumor suppressor PTEN by phosphoinositide-induced serine phosphorylation. J Immunol. 2002;169:286–291. doi: 10.4049/jimmunol.169.1.286. [DOI] [PubMed] [Google Scholar]
- Butler MG, Dasouki MJ, Zhou XP, Talebizadeh Z, Brown M, Takahashi TN, Miles JH, Wang CH, Stratton R, Pilarski R, Eng C. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet. 2005;42:318–321. doi: 10.1136/jmg.2004.024646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conner JM, Culberson A, Packowski C, Chiba AA, Tuszynski MH. Lesions of the basal forebrain cholinergic system impair task acquisition and abolish cortical plasticity associated with motor skill learning. Neuron. 2003;38:819–829. doi: 10.1016/s0896-6273(03)00288-5. [DOI] [PubMed] [Google Scholar]
- Davidson TJ, Harel S, Arboleda VA, Prunell GF, Shelanski ML, Greene LA, Troy CM. Highly efficient small interfering RNA delivery to primary mammalian neurons induces microRNA-like effects before mRNA degradation. J Neurosci. 2004;24:10040–10046. doi: 10.1523/JNEUROSCI.3643-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daya-Makin M, Sanghera JS, Mogentale TL, Lipp M, Parchomchuk J, Hogg JC, Pelech SL. Activation of a tumor-associated protein kinase (p40TAK) and casein kinase 2 in human squamous cell carcinomas and adenocarcinomas of the lung. Cancer Res. 1994;54:2262–2268. [PubMed] [Google Scholar]
- Domeniconi M, Hempstead BL, Chao MV. Pro-NGF secreted by astrocytes promotes motor neuron cell death. Mol Cell Neurosci. 2007;34:271–279. doi: 10.1016/j.mcn.2006.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahnestock M, Michalski B, Xu B, Coughlin MD. The precursor pro-nerve growth factor is the predominant form of nerve growth factor in brain and is increased in Alzheimer's disease. Mol Cell Neurosci. 2001;18:210–220. doi: 10.1006/mcne.2001.1016. [DOI] [PubMed] [Google Scholar]
- Farinelli SE, Greene LA, Friedman WJ. Neuroprotective actions of dipyridamole on cultured CNS neurons. J Neurosci. 1998;18:5112–5123. doi: 10.1523/JNEUROSCI.18-14-05112.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fombonne J, Rabizadeh S, Banwait S, Mehlen P, Bredesen DE. Selective vulnerability in Alzheimer's disease: amyloid precursor protein and p75(NTR) interaction. Ann Neurol. 2009;65:294–303. doi: 10.1002/ana.21578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman WJ. Neurotrophins induce death of hippocampal neurons via the p75 receptor. J Neurosci. 2000;20:6340–6346. doi: 10.1523/JNEUROSCI.20-17-06340.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman WJ, Greene LA. Neurotrophin signaling via Trks and p75. Exp Cell Res. 1999;253:131–142. doi: 10.1006/excr.1999.4705. [DOI] [PubMed] [Google Scholar]
- Friedman WJ, Ibanez CF, Hallbook F, Persson H, Cain LD, Dreyfus CF, Black IB. Differential actions of neurotrophins in the locus coeruleus and basal forebrain. Exp Neurol. 1993;119:72–78. doi: 10.1006/exnr.1993.1007. [DOI] [PubMed] [Google Scholar]
- Gary DS, Mattson MP. PTEN regulates Akt kinase activity in hippocampal neurons and increases their sensitivity to glutamate and apoptosis. Neuromol Med. 2002;2:261–269. doi: 10.1385/NMM:2:3:261. [DOI] [PubMed] [Google Scholar]
- Harrington AW, Leiner B, Blechschmitt C, Arevalo JC, Lee R, Morl K, Meyer M, Hempstead BL, Yoon SO, Giehl KM. Secreted proNGF is a pathophysiological death-inducing ligand after adult CNS injury. Proc Natl Acad Sci U S A. 2004;101:6226–6230. doi: 10.1073/pnas.0305755101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman GE, Henninger N, Ratliff-Schaub K, Pastore M, Fitzgerald S, McBride KL. Genetic testing in autism: how much is enough? Genet Med. 2007;9:268–274. doi: 10.1097/gim.0b013e31804d683b. [DOI] [PubMed] [Google Scholar]
- Ho CC, Rideout HJ, Ribe E, Troy CM, Dauer WT. The Parkinson disease protein leucine-rich repeat kinase 2 transduces death signals via Fas-associated protein with death domain and caspase-8 in a cellular model of neurodegeneration. J Neurosci. 2009;29:1011–1016. doi: 10.1523/JNEUROSCI.5175-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H, Wang J, Nika H, Hawke D, Keezer S, Ge Q, Fang B, Fang X, Fang D, Litchfield DW, Aldape K, Lu Z. EGF-induced ERK activation promotes CK2-mediated disassociation of alpha-Catenin from beta-Catenin and transactivation of beta-Catenin. Mol Cell. 2009;36:547–559. doi: 10.1016/j.molcel.2009.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston AL, Lun X, Rahn JJ, Liacini A, Wang L, Hamilton MG, Parney IF, Hempstead BL, Robbins SM, Forsyth PA, Senger DL. The p75 neurotrophin receptor is a central regulator of glioma invasion. PLoS Biol. 2007;5:e212. doi: 10.1371/journal.pbio.0050212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knobbe CB, Lapin V, Suzuki A, Mak TW. The roles of PTEN in development, physiology and tumorigenesis in mouse models: a tissue-by-tissue survey. Oncogene. 2008;27:5398–5415. doi: 10.1038/onc.2008.238. [DOI] [PubMed] [Google Scholar]
- Kwon CH, Zhu X, Zhang J, Knoop LL, Tharp R, Smeyne RJ, Eberhart CG, Burger PC, Baker SJ. Pten regulates neuronal soma size: a mouse model of Lhermitte-Duclos disease. Nat Genet. 2001;29:404–411. doi: 10.1038/ng781. [DOI] [PubMed] [Google Scholar]
- Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Li Y, Baker SJ, Parada LF. Pten regulates neuronal arborization and social interaction in mice. Neuron. 2006;50:377–388. doi: 10.1016/j.neuron.2006.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachyankar MB, Sultana N, Schonhoff CM, Mitra P, Poluha W, Lambert S, Quesenberry PJ, Litofsky NS, Recht LD, Nabi R, Miller SJ, Ohta S, Neel BG, Ross AH. A role for nuclear PTEN in neuronal differentiation. J Neurosci. 2000;20:1404–1413. doi: 10.1523/JNEUROSCI.20-04-01404.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee R, Kermani P, Teng KK, Hempstead BL. Regulation of cell survival by secreted proneurotrophins. Science. 2001;294:1945–1948. doi: 10.1126/science.1065057. [DOI] [PubMed] [Google Scholar]
- Lee SR, Yang KS, Kwon J, Lee C, Jeong W, Rhee SG. Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem. 2002;277:20336–20342. doi: 10.1074/jbc.M111899200. [DOI] [PubMed] [Google Scholar]
- Lewin GR, Barde YA. Physiology of the neurotrophins. Annu Rev Neurosci. 1996;19:289–317. doi: 10.1146/annurev.ne.19.030196.001445. [DOI] [PubMed] [Google Scholar]
- Li L, Liu F, Salmonsen RA, Turner TK, Litofsky NS, Di Cristofano A, Pandolfi PP, Jones SN, Recht LD, Ross AH. PTEN in neural precursor cells: regulation of migration, apoptosis, and proliferation. Mol Cell Neurosci. 2002;20:21–29. doi: 10.1006/mcne.2002.1115. [DOI] [PubMed] [Google Scholar]
- Maccario H, Perera NM, Davidson L, Downes CP, Leslie NR. PTEN is destabilized by phosphorylation on Thr366. Biochem J. 2007;405:439–444. doi: 10.1042/BJ20061837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- Maroney AC, Finn JP, Bozyczko-Coyne D, O'Kane TM, Neff NT, Tolkovsky AM, Park DS, Yan CY, Troy CM, Greene LA. CEP-1347 (KT7515), an inhibitor of JNK activation, rescues sympathetic neurons and neuronally differentiated PC12 cells from death evoked by three distinct insults. J Neurochem. 1999;73:1901–1912. [PubMed] [Google Scholar]
- Nykjaer A, Lee R, Teng K, Jansen P, Madsen P, Nielsen M, Jacobsen C, Kliemannel M, Schwarz E, Willnow T, Hempstead B, Petersen C. Sortilin is essential for proNGF-induced neuronal cell death. Nature. 2004;427:843–848. doi: 10.1038/nature02319. [DOI] [PubMed] [Google Scholar]
- Okumura K, Mendoza M, Bachoo RM, DePinho RA, Cavenee WK, Furnari FB. PCAF modulates PTEN activity. J Biol Chem. 2006;281:26562–26568. doi: 10.1074/jbc.M605391200. [DOI] [PubMed] [Google Scholar]
- Patapoutian A, Reichardt LF. Trk receptors: mediators of neurotrophin action. Curr Opin Neurobiol. 2001;11:272–280. doi: 10.1016/s0959-4388(00)00208-7. [DOI] [PubMed] [Google Scholar]
- Pedraza CE, Podlesniy P, Vidal N, Arevalo JC, Lee R, Hempstead B, Ferrer I, Iglesias M, Espinet C. Pro-NGF isolated from the human brain affected by Alzheimer's disease induces neuronal apoptosis mediated by p75NTR. Am J Pathol. 2005;166:533–543. doi: 10.1016/S0002-9440(10)62275-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng S, Wuu J, Mufson EJ, Fahnestock M. Increased proNGF levels in subjects with mild cognitive impairment and mild Alzheimer disease. J Neuropathol Exp Neurol. 2004;63:641–649. doi: 10.1093/jnen/63.6.641. [DOI] [PubMed] [Google Scholar]
- Perandones C, Costanzo RV, Kowaljow V, Pivetta OH, Carminatti H, Radrizzani M. Correlation between synaptogenesis and the PTEN phosphatase expression in dendrites during postnatal brain development. Brain Res Mol Brain Res. 2004;128:8–19. doi: 10.1016/j.molbrainres.2004.05.021. [DOI] [PubMed] [Google Scholar]
- Podlesniy P, Kichev A, Pedraza C, Saurat J, Encinas M, Perez B, Ferrer I, Espinet C. Pro-NGF from Alzheimer's disease and normal human brain displays distinctive abilities to induce processing and nuclear translocation of intracellular domain of p75NTR and apoptosis. Am J Pathol. 2006;169:119–131. doi: 10.2353/ajpath.2006.050787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichardt LF. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci. 2006;361:1545–1564. doi: 10.1098/rstb.2006.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux PP, Barker PA. Neurotrophin signaling through the p75 neurotrophin receptor. Prog Neurobiol. 2002;67:203–233. doi: 10.1016/s0301-0082(02)00016-3. [DOI] [PubMed] [Google Scholar]
- Schmid AC, Byrne RD, Vilar R, Woscholski R. Bisperoxovanadium compounds are potent PTEN inhibitors. FEBS Lett. 2004;566:35–38. doi: 10.1016/j.febslet.2004.03.102. [DOI] [PubMed] [Google Scholar]
- Seiler M, Schwab ME. Specific retrograde transport of nerve growth factor (NGF) from neocortex to nucleus basalis in the rat. Brain Res. 1984;300:33–39. doi: 10.1016/0006-8993(84)91338-6. [DOI] [PubMed] [Google Scholar]
- Teng HK, Teng KK, Lee R, Wright S, Tevar S, Almeida RD, Kermani P, Torkin R, Chen ZY, Lee FS, Kraemer RT, Nykjaer A, Hempstead BL. ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J Neurosci. 2005;25:5455–5463. doi: 10.1523/JNEUROSCI.5123-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troy CM, Friedman JE, Friedman WJ. Mechanisms of p75-mediated death of hippocampal neurons. Role of caspases. J Biol Chem. 2002;277:34295–34302. doi: 10.1074/jbc.M205167200. [DOI] [PubMed] [Google Scholar]
- Vazquez F, Ramaswamy S, Nakamura N, Sellers WR. Phosphorylation of the PTEN tail regulates protein stability and function. Mol Cell Biol. 2000;20:5010–5018. doi: 10.1128/mcb.20.14.5010-5018.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez F, Grossman SR, Takahashi Y, Rokas MV, Nakamura N, Sellers WR. Phosphorylation of the PTEN tail acts as an inhibitory switch by preventing its recruitment into a protein complex. J Biol Chem. 2001;276:48627–48630. doi: 10.1074/jbc.C100556200. [DOI] [PubMed] [Google Scholar]
- Volosin M, Song W, Almeida RD, Kaplan DR, Hempstead BL, Friedman WJ. Interaction of survival and death signaling in basal forebrain neurons: roles of neurotrophins and proneurotrophins. J Neurosci. 2006;26:7756–7766. doi: 10.1523/JNEUROSCI.1560-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volosin M, Trotter C, Cragnolini A, Kenchappa RS, Light M, Hempstead BL, Carter BD, Friedman WJ. Induction of proneurotrophins and activation of p75NTR-mediated apoptosis via neurotrophin receptor-interacting factor in hippocampal neurons after seizures. J Neurosci. 2008;28:9870–9879. doi: 10.1523/JNEUROSCI.2841-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Bauer JH, Li Y, Shao Z, Zetoune FS, Cattaneo E, Vincenz C. Characterization of a p75(NTR) apoptotic signaling pathway using a novel cellular model. J Biol Chem. 2001;276:33812–33820. doi: 10.1074/jbc.M010548200. [DOI] [PubMed] [Google Scholar]
- Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, Delon MR. Alzheimer's disease and senile dementia: loss of neurons in the basal forebrain. Science. 1982;215:1237–1239. doi: 10.1126/science.7058341. [DOI] [PubMed] [Google Scholar]
- Wu H, Friedman WJ, Dreyfus CF. Differential regulation of neurotrophin expression in basal forebrain astrocytes by neuronal signals. J Neurosci Res. 2004;76:76–85. doi: 10.1002/jnr.20060. [DOI] [PubMed] [Google Scholar]
- Xu J, Yeon JE, Chang H, Tison G, Chen GJ, Wands J, de la Monte S. Ethanol impairs insulin-stimulated neuronal survival in the developing brain: role of PTEN phosphatase. J Biol Chem. 2003;278:26929–26937. doi: 10.1074/jbc.M300401200. [DOI] [PubMed] [Google Scholar]