

Abstract
Classically, naive T cells recognize a specific peptide-MHC complex resulting in their activation and differentiation. However, it is known that T cells also have the ability to interact productively with variant ligands, indicating a flexibility in TCR Ag recognition. These altered peptide ligands have been shown to trigger responses ranging from complete activation to full inhibition of T cell responses, and thus may play an important role in initiating or sustaining T cell-mediated immunity. We have found that influenza virus-specific CD8+ TCR transgenic T cells differentially respond to a native (agonist) and variant viral epitope, differing in two amino acids that are thought to alter TCR recognition. In response to stimulation with the agonist epitope, these cells activate, proliferate, and differentiate into effector CTLs. Conversely, stimulation with the variant epitope results in activation, proliferation, and development of effector activity followed by rapid and extensive apoptotic cell death. Stimulation of the T cells with the altered ligand results in an inability to sustain the expression of the prosurvival molecules, Bcl-2 and Bcl-xL. These data suggest that the response to the agonist and variant epitopes may reflect TCR avidity-dependent differential signaling through the TCR, resulting either in activation-dependent T cell proliferative expansion and survival or in the accelerated death of acutely activated differentiating T cells. This process of CD8+ T cell activation, proliferation, and differentiation followed by rapid cell death may represent a novel mechanism of altered peptide ligand-induced apoptosis programmed by initial Ag receptor engagement.
The complete activation of CD8+ T lymphocytes requires multiple ligand-receptor interactions focused at the site of contact between the T cell and the APC. These initial receptor-ligand interactions trigger numerous signaling events within the T lymphocytes that culminate in a variety of cellular responses, including proliferation, activation, differentiation, persistence, and death. Optimal T cell stimulation triggers a differentiation program that is associated with the rapid and extensive proliferation of the cells and acquisition of effector activity. This expansion phase is followed by a contraction phase in which many of the activated T cells die, whereas others progress to long-lived memory cells (1, 2)
At least two signals are required for full T cell activation, a primary signal resulting from TCR recognition of specific peptide/MHC complexes and a second costimulatory signal, which often results from the interaction of CD28 on T cells binding the B7 family of molecules on APCs (3). However, additional and potentially equally important receptor:ligand pairs capable of providing costimulatory signals have also been described. Recently, costimulatory members of the tumor-necrosis factor receptor (TNFR) family (e.g., OX40) have been shown to be of particular importance in T cell responses. Studies indicate that costimulatory TNFR-related receptors can provide signals that allow the survival of newly generated effector cells at the peak of primary immune responses (4). Signaling through such receptors has been shown to lead to upregulation of anti-apoptotic members of the Bcl-2 family, such as Bcl-2 and Bcl-xL (5, 6).
The death of activated T cells during an immune response is important in maintaining T cell homeostasis and is also involved in peripheral T cell tolerance and the prevention of autoimmunity. In the regulation of lymphocyte fate, two distinct pathways leading to apoptosis have been described (7). One pathway involves signals that come from outside the T cell (extrinsic pathway) and is initiated by ligation of cell surface-bound death receptors, such as Fas and TNFR-1. Signaling through these receptors activates apoptosis through the recruitment and activation of caspase enzymes. Activated T cells also die through a pathway driven by signals generated within the T cell itself (intrinsic pathway). This pathway involves members of the Bcl-2 family, such as Bcl-2, Bcl-xL, and Bim. Although this intrinsic apoptotic pathway can also result in the activation of caspase enzymes, it is thought to be controlled by the balance between proapoptotic and anti-apoptotic members of the Bcl-2 protein family and is highly focused on the mitochondrion (8, 9).
The selective activation of CD8+ CTL is based on TCR recognition of specific peptide/MHC complexes presented on the surface of APCs. Previously, it was thought that the interaction of the TCRs with a particular peptide/MHC complex would result in an all or none response. It is now clear that the TCR operates not just as a simple on/off switch, but rather can differentially interpret a signal depending on the nature of the ligand. Studies have shown that T cells can interact productively with variant forms of antigenic peptides. These altered peptide ligands (APLs) have been shown to trigger responses ranging from complete activation to full abrogation of T cell responses (10). For example, variant peptides have been shown to stimulate cytokine production in the absence of proliferation in CD4+ T cells and a number of T cell clones (11, 12). Altered peptides have also been identified that act as antagonists of cytolysis and cytokine production by CD8+ T cells (13, 14). APLs can differ from the cognate Ag in all but the most critical TCR contact residues, which indicates a tremendous flexibility in Ag recognition. However, studies have also shown that the TCR is sensitive enough to detect subtle changes in peptide sequence, with as few as one or two amino acid substitutions triggering differential responses. Therefore, further investigation of APLs could provide insight into the mechanisms regulating T cell activation and differentiation.
We have found that naive CD8+ TCR transgenic (Tg) T cells specific for a nonamer peptide from the A/PR/8 strain of influenza virus show markedly different responses to a variant peptide, which differs in two amino acids, from the A/Japan strain of influenza virus. CD8+ Tg T cells stimulated with the agonist epitope activate and mature into effector CTL. In contrast, variant epitope-stimulated T cells undergo activation and maturation into effectors, followed by massive apoptotic cell death. The induction of cell death by the APL is associated with an inability to sustain upregulated expression of Bcl-2 and Bcl-xL. Vector-mediated transduction of either of these anti-apoptotic molecules into the T cells responding to the variant viral epitope suppressed apoptosis, restored cell viability, and continued proliferative expansion of the responding CD8+ T cells. The implications of these findings are discussed.
Materials and Methods
Mice
Clone-4 (CL-4) TCR Tg mice (H-2d; Thy-1.2) (15) were generously provided by Dr. R. W. Dutton (Trudeau Institute, Saranac Lake, NY). Thy-1.1+/+ BALB/c mice were a gift from Dr. R. I. Enelow (Yale University, New Haven, CT). These mice were bred and housed in a pathogen-free facility and used at 8–12 wk old.
Peptides
Synthetic A/PR/8/34 HA peptide spanning residues 533–541 (HA533–541) (IYSTVASSL) and A/Japan/305/57 HA peptide spanning residues 529–537 (HA529–537) (IYATVAGSL) were synthesized by the University of Virginia Biomolecular Research Facility (Charlottesville, VA).
Virus and virus inoculation
The preparation of influenza A/PR/8/34 (PR/8) virus (H1N1) and of influenza A/Japan/305/57 (Japan) virus (H2N2) and mouse inoculation protocol have been described.
CL-4 T cell stimulation
Naive CL-4 CD8+ T cells were purified to ~90% (data not shown) using CD8-conjugated magnetic beads and positive-selection columns from Miltenyi Biotech (Auburn, CA). In some experiments, purified cells were labeled with 5 μM CFDA-SE (CFSE; Molecular Probes, Eugene, OR) and then used for in vitro or in vivo assays.
For in vitro cultures, purified CD8+ T cells were stimulated with 20 Gyirradiated, syngeneic spleen cells that had been pulsed with the indicated doses of HA533–541 or HA529–537 peptide or infected with PR/8 or Japan virus. The cells were washed extensively and then cultured with CD8-purified CL-4 T cells at a stimulator to responder ratio of 20:1. Culture media consisted of IMEM (Invitrogen Life Technologies, Carlsbad, CA) supplemented with 10% FBS, 10 U/ml penicillin G, 10 μg/ml streptomycin sulfate, 2 mM L-glutamine, and 0.05% 2-ME. At the indicated times, cells were analyzed by flow cytometry for surface marker expression and/or functional activity. Adoptive transfer and preparation of tissue lymphocytes was performed as described (16).
Ab labeling and flow cytometry
For cell surface labeling experiments, ~1 × 106 cells were incubated with the following Abs in the presence of anti-CD16/32 (clone 2.4G2): anti-CD8α (clone 53-6.7), anti-CD25 (clone PC61), anti-CD69 (clone H1.2F3), anti-OX40 (clone OX-86), and anti-Thy1.2 (clone 53-2.1) from BD Biosciences (San Jose, CA). Cells were labeled for 45 min at 4°C in staining buffer (PBS with 2% FBS, 0.05% NaN3), washed extensively, and fixed in 2% paraformaldehyde.
To detect FasL (clone MFL3; BD Biosciences), TNFRI (clone 55R-170; Santa Cruz Biotechnology, Santa Cruz, CA), and TNFRII (clone TR75-89; Santa Cruz Biotechnology), cultured CL-4 T cells were stimulated for 6 h with 1 μM HA533–541 and then surface labeled with Ab as described.
To detect intracellular Bcl-xL (clone 54H6; Cell Signaling Technology, Beverly, MA), cultured CL-4 cells were fixed in 1.6% paraformaldehyde and permeabilized in 90% ice cold methanol. Cells were then labeled with Ab as described. Annexin V (BD Biosciences) labeling was performed according to the manufacturer’s protocol.
To detect intracellular cytokines, cultured CL-4 T cells were stimulated for 6 h with H-2Kd –expressing P815 mastocytoma targets in a 1:1 ratio in the presence or absence of 1 μM HA533–541 or HA529–537 peptide (except where noted) and 1 μg/ml brefeldin A. The cells were surface labeled with Ab, as described, and then intracellularly labeled with anti–IFN-γ (clone XMG1.2), anti–TNF-α (clone MP6-XT22), or anti–IL-2 (clone JES6-5H4; BD Biosciences) using BD Biosciences Cytofix/Cytoperm and perm/wash reagents. A similar procedure was used to detect intracellular granzyme B (clone GB12; Caltag Laboratories, Burlingame, CA) and Bcl-2 (clone 3F11; BD Biosciences).
Granule exocytosis (surface CD107a mobilization) was examined using a modification of the protocol described by Betts et al. (17). Activated CL-4 cells were cultured with P815 targets, specific peptide, and monensin (1 μg/ml). PE-conjugated anti-CD107a (1D4B; Santa Cruz Biotechnology) was included at a final concentration of 5 μg/ml during the culture period. Following T cell stimulation, the cells were labeled with Ab specific for surface molecules and fixed in 2% paraformaldehyde.
All samples were acquired on a FACSCalibur flow cytometer using CellQuest software (BD Biosciences). Data analysis was performed with FlowJo software (TreeStar, Ashland, OR).
Fas-induced apoptosis assay
Cultured CL-4 T cells were incubated with 20 μg/ml anti-Fas mAb (Clone Jo2) or Hamster IgG isotype control Ab (clone Ha4/8; BD Biosciences) and 2 μg/ml protein G for 6 h. Apoptosis was then analyzed by Annexin V labeling.
SDS-PAGE and Western blotting
Thy1.2-purified CL-4 T cells from agonist or variant-stimulated cultures were lysed in cell lysis buffer (Cell Signaling Technology). Sample buffer containing 2-ME was added, and cell lysates were boiled before loading equivalent cell numbers onto 12% polyacrylamide gels. Proteins were separated by gel electrophoresis and transferred to polyvinylidene difluoride using a semidry transfer system. Proteins were visualized by staining the blots with anti-Bim (Cell Signaling Technology) or anti–β-actin (Santa Cruz Biotechnology) Abs followed by staining with anti-rabbit IgG-HRP secondary Abs (Cell Signaling Technology). Blots were developed using ECL detection reagents (Amersham Biosciences) and Kodak x-ray film (Rochester, NY). Bands corresponding to BimEL were identified by m.w.
Retroviral transduction
Murine stem-cell virus IRES GFP (Mig) retroviral expression vectors containing cDNA for Bcl-2 or Bcl-xL were generously provided by Dr. Michael Croft (La Jolla Institute for Allergy and Immunology, San Diego, CA) and used to transduce cells as described. The virus was produced by calcium phosphate transfection of the Plat-E ecotropic packaging cell line. Supernatants were collected after 3 d, titered by measuring GFP expression, and used directly for transducing T cells; 2 × 105 T cells were stimulated with peptide and APCs in 24-well plates as described. After 1 d, the supernatant was replaced with 1 ml viral supernatant containing 5 μg/ml polybrene, and the cells were spun for 1.5 h at 32°C and then incubated at 32°C for 8 h. This procedure was repeated the following day. Viral supernatant was removed and replaced with fresh medium, and T cells were recultured. Expression of GFP was determined by flow cytometric gating on Thy1.2+ cells.
Results
As previously reported, the CL-4 Tg CD8+ T cells express a TCR that recognizes an epitope (residues 533–541) within the trans-membrane domain of the A/PR/8 influenza virus HA primary translation product in association with H-2Kd (15). Stimulation of this T cell population with syngeneic, irradiated splenocyte APCs infected with the A/PR/8 virus triggers robust proliferation that is exemplified by an increase in cell numbers, as determined by trypan blue counting (Fig. 1A). Stimulation of the CL-4 Tg T cells with splenocyte APCs infected with the A/Japan influenza virus, which contains a variant epitope (residues 529–537) that differs by two amino acids from the PR/8 epitope, triggers a similar proliferative response through day 3 poststimulation (Fig. 1A). On day 4 of culture, however, there was a dramatic reduction in the number of CL-4 T cells responding to Japan virus stimulation, with most of the cell loss occurring between days 3 and 4 (Fig. 1A). We considered the possibility that the differential response of the CL-4 T cells could be due to intrinsic properties of the different influenza viruses rather than to specific recognition of the viral epitopes. To address this, we stimulated CD8+ CL-4 T cells with splenocytes pulsed with synthetic PR8 HA533–541 or Japan HA529–537 peptide and examined cell counts by trypan blue exclusion at various times poststimulation (Fig. 1B). We found that stimulation with either the PR8 or Japan peptide induced an increase in cell numbers through day 3 poststimulation; however, there was a significant decrease in cell numbers in response to the Japan variant epitope between days 3 and 4, which was consistent with the results obtained when virus-infected splenocytes were used to stimulate the T cells (Fig. 1B). These data suggest that the differential response of the CL-4 T cells was due to specific recognition of the agonist PR8 and variant Japan viral epitopes, and that Japan epitope stimulation results in the death of the responding CL-4 T cells. It should be noted that the Japan HA529–537 epitope efficiently binds to H-2Kd and serves as an agonist to stimulate a vigorous in vivo primary anti-viral CD8+ effector T cell response (18)
FIGURE 1.
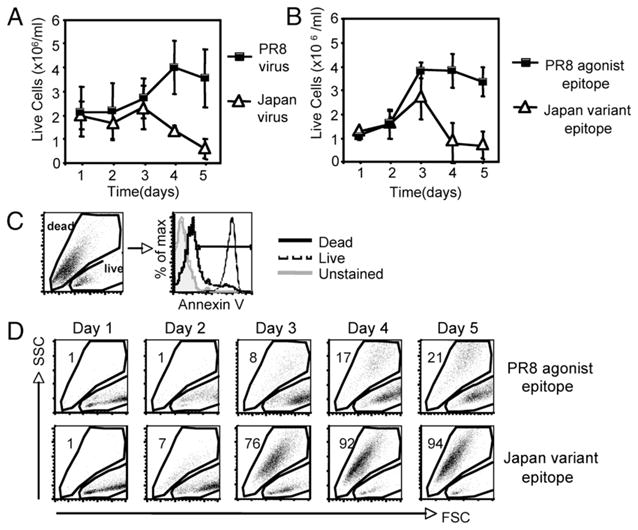
Stimulation with a variant viral epitope induces enhanced apoptosis in CD8+ CL-4 T cells. CL-4 T cells were stimulated in vitro with A/PR/8/34 or A/Japan/305/57 influenza virus-infected splenocytes (A) or with 10−5 M (B) or 10−6 M (C, D) PR/8 HA533–541 or Japan HA529–537 peptide-pulsed splenocytes. A and B, Viable cell counts were calculated using trypan blue exclusion at the indicated times poststimulation. Error bars represent SD. C, Detection of apoptotic cells by annexin V labeling of CL-4 T cells stimulated with Japan HA529–533 peptide. Histograms indicate annexin V staining on cells within the indicated gate at day 3 poststimulation. D, Forward and side scatter properties of CL-4 T cells poststimulation. Plots represent CD8+/CFSE+ cells. Numbers indicate percentage of cells within the dead gate.
We next assessed the viability of the CD8+ CL-4 T cells stimulated with the agonist or variant epitopes by flow cytometric detection of their forward and side light-scattering properties. To first establish the scatter profile of live and dead cell populations, fluorescently conjugated Annexin V was used to label apoptotic cells. As shown in Fig. 1C, all of the T cells within a traditional lymphocyte or live cell gate were not efficiently bound by annexin V, confirming that they were live lymphocytes. In contrast, cells with reduced forward scatter (smaller size) and increased side scatter (higher granularity) bound annexin V well. We termed this a dead gate (Fig. 1C). Using this defined live and dead cell gating strategy, we then examined the survival of the CD8+ CL-4 T cells after stimulation with either the PR8 HA533–541 or Japan HA529–537 peptide (Fig. 1D). At early times poststimulation (days 1–2), T cells responding to either the agonist or variant peptide were present within the live lymphocyte gate. However, obvious differences in the culture were observed at later time points. Although the majority of agonist epitope-stimulated T cells remained in the live lymphocyte gate until day 5 poststimulation, >90% of variant epitope-stimulated T cells accumulated in the dead cell gate by day 4 (Fig. 1D). These initial observations suggest that stimulation with the variant viral epitope induces accelerated and enhanced apoptosis of CD8+ CL-4 T cells.
Because these initial in vitro observations could be related to the nonphysiologic stimulation of CL-4 T cells with high doses of synthetic peptide or virus, we next sought to determine whether CL-4 T cells would undergo extensive cell death in response to natural virus infection in vivo. To this end, we adoptively transferred CD8-purified, CFSE-labeled Thy1.2+ CL-4 T cells into naive Thy1.1+ recipient mice, allowing early detection and quantitation of donor CD8+ T cells in vivo based on Thy1.2 iso-form expression. One day after cell transfer, recipient mice were infected intranasally with a sublethal dose of PR/8 or Japan influenza virus. The total numbers of transferred CD8+ Tg T cells in the draining mediastinal lymph nodes (MLNs), lung, and spleen were determined on successive days postinfection (Fig. 2). As previously reported, donor T cells were detectable throughout the lymphoid tissue of recipient mice, including the lung-draining MLNs, peripheral nondraining lymph nodes, and spleen after transfer (data not shown) (16). The onset of donor T cell proliferation in the MLNs occurred at day 3 postinfection and continued through days 4–5. Although both the Japan and PR8 viruses triggered similar CL-4 T cell proliferative responses in the MLNs at day 4 postinfection, there was a dramatic reduction in the total number of CL-4 T cells in the MLN 1 d later in Japan virus-infected compared with PR8 virus-infected mice (Fig. 2). We reasoned that the loss of CL-4 T cells in the MLNs of Japan-infected mice could result from the migration of these cells to other organs, such as the lung or spleen; however, the low numbers of Tg T cells present in these organs at day 5 postinfection could not account for the significant loss of cells from the MLN between days 4 and 5 postinfection (Fig. 2). These results demonstrating a selective loss of CD8+ CL-4 T cells responding to natural Japan virus infection in vivo suggest that the enhanced apoptosis we observed in vitro is a physiologic response of the these cells to stimulation with the variant Japan epitope and not an artifact of high-dose peptide stimulation.
FIGURE 2.
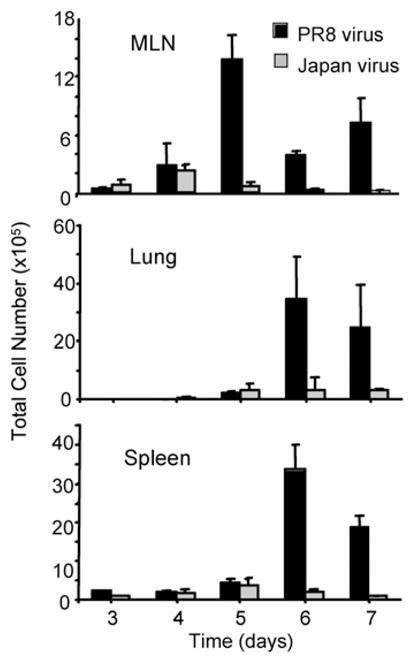
Selective loss of CD8+ CL-4 T cells in response to the altered Japan epitope following natural virus infection in vivo. Purified CD8+ Thy1.2+ splenic CL-4 T cells were labeled with CFSE and injected i.v. into Thy1.1+ recipient mice that were infected i.n. with PR/8 or Japan virus 1 d later. At the indicated times postinfection, MLNs, lungs, and the spleen were harvested, and the absolute numbers of donor CL-4 T cells were calculated from organ cell counts and CD8+ Thy1.2+ percentages. Error bars represent SD.
Studies have suggested that some APLs may alter the outcome of T cell priming by providing a weak or suboptimal TCR signal. Therefore, we considered the possibility that the marked difference in viability of the CL-4 T cells stimulated with the agonist and variant viral peptides might result simply from the Japan peptide triggering suboptimal TCR stimulation. If this were the case, we hypothesized that stimulation of the CL-4 T cells with lower doses of the agonist PR8 peptide should mimic the phenotype of variant epitope-stimulated cells. To address this, we examined the response of CD8+ CL-4 T cells after stimulation with splenocytes pulsed with a range of PR8 HA533–541 or Japan HA529–537 peptide concentrations (10−9–10−5 M; Fig. 3A).
FIGURE 3.

Peptide dose response of CD8+ CL-4 T cells. A, CL-4 T cells were stimulated in vitro with the indicated concentrations of PR/8 HA533–541 or Japan HA529–537 peptide-pulsed splenocytes. At the indicated times poststimulation, viable cell counts were calculated using trypan blue exclusion. B, CL-4 T cells were stimulated in vitro with PR/8 HA533–541 for 5 d, then the effector CL-4 T cells were washed and restimulated with indicated concentrations of PR/8 HA533–541 or Japan HA529–537 peptide in the presence of Golgi-Stop for 5 h. The production of IFN-γ by CL-4 cells was then measured by intracellular staining. The percentages of IFN-γ+ cells in the total CL-4 cells are depicted. C, CL-4 T cells were stimulated in vitro with 10−6 M Japan HA529–537 peptide-pulsed splenocytes in the absence or presence of hIL-2 (40 U/ml). Forward and side scatter properties of CL-4 T cells poststimulation are depicted. Numbers indicate percentage of cells within the dead gate.
Although lower doses (10−9–10−8M) of the variant peptide appear to be less efficient at triggering the CL-4 T cells than the agonist peptide at these same concentrations, we were unable either to reverse the accelerated apoptosis observed following stimulation with the variant epitope by increasing variant peptide concentrations used for stimulation or to accelerate the apoptosis of naive CL-4 T cells stimulated with the agonist epitope at low peptide concentrations (Fig. 3A). At peptide concentrations <10−9 M, the variant Japan epitope was minimally stimulatory, whereas the agonist PR8 epitope decreased in stimulation efficiency (i.e., the fraction of CL-4 T cells responding at progressively lower peptide concentrations) (data not shown).
As discussed below, CL-4 T cells stimulated with the agonist PR8 epitope differentiated into effector T cells. This provided an opportunity to determine whether these activated CL-4 effector cells differed in their functional avidity for the agonist and variant epitopes. To this end, we evaluated the peptide dose-dependent of IFN-γ production by the activated CL-4 effector cells in the intracellular cytokine assay. As Fig. 3B demonstrates, whereas both ligands elicited comparable IFN-γ production at higher peptide doses, the efficiency of recognition of the variant Japan epitope decreased significantly in the triggering of IFN-γ production at peptide concentrations below 10−9 M. This finding suggested that the CL-4 T cells differed in functional avidity for the agonist and variant epitopes.
IL-2 has been well recognized to have both pro-proliferative and anti-apoptotic effects on responding T cells (as well as the capacity to prevent or reverse APL-induced anergy) following engagement of the high affinity IL-2 receptor. We asked whether accelerated apoptosis induced by stimulation of naive CL-4 T cells with variant epitopes could be reversed by stimulation of the T cells with the variant epitope in medium supplemented with exogenous IL-2. As Fig. 3C demonstrates, IL-2 supplementation of cultures at 40 U/ml had no effect on T cell viability or the tempo of induction of apoptosis. Similar results were obtained over a range of IL-2 concentrations (10–100 U/ml, data not shown).
The proliferation of naive T cells responding to specific Ag is thought to be linked to their differentiation and acquisition of effector activity. To characterize the response of CD8+ CL-4 T cells to agonist and variant epitope stimulation in vitro, we first analyzed their proliferative capacity by CFSE dilution. The agonist and variant peptides triggered a similar onset and rate of cell division in CD8+ CL-4 T cells (Fig. 4). In both cases, the T cells began proliferating at day 2 and had undergone 6 divisions by 3 d poststimulation. However, there was a significant loss of divided (CFSE-low) CL-4 T cells at days 3–4 in cultures stimulated with the variant peptide, whereas cultures stimulated with the agonist peptide continued to accumulate cells (Fig. 4A). Similar data were obtained using virus-infected splenocytes as stimulators (Fig. 4B). These results are consistent with the trypan blue cell count data in Fig. 1 and suggest that although the initial proliferation of CD8+ CL-4 T cells responding to the altered influenza epitope is normal, the survival of these cells is markedly impaired.
FIGURE 4.

CD8+ CL-4 T cells proliferate comparably in response to agonist and variant epitope stimulation. CFSE-labeled CD8+ CL-4 T cells were stimulated in vitro with 10−6 M PR/8 HA533–541 or Japan HA529–537 peptide-pulsed splenocytes (A) or with A/PR/8/34 or A/Japan/305/57 influenza virus-infected splenocytes (B). Division was analyzed by flow cytometry and CFSE dilution at the indicated times poststimulation. Histograms represent CD8+ cells within the live lymphocyte gate.
To further evaluate the effects of agonist and variant epitope stimulation on CL-4 T cells, the cells were examined for their expression of proteins whose expression is tied to the activation/differentiation state of the cells following antigenic stimulation. As shown in Fig. 5A, CD8+ CL-4 T cells from both agonist and variant epitope-stimulated cultures upregulated the critical activation markers, CD25 and CD69, to a similar extent at days 1–2 post-stimulation. Likewise, agonist and variant peptide-stimulated CL-4 T cells were able to rapidly produce IFN-γ, TNF-α, and IL-2 early after stimulation, suggesting that these cells had become mature effectors (Fig. 5A). Cytotoxicity is another hallmark feature of mature effector CD8+ T cell differentiation, with cytolysis linked to the expression of lytic granule-associated molecules, such as granzyme B (19). The agonist and variant viral peptides triggered similar increases in the expression of intracellular granzyme B in CD8+ CL-4 T cells early after stimulation (Fig. 5A). In addition, CD8+ CL-4 T cells responding to either the agonist or variant epitope upregulated cell surface CD107a (LAMP-1a), which is mobilized from intracellular granule stores to the cell surface of CD8+ T cells in response to acute TCR engagement and serves as a surrogate for granule exocytosis-dependent cytotoxicity (Fig. 5B). These data indicate that both the agonist and variant influenza epitopes are capable of driving the differentiation of CD8+ CL-4 T cells, culminating in the acquisition of effector activity.
FIGURE 5.
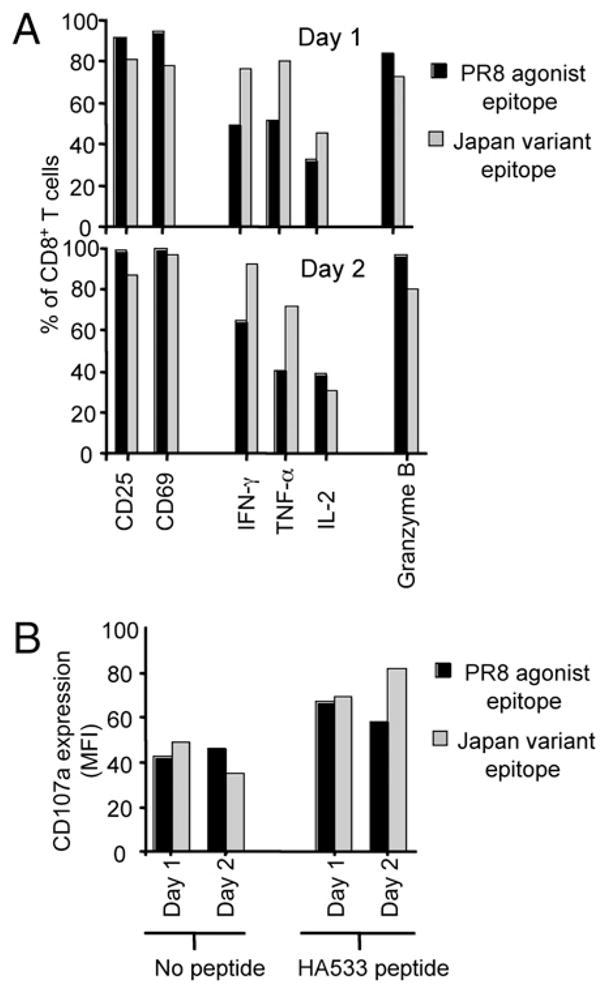
Agonist and variant epitope-stimulated CD8+ CL-4 T cells differentiate into mature effector CTLs. CD8+ CL-4 T cells were cultured with 10−6 M PR/8 agonist or Japan variant peptide-pulsed splenocytes and analyzed by flow cytometry for the expression of surface and intracellular markers at day 1 and day 2 poststimulation. CD69, CD25, IFN-γ, TNF-α, IL-2, and Granzyme B production (A) and surface CD107a upregulation (B) was assayed following 6 h stimulation with 1 μM PR/8 HA533–541 peptide. The y-axis scale indicates percentage of live-gated CD8+ T cells.
Our findings to this point indicate that variant epitope stimulation of CD8+ CL-4 T cells results in activation, proliferation, and differentiation into mature effector CTLs followed by rapid and extensive apoptotic cell death. We next wanted to investigate the possible causes of the enhanced apoptosis of variant ligand-stimulated CL-4 T cells. Members of the TNFR family known as death receptors, including FasL and TNFRI, have been demonstrated to mediate apoptotic signals in lymphocytes (20). We had previously implicated Fas/FasL-dependent interactions between responding CD8+ T cells and dendritic cells in the control of effector T cell numbers in vivo following high-dose lethal influenza virus infection (21). To determine whether death receptor members of the TNFR superfamily may be playing a role in the marked apoptosis of CD8+ CL-4 T cells, we examined the surface expression of Fas, FasL, TNFRI, and TNFRII at various time points following antigenic stimulation (Fig. 6A). We observed no difference in the level of expression of any of these molecules on CL-4 T cells stimulated with either agonist or variant peptide (Fig. 6A, and data not shown). Although the expression levels were similar, it was possible that a differential sensitivity to signaling through these receptors might explain the enhanced apoptosis. Therefore, we examined the sensitivity of agonist and variant epitope-stimulated CL-4 T cells to Fas-induced cell death after incubation with an anti-Fas mAb (Clone Jo2; Fig. 6B). Using annexin V as a marker for apoptotic cells, no difference in the sensitivity of agonist or variant epitope-stimulated Tg T cells to Fas-induced apoptosis was observed (Fig. 6B). These data suggest that death receptor/ligand interactions on agonist and variant epitope-stimulated CL-4 T cells may not be involved in the enhanced apoptosis of variant ligand-stimulated CD8+ T cells.
FIGURE 6.
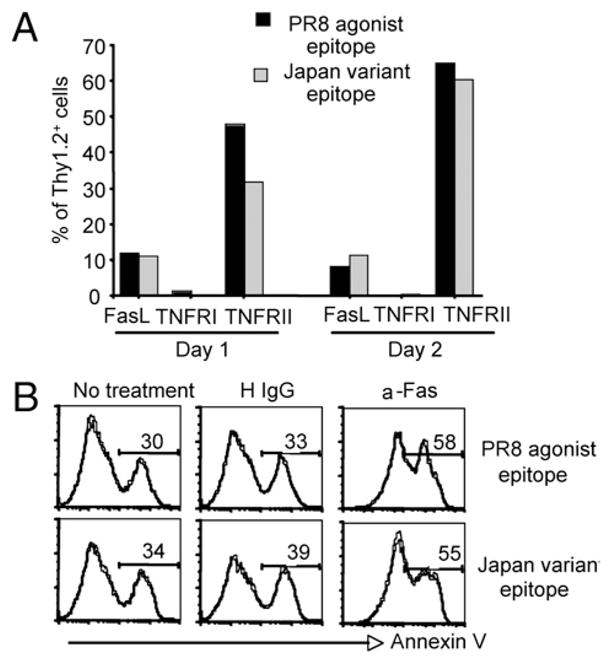
Role of death receptors in the enhanced apoptosis of CD8+ CL-4 T cells responding to variant epitope stimulation. CD8+ CL-4 T cells were stimulated in vitro with 10−6 M PR/8 agonist or Japan variant peptide-pulsed splenocytes. A, At day 1 and day 2 poststimulation FasL, TNFRI, and TNFRII surface expression was analyzed by flow cytometry after 6 h stimulation with 1 μM PR/8 HA533–541 peptide. The y-axis scale indicates percentage of live-gated Thy1.2+ cells. B, At day 2 post-stimulation, sensitivity to Fas-induced cell death was assessed by annexin V labeling following 6 h incubation with 20 μg/ml of anti-Fas mAB (Jo-2) and 2 μg/ml protein G. Histograms represent Thy1.2+ cells. Numbers indicate percentage of annexin V positive cells.
It has been reported that several costimulatory members of the TNFR superfamily may be important regulators of Ag-driven CD4+ and CD8+ T cell survival (4). We surveyed the expression of several of these costimulatory receptors (including CD27, 4-1 BB, and OX40) on CL-4 T cells responding to agonist and variant peptide stimulation. We observed no difference in the expression of the majority of these costimulatory receptors (e.g., CD27, 4-1BB) between the two responding T cell populations (data not shown). However, we found that T cells stimulated with the agonist peptide upregulated expression of OX40 by day 1 post-stimulation and maintained elevated OX40 expression until day 4 of culture (Supplemental Fig. 1, data not shown). By contrast, OX40 was only marginally upregulated on the majority of variant peptide stimulated CL-4 T cells, and expression was not sustained (Supplemental Fig. 1). This result raised the possibility that differences in OX40 expression on the agonist and variant peptide-stimulated T cells could account for the difference in cell viability between the two activated T cell populations and extent of T cell apoptosis observed. To further explore this possibility, we asked whether blocking the interaction of OX40 displayed on the agonist epitope stimulated T cells with its ligand (OX40L) would enhance apoptosis of the agonist stimulated T cells. We found that the addition of saturating concentrations of a blocking Ab to OX40L did not increase apoptosis of the responding T cells (data not shown). Likewise, our attempt to enhance survival of variant peptide stimulated CL-4 T cells by signaling through the OX40 molecule on these cells also had no effect on the development of accelerated apoptosis (data not shown).
Because survival of activated T cells has been directly linked to level expression of the anti-apoptotic members of the Bcl-2 gene family, we evaluated the kinetics of expression of two critical anti-apoptotic gene products Bcl-2 and Bcl-xL in agonist and variant peptide-stimulated CL-4 T cells by flow-based analysis. We found that agonist-stimulated CL-4 T cells strongly upregulated Bcl-2 expression beginning at day 2 and maintained this elevated level until day 4 poststimulation (Fig. 7A, data not shown). In contrast, variant stimulated CL-4 T cells, which expressed considerably lower levels of OX40, also had significantly reduced levels of Bcl-2 for the duration of culture (Fig. 7A). Although both agonist and variant epitope-stimulated CL-4 T cells expressed comparable amounts of Bcl-xL at day 1 poststimulation, only PR8-stimulated cultures maintained expression of this molecule through day 3 poststimulation (Fig. 7A). These findings suggest that the inability of variant epitope-stimulated CL-4 T cells to upregulate and maintain Bcl-2/Bcl-xL expression may lead to defective survival signaling resulting in the enhanced apoptosis of these cells.
FIGURE 7.

Bcl-2 family member expression in agonist and variant epitope-stimulated CD8+ CL-4 T cells. CD8+ CL-4 T cells were stimulated in vitro with 10−6 M PR/8 agonist or Japan variant viral epitopes. A, Cells were analyzed for intracellular Bcl-2 and Bcl-xL expression by flow cytometry. Histograms represent live-gated Thy1.2+ cells. Shaded histograms represent isotype controls. Numbers indicate mean fluorescence intensity. B, At day 1 and day 2 poststimulation, Thy1.2+ cells were purified from culture and lysed in lysis buffer. Equivalent numbers of cells were loaded onto 12% SDS-PAGE gels, and separated proteins were transferred to polyvinylidene difluoride. Proteins were visualized by staining blots with Abs to Bim or β-actin, followed by staining with anti-rabbit IgG-HRP Abs, and developed with ECL substrate. Intensities of Bim-specific bands were measured by densitometry and expressed as arbitrary units normalized to the actin loading controls.
The BH3-only proteins are one of the main proapoptotic subgroups in the Bcl-2 family (22). Because recent evidence has described an important role for the BH3-only protein, Bim, in the death of activated T cells (23, 24), we sought to determine whether Bim levels were changed in CD8+ CL-4 T cells responding to agonist or variant epitope stimulation. To this end, Thy1.2+ CL-4 T cells (the splenocyte stimulators are Thy1.1+) were purified from agonist or variant epitope-stimulated cultures at days 1 and 2 poststimulation, whole-cell lysates were prepared and subjected to SDS-PAGE and Western blotting for Bim. Although alternative splicing can give rise to three Bim variant proteins, T cells express predominantly BimEL, which was the isoform detected in this assay (Fig. 7B). At day 1 poststimulation, both agonist and variant epitope-stimulated CL-4 T cells expressed similar levels of Bim (Fig. 7B). Although Bim expression remained relatively unchanged in variant-stimulated T cells at day 2 poststimulation, there was a significant reduction in the level of Bim expressed in agonist-stimulated cells at the same time interval (Fig. 7B). This decreased level of Bim, in combination with the increased expression of Bcl-2 and Bcl-xL, might provide a selective survival advantage in agonist-stimulated CL-4 T cells.
If anti-apoptotic members of the Bcl-2 family are responsible for the reduced survival of variant epitope-stimulated CL-4 T cells, it should be possible to rescue the rapid apoptosis of these cells by increasing Bcl-2 or Bcl-xL expression. To this end, peptidestimulated CL-4 T cells were transduced with a retrovirus vector expressing Bcl-2 or Bcl-xL and GFP and examined for GFP expression. We would expect that if expression of Bcl-2 or Bcl-xL provided a survival advantage, the proportion of GFP+ cells within the live gate would increase. In CL-4 T cell cultures stimulated with the agonist PR8 epitope and transduced with the control vector, the percentage of GFP+ cells remained constant between days 3 and 4, and there was no significant increase in the proportion of GFP+ cells in cultures transduced with Bcl-2 or Bcl-xL–expressing retrovirus (Fig. 8). In contrast, whereas the percentage of GFP+ cells also remained constant with control vector transduction in variant Japan epitope-stimulated cultures, there was a dramatic increase in the proportion of GFP+ cells between days 3 and 4 after retroviral expression of Bcl-2 and Bcl-xL (Fig. 8). These data support the idea that defective survival of variant epitope-stimulated CL-4 T cells is associated with reduced levels of the anti-apoptotic members of the Bcl-2 family.
FIGURE 8.
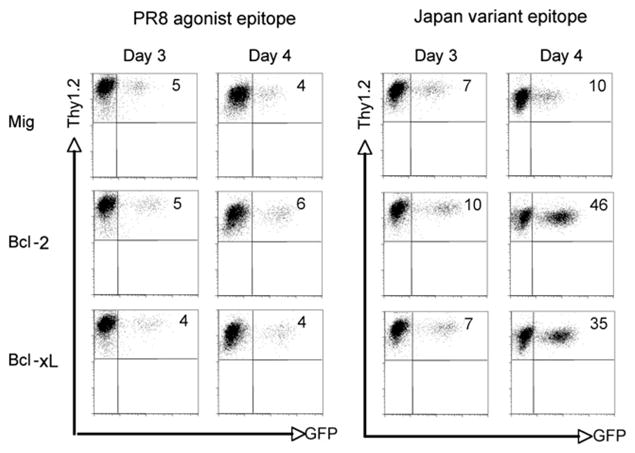
Retroviral expression of Bcl-2 and Bcl-xL suppresses apoptosis of variant epitope-stimulated CD8+ CL-4 T cells. CD8+ CL-4 T cells were stimulated in vitro with 10−6 M PR/8 agonist or Japan variant peptide. On day 1 and 2, cells were transduced with retroviral vectors expressing GFP alone (Mig) or GFP with Bcl-2 or Bcl-xL. T cells were recultured and analyzed for survival by flow cytometry. Numbers represent Thy1.2+/GFP+ cells within the live gate.
Discussion
In this study, we sought to characterize the response of Tg CD8+ CL-4 T cells to stimulation with agonist and variant influenza epitopes. CD8+ CL-4 T cells responding to the variant influenza epitope are fully capable of activating and differentiating into functional effector cells. Like the effector T cells responding to the agonist stimulus, these activated T cells express normal levels of markers of early and late T cell activation and displayed effector activity (e.g., effector cytokine synthesis) characteristic of fully differentiated CD8+ cytolytic T cells. As observed for variant peptides that serve as APLs, activated/effector CL-4 had a lower functional avidity for the variant epitope than for the agonist epitope. However, unlike many characterized APLs that act as partial agonists driving the expression of some but not other effector activities (25), the response of CL-4 T cells to this viral variant epitope not only results in the development of fully differentiated effector cells but also results in the accelerated and enhanced apoptosis of the responding T cells. Furthermore, the development of this enhanced cell death following TCR engagement by the variant epitope is linked to the lack of sustained expression of the anti-apoptotic members of the Bcl-2 gene family, Bcl-2, and Bcl-xL in the T cells.
Variant peptide ligands capable of inducing apoptosis in both CD4+ and CD8+ T cells have been identified; but in the main, these APLs act as partial agonists capable of inducing apoptosis without the concomitant expression of effector functions (25). Our findings and recent results with another APL set (26, 27) represent notable exceptions wherestimulation by an epitopewith low avidity for the TCR results in T cell activation, proliferation, differentiation, and the generation of functional effector cells with extremely limited life spans.
To determine the mechanism to account for the accelerated apoptosis triggered by the variant HA epitope, we first examined the potential contribution of proapoptotic members of the TNFR family. Using the in vivo CL-4 adoptive transfer system and infection with the A/Japan virus expressing the variant epitope, we previously reported that activated effector CL-4 T cells responding to a high-dose lethal infection were more susceptible to Fas/FasL-mediated apoptosis than animals undergoing sublethal infection with this influenza strain (21). When we analyzed proapoptotic TNFR expression on CL-4 T cells induced by the agonist and variant epitope, we found no difference in expression of these receptors or in the susceptibility of either activated T population to death receptor mediated apoptosis. We believe that, in our previous report, the Fas/FasL-mediated apoptosis observed after high-dose lethal infection, which was orchestrated by FasL expressed on respiratory dendritic cells, was likely an infectious virus dose-dependent effect beyond the basal level of apoptosis described in this study and observed after sublethal infection with the virus expressing the variant HA epitope.
Two other potential mechanisms to account for the extensive and accelerated apoptosis demonstrated by the variant epitope-stimulated T cells emerged from the analysis of prosurvival TNFR family members and pro- and anti-apoptotic Bcl-2 family gene products. We observed that OX40 was only minimally and transiently expressed on variant epitope-stimulated CL-4 cells. However, we were unable to demonstrate an effect of OX40 blockade on the development of apoptosis in agonist peptide-stimulated T cells, nor enhanced survival of variant epitopes stimulated T cells with OX40 activation. By contrast, we observed upregulated and sustained expression of the anti-apoptotic Bcl-2 and Bcl-xL gene products after TCR engagement by the agonist epitope with only modest upregulation and transient expression of these anti-apoptotic gene products in the T cells stimulated by the variant epitope. We directly established a role for these two Bcl-2 family members as regulators of apoptosis by retrovirus mediated transduction. Expression of either one of these transduced anti-apoptotic gene products inhibited apoptosis and promoted survival of variant epitope-stimulated T cells, but had no effect on the viability of CL-4 stimulated by the agonist peptide. It is also noteworthy that the proapoptotic (BH3-only) Bcl-2 family member protein, Bim (23), remained elevated in variant epitope stimulated cells, but progressively decreased over 48 h of stimulation with the agonist peptide.
Two recent reports, which analyze the effect of affinity or avidity of TCR-peptide/MHC interaction in vitro and in vivo on T cell signaling efficiency and T cells survival in the OT-1 CD8+ TCR Tg model system, are relevant to our findings (26, 27). In both reports, triggering of naive T cells with peptide epitopes with low functional avidity for the TCR (and where measured, a low affinity of the TCR for the peptide/MHC complex) resulted in T cell activation proliferation and effector cell differentiation, but also accelerated death of the effector cells. As observed in this study, the accelerated death of the variant peptide stimulated T cells was unaffected by the peptide concentration used for stimulation. Similarly, stimulation of the T cells with suboptimal concentrations of the agonist peptide, while decreasing the efficiency of T cell activation, did not result in enhanced cell death. In both the OT-1 model and the CL-4 model examined in this study, the variant epitope like the agonist epitope could trigger efficient initial proliferation of stimulated T cells both in vivo and in vitro. However, we observed the onset of apoptosis after relatively fewer cell divisions (i.e., 4–6) than noted in these reports. As suggested by the findings of Zehn et al. (27), this difference may stem from a relatively low functional avidity of the CL-4 TCR for the variant Japan HA epitope than for the APL used in the OT-1 TCR Tg model. Although neither of these reports formally addresses the mechanism of accelerated or enhanced cell death, our findings suggest that the balance between the expression of pro- and anti-apoptotic Bcl-2 family members might play a critical role in dictating the outcome of T cell stimulation with low-avidity epitopes.
Our results and other related findings (26, 27) are most compatible with the models for T cell recognition and sensing of TCR affinity or avidity in which the duration of TCR engagement of the ligand is sensed and translated as the summation of downstream positive and negative signaling events (28), culminating in T cell activation, proliferation, differentiation, and sustained effector cell viability or, as observed in some instances, accelerated apoptosis. Although not formally established by measurements of the intrinsic affinity of the CL-4 TCR for the variant peptide epitope complex, we believe that the low functional avidity interaction between the T cell and its ligand culminates in a signal of sufficient intensity and duration to allow the signaling associated activation and differentiation events required for effector T cell generation to occur. However, this signaling cascade fails to activate the transcriptional machinery necessary for elevated and sustained expression of anti-apoptotic Bcl-2 family members within the cell (and possibly signaling intermediates required for the inactivation of proapoptotic BH3-only members, such as Bim) (23). Although it is simplest to consider the expression of the anti-apoptotic Bcl-2 gene products to be controlled by signaling events directly linked to TCR engagement, we cannot exclude a role for prosurvival molecules, such as OX40, as direct regulators of the Bcl-2 gene family expression, because their expression mght also be dependent on TCR avidity and TCR signaling efficiency (5).
In conclusion, our results in this viral model support the view that the overall avidity of the TCR interaction with Ag can regulate the differentiation of the mature precursor T cells into cells with effector activity and control the relative contribution of individual T cell clonotypes to the overall response through the early or premature induction of apoptosis. Apoptosis of the responding T cell clones is regulated, in part at least, by the level and duration of expression of anti-apoptotic Bcl-2 family members. Whether high-level, sustained expression of these molecules occurs directly as a consequence of high-avidity TCR binding to its ligand—or indirectly as a result of the engagement of one or more prosurvival signaling molecules whose expression on the activated T cell surface requires high avidtity TCR engagemnent—remains to be determined.
Supplementary Material
Acknowledgments
We thank the members of the Braciale Lab for critical comments and B. Small and T. Gill for technical assistance.
This work was supported by the U.S. National Institutes of Health Grants AI-15608, HL-33391, and AI37293 (to T.J.B.). J.S. is a recipient of a Senior Research Training Fellowship from the American Lung Association.
Abbreviations in this paper
- APL
altered peptide ligand
- MLN
mediastinal lymph node
- Tg
transgenic
- TNFR
tumor-necrosis factor receptor
Footnotes
The online version of this article contains supplemental material.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Kaech SM, Wherry EJ, Ahmed R. Effector and memory T-cell differentiation: implications for vaccine development. Nat Rev Immunol. 2002;2:251–262. doi: 10.1038/nri778. [DOI] [PubMed] [Google Scholar]
- 2.Williams MA, Bevan MJ. Effector and memory CTL differentiation. Annu Rev Immunol. 2007;25:171–192. doi: 10.1146/annurev.immunol.25.022106.141548. [DOI] [PubMed] [Google Scholar]
- 3.Peggs KS, Allison JP. Co-stimulatory pathways in lymphocyte regulation: the immunoglobulin superfamily. Br J Haematol. 2005;130:809–824. doi: 10.1111/j.1365-2141.2005.05627.x. [DOI] [PubMed] [Google Scholar]
- 4.Croft M. Co-stimulatory members of the TNFR family: keys to effective T-cell immunity? Nat Rev Immunol. 2003;3:609–620. doi: 10.1038/nri1148. [DOI] [PubMed] [Google Scholar]
- 5.Rogers PR, Song J, Gramaglia I, Killeen N, Croft M. OX40 promotes Bcl-xL and Bcl-2 expression and is essential for long-term survival of CD4 T cells. Immunity. 2001;15:445–455. doi: 10.1016/s1074-7613(01)00191-1. [DOI] [PubMed] [Google Scholar]
- 6.Song A, Tang X, Harms KM, Croft M. OX40 and Bcl-xL promote the persistence of CD8 T cells to recall tumor-associated antigen. J Immunol. 2005;175:3534–3541. doi: 10.4049/jimmunol.175.6.3534. [DOI] [PubMed] [Google Scholar]
- 7.Rathmell JC, Thompson CB. Pathways of apoptosis in lymphocyte development, homeostasis, and disease. Cell. 2002;109(Suppl):S97–S107. doi: 10.1016/s0092-8674(02)00704-3. [DOI] [PubMed] [Google Scholar]
- 8.Hildeman DA, Zhu Y, Mitchell TC, Kappler J, Marrack P. Molecular mechanisms of activated T cell death in vivo. Curr Opin Immunol. 2002;14:354–359. doi: 10.1016/s0952-7915(02)00335-7. [DOI] [PubMed] [Google Scholar]
- 9.Marrack P, Kappler J. Control of T cell viability. Annu Rev Immunol. 2004;22:765–787. doi: 10.1146/annurev.immunol.22.012703.104554. [DOI] [PubMed] [Google Scholar]
- 10.Evavold BD, Sloan-Lancaster J, Allen PM. Tickling the TCR: selective T-cell functions stimulated by altered peptide ligands. Immunol Today. 1993;14:602–609. doi: 10.1016/0167-5699(93)90200-5. [DOI] [PubMed] [Google Scholar]
- 11.Evavold BD, Sloan-Lancaster J, Hsu BL, Allen PM. Separation of T helper 1 clone cytolysis from proliferation and lymphokine production using analog peptides. J Immunol. 1993;150:3131–3140. [PubMed] [Google Scholar]
- 12.Racioppi L, Ronchese F, Matis LA, Germain RN. Peptide-major histocompatibility complex class II complexes with mixed agonist/antagonist properties provide evidence for ligand-related differences in T cell receptor-dependent intracellular signaling. J Exp Med. 1993;177:1047–1060. doi: 10.1084/jem.177.4.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bachmann MF, Speiser DE, Zakarian A, Ohashi PS. Inhibition of TCR triggering by a spectrum of altered peptide ligands suggests the mechanism for TCR antagonism. Eur J Immunol. 1998;28:3110–3119. doi: 10.1002/(SICI)1521-4141(199810)28:10<3110::AID-IMMU3110>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 14.Jameson SC, Carbone FR, Bevan MJ. Clone-specific T cell receptor antagonists of major histocompatibility complex class I-restricted cytotoxic T cells. J Exp Med. 1993;177:1541–1550. doi: 10.1084/jem.177.6.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morgan DJ, Liblau R, Scott B, Fleck S, McDevitt HO, Sarvetnick N, Lo D, Sherman LA. CD8+ T cell-mediated spontaneous diabetes in neonatal mice. J Immunol. 1996;157:978–983. [PubMed] [Google Scholar]
- 16.Lawrence CW, Braciale TJ. Activation, differentiation, and migration of naive virus-specific CD8+ T cells during pulmonary influenza virus infection. J Immunol. 2004;173:1209–1218. doi: 10.4049/jimmunol.173.2.1209. [DOI] [PubMed] [Google Scholar]
- 17.Betts MR, Brenchley JM, Price DA, De Rosa SC, Douek DC, Roederer M, Koup RA. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods. 2003;281:65–78. doi: 10.1016/s0022-1759(03)00265-5. [DOI] [PubMed] [Google Scholar]
- 18.Lawrence CW, Ream RM, Braciale TJ. Frequency, specificity, and sites of expansion of CD8+ T cells during primary pulmonary influenza virus infection. J Immunol. 2005;174:5332–5340. doi: 10.4049/jimmunol.174.9.5332. [DOI] [PubMed] [Google Scholar]
- 19.Lieberman J. The ABCs of granule-mediated cytotoxicity: new weapons in the arsenal. Nat Rev Immunol. 2003;3:361–370. doi: 10.1038/nri1083. [DOI] [PubMed] [Google Scholar]
- 20.Lenardo M, Chan KM, Hornung F, McFarland H, Siegel R, Wang J, Zheng L. Mature T lymphocyte apoptosis—immune regulation in a dynamic and unpredictable antigenic environment. Annu Rev Immunol. 1999;17:221–253. doi: 10.1146/annurev.immunol.17.1.221. [DOI] [PubMed] [Google Scholar]
- 21.Legge KL, Braciale TJ. Lymph node dendritic cells control CD8+ T cell responses through regulated FasL expression. Immunity. 2005;23:649–659. doi: 10.1016/j.immuni.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 22.Strasser A. The role of BH3-only proteins in the immune system. Nat Rev Immunol. 2005;5:189–200. doi: 10.1038/nri1568. [DOI] [PubMed] [Google Scholar]
- 23.Hildeman DA, Zhu Y, Mitchell TC, Bouillet P, Strasser A, Kappler J, Marrack P. Activated T cell death in vivo mediated by proapoptotic bcl-2 family member bim. Immunity. 2002;16:759–767. doi: 10.1016/s1074-7613(02)00322-9. [DOI] [PubMed] [Google Scholar]
- 24.Pellegrini M, Belz G, Bouillet P, Strasser A. Shutdown of an acute T cell immune response to viral infection is mediated by the proapoptotic Bcl-2 homology 3-only protein Bim. Proc Natl Acad Sci USA. 2003;100:14175–14180. doi: 10.1073/pnas.2336198100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Katsara M, Minigo G, Plebanski M, Apostolopoulos V. The good, the bad and the ugly: how altered peptide ligands modulate immunity. Expert Opin Biol Ther. 2008;8:1873–1884. doi: 10.1517/14712590802494501. [DOI] [PubMed] [Google Scholar]
- 26.Hommel M, Hodgkin PD. TCR affinity promotes CD8+ T cell expansion by regulating survival. J Immunol. 2007;179:2250–2260. doi: 10.4049/jimmunol.179.4.2250. [DOI] [PubMed] [Google Scholar]
- 27.Zehn D, Lee SY, Bevan MJ. Complete but curtailed T-cell response to very low-affinity antigen. Nature. 2009;458:211–214. doi: 10.1038/nature07657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. 2009;27:591–619. doi: 10.1146/annurev.immunol.021908.132706. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.