

Abstract
Thiazoles, oxazole and their corresponding reduced derivatives, thiazolines and oxazolines, are found in marine sources exhibiting significant biological activities. The isolation, synthetic, and biological studies of these natural products, covering literature from January 2007 to June 2010, are summarized.
Keywords: thiazole, oxazole, marine
1. Introduction
The role of natural products in drug discovery has undergone many changes over the past two decades, from a decline in participation by pharmaceutical companies in the 1990s, to a renaissance in recent years. In addition, the expectations from combinatorial libraries in drug screening have not been fulfilled and more than 60% of prescription drugs are of natural product origin. As a consequence, the natural product-inspired drug discovery and development has received renewed attention in recent years.
A large number of natural products, in particular from the marine environment, contain thiazole, oxazole, thiazolines or oxazolines heterocycles. In many cases, promising anti-tumor, antibacterial, anti-viral, anti-malaria and anthelmintic activities have been identified for these compounds. This review focusses on the isolation and structural determination, biological activities and synthetic studies of these marine products covering literature from January 2007 to June 2010.
2. New Thiazoles Isolated from Marine Sources
The known algaecide bacillamide A (1) and two new analogues, bacillamides B (2) and C (3), Figure 1, were isolated from Bacillus endophyticus obtained from a Bahamian hypersaline microbial mat [1]. The detection of these metabolites containing tryptamide thiazole motif, was performed using HPLC-UV-MS bioassay technique. Due to a lack of sufficient material and the appropriate test organisms, bacillamides B and C were not tested for algicidal activity. None of the purified bacillamides demonstrated antibiotic activity against target isolates of hypersaline pond Bacillus sp. at concentrations less than 500 μM.
Figure 1.

Structures of bacillamide A, B and C and neobacillamide A.
A related compound, neobacillamide A (4), Figure 1, together with bacillamide C, was isolated from the bacterium Bacillus vallismortis C89 associated with the sponge Dysidea avara [2]. Neobacillamide A represents the first example of a thiazole-carboxamide bearing a 2-phenylethylamine moiety. Both compounds, Neobacillamide and Bacillamide C were inactive as cytotoxic against HL60 human leukemia cells and A549 human lung cancer cells.
A thiopeptide antibiotic, urukthapelstatin A (5), Figure 2, was isolated from a culture of Thermoactinomycetaceae bacterium Mechercharimyces asporophorigenens YM11-542 [3]. Its structure was determined by spectroscopic methods and chemical degradation, and confirmed by X-ray crystallographic analysis. It is related to 24 membered cyclic compounds mechercharstatin and telomestatin. Urukthapelstatin A inhibited the growth of human lung cancer A549 cells with an IC50 value of 12 nM and also showed potent cytotoxic activity against a human cancer cell line panel.
Figure 2.

Structures of urukthapelstatin A.
Two new cyclic hexapeptides, venturamides A (6) and B (7), Figure 3, were isolated from the anamanian marine cyanobacterium Oscillatoria sp. [4]. Venturamide A and B showed in vitro antimalarial activity against Plasmodium falciparum (8.2 and 5.6 μM respectively), with only mild cytotoxicity to mammalian Vero cells (86 and 56 μM respectively). They exhibited only mild activity against Trypanasoma cruzi and Leishmania donovani.
Figure 3.

Structures of venturamide A and B.
Four new modified hexacyclopeptides, aerucyclamides A (8), B (9), C (10) and D (11), Figure 4, were isolated from the toxic freshwater cyanobacterium Microcystis aeruginosa PCC 7806 [5,6]. These compounds encompass hexameric cyclopeptides alternating in hydrophobic aminoacids and oxazoline, thiazoline, thiazole or oxazole heterocycles derived from hydrophilic aminoacids (Ser, Thr, Cys). Aerucyclamide B displayed a submicromolar (0.7 μM) IC50 value against the chloroquine-resistant strain K1 of P. falciparum. In addition, this compound displays a large selectivity for the parasite with respect to the L6 rat myoblast cell line. Aerucyclamides A, C y D displayed low micromolar activity against P. falciparum. The most active compound against T. brucei rhodesiense, is aerucyclamide C (10), albeit with moderate activity.
Figure 4.

Structures of aerucyclamides A, B, C and D.
A new hexapeptide, Hexamollamide (12), Figure 5, was isolated from an Okinawan ascidian Didemnum molle [7]. The relative stereostructure was confirmed by X-ray crystallographic analysis. Hexamollamide contains a thiazoline ring and a threonine amino acid, which is modified by attachment to a dimethylallyl ether. It is structurally similar to patellin 2 isolated from L. patella [8,9]. Hexamollamide showed moderate cytotoxicity against HeLa S3 cells, with an IC50 value of 17 μg/mL.
Figure 5.

Structure of hexamollamide.
Two new cyclic hexapeptides, mollamides B (13) and C (14), Figure 6, were isolated from Didemnum molle collected from Manado Bay [10]. They contain threonine or serine amino acids, modified by attachment to a dimethylallyl ether. The relative configuration of mollamide B at the thiazoline moiety was determined using molecular modeling coupled with NMR-derived restraints. Their absolute configuration was determined using Marfey’s method. Mollamide B (13) exhibited moderate antimalarial activity against Plasmodium falciparum (D6 clone and W2 clone), with IC50 values of 2.0 and 2.1 μg/mL, respectively. Mollamide B also exhibited marginal activity against Leishmania donovani, with IC50 and IC90 values of 18 and 35 μg/mL and cytotoxicity against several cancer cell lines. It showed neither antimicrobial nor anti-inflammatory activities. Mollamide C has not shown antiinflamatory activity and it was not considered to be solid tumor selective.
Figure 6.

Structures of mollamide B and C.
In 2008, a new metabolite with novel chemical scaffold and nanomolar antiproliferative activity, largazole (15), Figure 7, was isolated from a sample of a cyanobacterium of the genus Symploca collected from Key Largo, Florida Keys, U.S. [11]. It potently inhibited the growth of highly invasive transformed human mammary epithelial cells (MDA-MB-231) (GI50 = 7.7 nM). In contrast, nontransformed murine mammary epithelial cells (NMuMG) were less susceptible (GI50 = 122 nM). The authors suggested that cancer cells are preferentially targeted by largazole. It possesses a combination of unusual structural features, including a substituted 4-methylthiazoline fused to a thiazole, a thioester moiety that has not been reported in metabolites from cyanobacteria and the unprecedented in marine natural products 3-hydroxy-7-mercaptohept-4-enoic acid unit.
Figure 7.

Structure of largazole.
From an environmental assemblage of the marine cyanobacteria Lyngbya majuscula and Phormidium gracile collected in Papua New Guinea, a novel bioactive cyclic depsipeptide, hoiamide A (16), Figure 8, was isolated [12]. The structure was then corrected by the authors in an erratum, showing R configuration at C24 (instead of the S configuration pictured in the original paper) [13]. This metabolite possesses an unusual structure derived from a mixed peptide-polyketide biogenetic origin, and includes three fragments: a triheterocycle bearing two methylated thiazolines and one thiazol, a peptide and an oxygenated and methylated C15-polyketide. Hoiamide A exhibited modest cytotoxicity to cancer cells, but potently inhibited [3H]batrachotoxin binding to voltage-gated sodium channels (IC50 = 92.8 nM) and activated sodium influx (EC50 = 2.31 μM) in mouse neocortical neurons.
Figure 8.

Structure of hoiamide A.
Eight novel thiazole and oxazole containing cyclic peptides, microcyclamides, GL616 (17), GL582 (18), GL614A (19), GL614B (20), GL546A (21), GL546B (22), GL628 (23), GL614C (24), Figure 9, were isolated from the hydrophilic extract of a Mycrocystis sp. water-bloom collected in Gelbea reservoir, Valley of Armagedon, Israel [14]. It is the first example where acidic and modified aminoacids are incorporated in this group of ribosomally biosynthesized metabolites. The compounds were assayed against one solid tumor (A549-lung) and one leukemis (Molt-4) cell lines. Microcyclamides GL582 (18) displayed weak cytotoxicity (20% grow inhibition) against Molt-4 cell line at 10 μM/mL.
Figure 9.

Structures of microcyclamides.
3. Thiazolines Isolated from Marine Sources
A collection of the cyanobacterium Lyngbya confervoides off Grassy Key in Florida, U.S., yielded grassypeptolide (25), Figure 10, a 31-membered macrocyclic depsipeptide with unusually high d-amino acid content, two thiazolines, and one β-amino acid [15]. The structure was confirmed by X-ray crystallography. The antiproliferative activity was evaluated in four cell lines derived from human osteosarcoma (U2OS), cervical carcinoma (HeLa), colorectal adenocarcinoma (HT29), and neuroblastoma (IMR-32). Grassypeptolide showed moderate broad-spectrum activity with IC50 values of 2.2, 1.0, 1.5, and 4.2 μM, respectively.
Figure 10.

Structure of grassypeptolide.
A new thiazoline metabolite, bisebromoamide (26), Figure 11, was isolated from the marine cyanobacterium Lyngbya sp. [16]. Its planar structure was determined by 1D and 2D NMR spectroscopy. The absolute stereostructure was determined by chemical degradation followed by chiral HPLC analysis. It contains a significant number of d-amino acids and N-methylated amino acids along with several other modified amino acid residues. Bisebromoamide exhibited antiproliferative activity at nanomolar levels and potent kinase inhibition.
Figure 11.

Proposed structure in the isolation report of bisebromoamide.
4. Synthesis of Thiazoles of Marine Origin and Analogues
The synthesis of the C1-N15 fragment of the originally proposed scleritodermin A (27), Figure 12, has been reported in good overall yield through a short and stereocontrolled sequence [17]. Highlights of the route include the synthesis of the novel ACT fragment (28) and the formation of the α-ketoamide linkage by the use of a highly activated α,β-ketonitrile.
Figure 12.

Retrosynthetic analysis of scleritodermin A.
In 2008, the first total synthesis of the originally proposed scleritodermin A (27) along with two isomers (29) and (30) were achieved [18]. The application of a transamination reaction was used for the formation of α-ketoamide. The authors reported that 1H and 13C NMR spectra of molecule 27 did not match those reported for natural scleritodermin. In contrast, the spectra of compound 29 and those reported for the isolated marine product were identical. Thus, compound 29 was proposed as the revised structure of scleritodermin A.
Recently, the synthesis of key fragments and open analogs of scleritodermin A and their biological evaluation as cytotoxic and anthelmintic were reported. Open and simplified analogs (31), Figure 13, were obtained in very good yield using a convergent strategy [19]. The compound with the highest antihelminthic activity is the open analog of 27 (LC50 = 9.6 μM). The presence, in some obtained compounds, of α,β-ketoamide function improves the cytotoxic activity on HCT-15 cells.
Figure 13.

Synthesized analogs of scleritodermin A.
Pattellamide A (32), Figure 14, was synthesized from thiazole 33 and protected aminoacids using Burgess reagent to obtain the oxazolines rings. Macrocyclization was performed using PyBOP, DMAP, DIEA in CH2Cl2/DMF [20].
Figure 14.

Synthetic route to patellamide A.
The first total synthesis of Largazole (15), Figure 7, was reported four months after the publication of its isolation [21]. The synthesis (eight steps, 19% overall yield), involved a macrocyclization reaction for formation of the strained 16-membered depsipeptide core followed by an olefin cross-metathesis reaction for installation of the thioester, Figure 15. The biological evaluation of largazole and key analogues, suggested that histone deacetylases (HDACs) are molecular targets of largazole. The authors concluded that the cell growth inhibition is a functional consequence of I HDAC inhibition. In addition, structure-activity relationship (SAR) studies revealed that the thiol group is indispensable for both activities.
Figure 15.

Retrosynthetic analysis of the first total synthetis of largazole.
During 2008 seven more syntheses of largazole were reported [22–28]. Some of the used methodologies differ in the formation of the macrocycle or in the precursor of the 3-hydroxy-7-mercaptohept-4-enoic acid. In addition, some structure activity relationships studies were performed. It was demonstrated that largazole is a pro-drug that is activated by removal of the octanoyl residue from the 3-hydroxy-7-mercaptohept-4-enoic acid moiety to generate the active metabolite 34, Figure 16, which is an extraordinarily potent Class I histone deacetylase inhibitor (Ki = 0.07 nM) [22].
Figure 16.
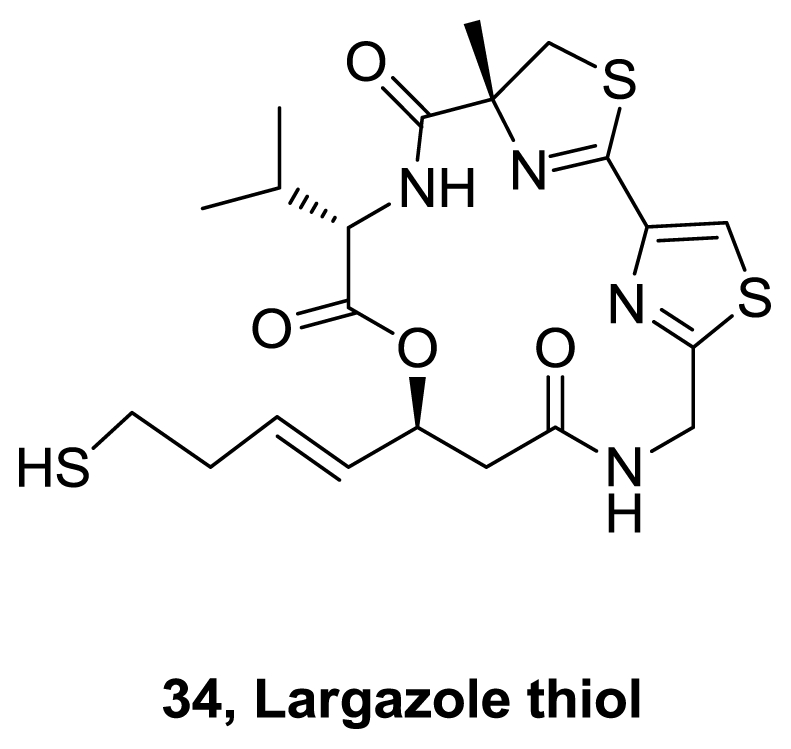
Active metabolite of largazole.
During 2009, six reports related to the synthesis of largazole and/or analogs were published [29–34]. A significant increase in potency with a pyridine substitution of the thiazole was reported. In addition, it was demonstrated that the methyl substituent of the thiazoline ring is nonessential for the dramatic potency of the natural product. In contrast, substitution of the thiazoline by thiazol, diminishes the activity [30].
Recently, new analogs of largazole were synthesized [35,36]. Structure-activity relationship studies suggested that the geometry of the alkene in the side chain is critical. While the largazole’s analogues with trans-alkene are potent for the antiproliferative effect, those with cis-alkene are completely inactive. In addition, replacement of valine by tyrosine increased selectivity toward human cancer cells over human normal cells more than 100-fold [35].
Starting from d-alanine, bacillamide C (3), was obtained in 22% overall yield [37]. The key intermediate, thiazole 35, Figure 17, was synthesized using Hantzsch reaction.
Figure 17.

Thiazole precursor of bacillamide C.
A thiazole Ugi multicomponent reaction and an aminolisis reaction were used to obtain racemic Bacillamide C, Figure 18 [38]. Additionally, several analogs of this natural product and a fluoro derivative of neobacillamide (4), Figure 1, were synthesized.
Figure 18.

Synthesis of bacillamide C.
Recently, the enantioselective total synthesis of trichloroleucine-derived marine natural product neodysidenin (36), Figure 19, was accomplished. The strategy involves a direct ruthenium-catalyzed radical chloroalkylation capitalizing on the valence tautomerism of titanium enolates [39].
Figure 19.

Retrosynthetic analysis of neodysidenin.
5. Synthesis of Thiazolines of Marine Origin and Analogues
The cytotoxic thiazoline, apratoxin A (37), Figure 20, was synthesized in 18 steps and 18% overall yield. In addition, an oxazoline analogue (38) and 34-epiapratoxin A (39) were obtained [40]. The synthetic strategy to obtain apratoxin A involves the preparation of fragment 40 through a stereoselective route based in three asymmetric reactions. Thiazoline formation was successfully accomplished from a Cys amide using Ph3PO/Tf2O and macrolactamization was performed between N-methylisoleucine and proline residues using HATU.
Figure 20.

Apratoxin A analogs and retrosynthetic analysis of apratoxin A.
Recently, the first total synthesis and a revised configurational assignment of bisebromoamide were reported [41]. The authors synthesized the originally proposed structure (26), Figure 11, but, neither NMR spectra nor optical rotation for the obtained compound were identical with those of the natural product. The authors elected to synthesize the epimer with R-configuration of the stereogenic center at the thiazoline ring of the proposed structure (41), Figure 21, from different fragments as is shown in the retrosynthetic analysis. The spectral data and optical rotation for 41 were identical to those of the natural product leading to a revision of the reported stereochemistry of bisebromoamide.
Figure 21.

Retrosynthetic analysis of bisebromoamide.
6. New Oxazoles Isolated from Marine Sources
A new marine-derived macrolide designated as neopeltolide (42), Figure 22, was isolated from a deep-water sponge of the family Neopeltidae. Its structure was elucidated on the basis of spectroscopic data interpretation. Neopeltolide is a potent inhibitor of the in vitro proliferation of the A-549 human lung adenocarcinoma, the NCI-ADR-RES human ovarian sarcoma, and the P388 murine leukemia cell lines, with IC50 of 1.2, 5.1, and 0.56 nM, respectively. 42 also inhibited the growth of the fungal pathogen Candida albicans with a minimum inhibitory concentration of 0.62 μg/mL [42].
Figure 22.

Structure of neopeltolide.
Ariakemicins A (43) and B (44), Figure 23, unusual linear hybrid polyketide-nonribosomal peptide antibiotics, were discovered from the fermentation extract of the marine gliding bacterium Rapidithrix sp. [43]. These metabolites were positional isomers with regard to a double bond and chromatographically inseparable, rendering the structure study on a mixture basis. The antibiotics selectively inhibited the growth of Gram-positive bacteria.
Figure 23.

Structure of ariakemicins A and B.
Bioassay-guided fractionation of extract of the sponge Dorypleres splendens has led to the discovery of a new natural source of bengazole A (45), B (46), and E (47), Figure 24, together with bengamide A. It had been thought that these compounds were confined to Jaspis species. This is the first time that bengazoles were isolated from another genus. Those compounds showed growth inhibitory activity to seven murine and human cancer cell lines [44].
Figure 24.

Structure of bengazoles A, B and E.
An oxazole lipodepsipeptide, taumycins A (49), Figure 25, were isolated from the Madagascar sponge Fascaplysinopsis sp. with other closely related no oxazole containing metabolite taumycine B. The two compounds have the same 12-membered oxodepsipeptide ring system in common. Both were toxic to brine shrimp larvae, and taumycin A (1 μM), but not taumycin B, inhibited growth of the human UT-7 leukemic cell line. The structure of the two compounds, likely to be derived from microorganisms, was established by MS and 1D and 2D NMR data [45].
Figure 25.

Structure of taumycin A.
A novel oxazole macrolide, salarin C (50), Figure 26, was isolated from the Madagascar sponge Fascaplysinopsis sp. The structure of the compound was elucidated by interpretation of MS and 1D and 2D NMR spectra. Salarin C (50) is closely related to salarin A and is considered to be the precursor of salarins A and B. Air oxidation was found to transform 50 to salarin A. 50 was found to inhibit cell proliferation of human leukeamic cell lines UT-7 and K562, and the murine pro-B cell line Ba/F3 at concentrations of 0.0005–0.5 μg/mL [46].
Figure 26.

Structure of salarins C, F and I.
Recently, seven new nitrogenous macrolides, designated salarins D-J, closely related to salarins A-C, were isolated from the same source. Salarin F (51) and I (52), Figure 26, have the same oxazole macrolide that 50. All compounds were evaluated for their cytotoxicity against K562 and UT-7 human leukemia cells. While salarins D, E, H, and J displayed dose- and time-dependent inhibition of proliferation, 51 and 52 were not active in these assays [47].
Two new trisoxazole macrolides, 9-O-desmethylkabiramide B (53) and 33-methyltetrahydrohalichondramide (54), Figure 27, were isolated in submicromolar amounts together with two new thiazole-containing cyclic peptides from a single specimen of Hexabranchus sanguineus, a nudibranch from the Indo-Pacific. The structure elucidation of these very minor metabolites was preformed using a 1 mm 1H NMR high-temperature superconducting microcryoprobe with unmatched sensitivity [48].
Figure 27.

Structure of trisoxazole macrolides.
Three new macrocyclic peptides, diazonamides C–E (55–57), Figure 28, were isolated together with the previously reported diazonamides A and B from samples of the marine ascidian Diazona sp. collected in Indonesia. The cytotoxic activity of the new compounds and diazonamide A was evaluated against a panel of three human tumor cell lines, including lung (A549), colon (HT29), and breast (MDA-MB-231). Moderate cytotoxicity with GI50 values in the micromolar range was found for the new compounds; whereas the potent activity previously reported for diazonamide A was confirmed with values in the low nanomolar range [49].
Figure 28.

Structure of diazonamides.
Enigmazole A (58), a novel phosphate-containing macrolide, was isolated from a Papua New Guinea collection of the marine sponge Cinachyrella enigmatica [50]. Compound 58 is comprised of an 18-membered phosphomacrolide that contains an embedded exomethylene-substituted tetrahydropyran ring and an acyclic portion that spans an oxazole moiety. Two additional analogues, 15-O-methylenigmazole A and 13-hydroxy-15-O-methylenigmazole A, were also isolated. The enigmazoles are the first phosphomacrolides from a marine source and 58 exhibited significant cytotoxicity in the NCI 60-cell line antitumor screen, with a mean GI50 of 1.7 μM.
7. Synthesis of Oxazoles of Marine Origin and Analogues
The relative stereochemistry of the metabolite (+)-neopeltolide (42), Figure 22, was originally proposed based on 1D and 2D NOESY spectra, which ultimately led to the incorrect structural assignment. The enantioselective total synthesis, with reassigned stereochemical and absolute configuration for neopeltolide (59), Figure 30, and their diastereomers has been reported almost simultaneously by Panek [51] and Scheidt [52].
Figure 30.

Revised structure of neopeltolide.
Retrosynthetic strategy by Panek, Figure 31, began with disconnection of the C19–C20 double bond to reveal the macrolide 60 and the oxazole side chain 61. Synthetic highlights of this route include a modified Evans-Tishchenko reduction to introduce the C11 stereocenter, [4 + 2] annulation to construct the pyran system, and a Still-Gennari olefination to install the oxazole side chain. Macrocyclization was performed through Yamaguchi esterification of the seco acid intermediate to form macrolide 60 in 44% yield. The longest linear sequence required 19 steps with an overall yield of 1.3%.
Figure 31.

Retrosynthetic analysis of neopeltolide by Panek.
Scheidt and co-workers performed a macrocyclization of the linear precursor 62, Figure 32, constructed from dioxinone acid 63 and alcohol 64. In this key step, scandium-(III) triflate promoted the macrocyclization of 62 in a Prins type transformation. The synthetic neopeltolide inhibited proliferation of the murine leukemia line P388 with an IC50 of 0.6 nM and the human breast adenocarcinoma line MCF-7 with an IC50 value of 2.2 nM. Validating the revised structure as the correct one; diastereomer which corresponded to the originally proposed structure, was approximately 100-fold less active in both cell lines [53].
Figure 32.

Retrosynthetic analysis of neopeltolide by Scheidt group.
Five total syntheses of 59 have been reported in 2008 [54–58] and they were reviewed by Sasaki [59]. Since then, another four total syntheses were reported [60–64].
The first total synthesis of the cytotoxic marine macrolide enigmazole A (58), Figure 29, was completed in 22 steps (longest linear sequence). The sensitive, densely functionalized 2,4-disubstituted oxazole fragment (64), Figure 33, was constructed using a Negishi-type coupling of an oxazol-2-ylzinc reagent formed directly from the parent ethyl 2-iodooxazole-4-carboxylate by zinc insertion. Other key steps include a hetero-Diels-Alder cycloaddition to form the central embedded pyran ring, a Wittig reaction to unite the hemispheres, and a ring size-selective Keck macrolactonization [65].
Figure 29.

Structure of enigmazole A.
Figure 33.

Retrosynthetic analysis of enigmazole A by Molinski.
Stereochemistry of (−)-ulapualide A (65), Figure 34, was revised by X-ray analysis of its complex with the protein actin. A new total synthesis of 65 was reported, the first to yield a product that corresponds with the natural product. The synthesis involved the macrolactamization of the intermediate 66, followed by elaboration of the central oxazole ring in 65 as a late step in the overall synthesis [66,67].
Figure 34.

Structure of ulapualide A and their retrosynthetic analysis.
An enantioselective, convergent, total synthesis of the antiviral marine natural product (−)-hennoxazole A (67), Figure 35, was completed in 17 steps, longest linear sequence, from serine methyl ester and in nine steps from an achiral bis-oxazole intermediate. Elaboration of a thiazolidinethione allowed for rapid assembly of the pyran-based ring system. Key late-stage coupling was effected by deprotonation of the bisoxazole methyl group, followed by alkylation with an allylic bromide side chain segment [68,69].
Figure 35.

Structure of hennoxazole A.
Siphonazoles A (68) and B (69), Figure 36, are structurally novel natural products isolated from a Herpetosiphon species [70]. They are the first, and so far the only, naturally occurring substances known that incorporate oxazole subunits connected by a two-carbon tether.
Figure 36.

Structure of siphonazoles A–B and their retrosynthetic analysis by Ciufoloni group.
The first synthesis of a siphonazole was performed by Moody and Linder [71,72]. Key aspects of this work are the assembly of oxazole units via the reaction of rhodium carbenes with an amide or a nitrile, and the construction of the central (2-oxazolyl)methyl ketone motif by acylation of an organozinc agent derived from 4-carbomethoxy-2-iodomethyl-5-methyloxazole.
Other synthesis of siphonazoles was performed by Ciufoloni and co-workers involving the iterative use of a conjunctive oxazole building block that acts as a carrier of synthon 70, Figure 36. Synthetic siphonazoles were devoid of antimicrobial activity, but they showed appreciable and selective cytotoxicity against human breast carcinoma HTB-129 (IC50 of 20 μg/mL) and acute T-cell leukemia TIB-152 (IC50 value equal to 16 μg/mL), with 68 being significantly more active than 69 [73].
The bengazoles are a family of metabolites isolated from a Jaspis sponge that display potent antifungal and anthelmintic activity, and have a unique structure containing two oxazole rings flanking a single carbon atom. Three new total synthesis of bengazoles [74–76] and analogs [77–79] were reported.
The marine alkaloid almazole C (71), Figure 37, isolated from the red seaweed Haraldiophylum sp. was synthesized in four steps. The key step, construction of the central 2,5-disubstituted oxazole ring, was based on the aza-Wittig reaction of the iminophosphorane derived from the α-azidoacetyl indole and (S)-N-phthaloylphenylalanyl chloride [80].
Figure 37.

Structure of almazoles.
The structure of Almazole D (72), Figure 37, antibacterial metabolite isolated from a red seaweed from the Dakar coast [81], was revised by Horne and co-workers to 73. Synthesis of 73 was performed by coupling the oxotryptophan methyl ester with N,N-dimethyl-L-phenylalanine and subsequent treatment with POCl3 [82].
The complex structure of phorboxazoles and their unique biological activity is a driving force for continued interest from the synthetic community. The phorboxazoles are among the most potent discovered cytostatic agents, exhibiting a mean GI50 < 1.58 × 10−9 M against the NCI panel of 60 tumor cell lines. Phorboxazole B (74), Figure 38, was prepared in 55 steps (longest linear sequence 28 steps) using a stereoselective hetero-Diels-Alder reaction to construct the key C33–C39 segment. The stereoselective hetero-Diels-Alder reaction of (R)-2,3-dibenzyloxypropanal and the Brassard diene EtO(Me3SiO)C:CHC(OMe):CH2 in presence of Eu(fod)3 provided dihydropyranone I, enabling the synthesis of the C18–C46 fragment of phorboxazole B. Coupling of the C18–C46 fragment with a suitable C3–C17 partner, followed by the late-stage formation of the C16–C18 oxazole unit and stereoselective macrocyclization and deprotection, rendered synthetic phorboxazole B [83].
Figure 38.

Structure of phorboxazoles.
Highly convergent syntheses of eight phorboxazole congeners and their evaluation against a diverse panel of human solid tumor cancer cell lines have been achieved. Specifically, the C(45–46) alkyne, alkene, and alkane phorboxazole A analogs were constructed and found to display single digit nanomolar cell growth inhibitory activities in a series of human cancer cell lines. The structurally simplified C(11–15)-acetal congener also proved potent, albeit reduced, when evaluated against the same cell line panel. Importantly, (+)-C(46)-chlorophorboxazole A displayed picomolar (pM) inhibitory activity in several cell lines [84].
A highly convergent second-generation synthesis of (+)-phorboxazole A (75), Figure 38, was achieved. Highlights of the synthetic strategy include improved Petasis-Ferrier union/rearrangement conditions on a scale to assemble multigram quantities of the C(11–15) and C(22–26) cis-tetrahydropyrans inscribed with the phorboxazole architecture, a convenient method to prepare E and Z-vinyl bromides from TMS-protected alkynes utilizing radical isomerization of Z-vinylsilanes, and a convergent late-stage Stille union to couple a fully elaborated C(1–28) macrocyclic iodide with a C(29–46) oxazole stannane side chain, to establish the complete phorboxazole skeleton. The synthesis, achieved with a longest linear sequence of 24 steps, resulted in 4.6% overall yield [85].
8. Synthesis of Oxazolines of Marine Origin and Analogues
The synthesis of westiellamide analogues of type 76, Figure 39, wherein the oxazoline units were replaced by oxazole or thiazole units, was reported [86]. The strategy involved a one-pot macrocyclization of the monomer building blocks using diphenyl phosphorazidate (DPPA) in the presence of an excess of Hünig’s base under high dilution conditions at room temperature. The trimers 76 where X = O or S were obtained in 35% and 25% yield, respectively. The structures of these analogues, and also of an imidazole-based analogue, were investigated using X-ray diffraction and DFT-based molecular modeling calculations. The flexibility of the systems essentially depends on the type of the azole building block. The oxazole-based peptide is almost coplanar, whereas in the case of the thiazole and the imidazole cyclicpeptides the azole moieties form cone-like structures. Then, the copper(II) coordination chemistry of westiellamide, as well as of the three synthetic analogues (76) with an [18]azacrown-6 macrocyclic structure was reported [87]. The authors emphasized that, as in the larger patellamide rings, the N-heterocycle-N-peptide-N-heterocycle binding site is highly preorganized for copper(II) coordination. The macrocyclic peptides have been found to form stable mono- and dinuclear copper(II) complexes.
Figure 39.

Westiellamide analogs and monomers building blocks.
Acknowledgements
The authors acknowledge financial support from UdelaR (Universidad de la República) and PEDECIBA (Programa de Desarrollo de las Ciencias Básicas).
References
- 1.Aaron MS, Richard AL, David CR. Bacillamides from a hypersaline microbial mat bacterium. J Nat Prod. 2007;70:1793–1795. doi: 10.1021/np070126a. [DOI] [PubMed] [Google Scholar]
- 2.Yu L, Li Z, Peng C, Li Z, Guo Y. Neoobacillamide A, a novel thiazole-containing alkaloid from the marine bacterium bacillus vallismortis C89, associated with south china sea sponge Dysidea avara. Helv Chim. 2009;92:607–612. [Google Scholar]
- 3.Matsuo Y, Kanoh K, Imagawa H, Adachi K, Nishizawa M, Shizuri Y. Urukthapelstatin A, a novel cytotoxic substance from marine-derived Mechercharimyces asporophorigenens YM11-542. J Antib. 2007;60:256–260. doi: 10.1038/ja.2007.31. [DOI] [PubMed] [Google Scholar]
- 4.Linington RG, González J, Urena L, Romero LI, Ortega-Barría E, Gerwick WH. Venturamides A and B: antimalarial constituents of the panamanian marine cyanobacterium Oscillatoria sp. J Nat Prod. 2007;70:397–401. doi: 10.1021/np0605790. [DOI] [PubMed] [Google Scholar]
- 5.Portmann C, Blom JF, Gademann K, Jüttner F. Aerucyclamides A and B: isolation and synthesis of toxic ribosomal heterocyclic peptides from the cyanobacterium Microcystis aeruginosa PCC 7806. J. Nat. Prod. 2008;71:1193–1196. doi: 10.1021/np800118g. [DOI] [PubMed] [Google Scholar]
- 6.Portmann C, Blom JF, Kaiser M, Brun R, Jüttner F, Gademann K. Isolation of Aerucyclamides C and D and structure revision of Microcyclamide 7806A: heterocyclic ribosomal peptides from Microcystis aeruginosa PCC 7806 and their antiparasite evaluation. J Nat Prod. 2008;71:1891–1896. doi: 10.1021/np800409z. [DOI] [PubMed] [Google Scholar]
- 7.Teruya T, Sasaki H, Suenaga K. Hexamollamide, a hexapeptide from an Okinawan ascidian Didemnum molle. Tetraedron Lett. 2008;49:5297–5299. [Google Scholar]
- 8.Zabriskie TM, Foster MP, Stout TJ, Clardy J, Ireland CM. Studies on the solution- and solid-state structure of patellin 2. J Am Chem Soc. 1990;112:8080–8084. [Google Scholar]
- 9.Carroll AR, Coll JC, Bourne DJ, MacLeod JK, Ireland CM, Bowden BF. Patellins 1–6 and trunkamide A: novel cyclic hexa-, hepta- and octa-peptides from colonial ascidians, Lissoclinum sp. Aust J Chem. 1996;49:659–667. [Google Scholar]
- 10.Donia MS, Wang B, Dunbar DC, Desai PV, Patny A, Avery M, Hamann MT. Mollamides B and C, Cyclic Hexapeptides from the Indonesian Tunicate Didemnum molle. J Nat Prod. 2008;71:941–945. doi: 10.1021/np700718p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taori K, Paul VJ, Luesch H. Structure and activity of largazole, a potent antiproliferative agent from the floridian marine cyanobacterium Symploca sp. J Am Chem Soc. 2008;130:1806–1807. doi: 10.1021/ja7110064. [DOI] [PubMed] [Google Scholar]
- 12.Pereira A, Cao Z, Murray TF, Gerwick WH. Hoiamide A, a sodium channel activator of unusual architecture from a consortium of two papua new guinea cyanobacteria. Chem Biol. 2009;16:893–906. doi: 10.1016/j.chembiol.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pereira A, Cao Z, Murray TF, Gerwick WH. Hoiamide A, a sodium channel activator of unusual architecture from a consortium of two papua new guinea cyanobacteria. Chem Biol. 2009;16:1208. doi: 10.1016/j.chembiol.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raveh A, Moshe S, Evron Z, Flescher E, Carmeli S. Novel thiazole and oxazole containing cyclic hexapeptides from a waterbloom of the cyanobacterium Microcystis sp. Tetrahedron. 2010;66:2705–2712. [Google Scholar]
- 15.Kwan J, Rocca JR, Abboud KA, Paul VJ, Luesch H. Total structure determination of grassypeptolide, a new marine cyanobacterial cytotoxin. Org Lett. 2008;10:789–792. doi: 10.1021/ol702946d. [DOI] [PubMed] [Google Scholar]
- 16.Teruya T, Sasaki H, Fukazawa H, Suenaga K. Bisebromoamide, a potent cytotoxic peptide from the marine cyanobacterium Lyngbya sp. Isolation, stereostructure, and biological activity. Org Lett. 2009;11:5062–5065. doi: 10.1021/ol9020546. [DOI] [PubMed] [Google Scholar]
- 17.Sellanes D, Manta E, Serra G. Toward the total synthesis of scleritodermin A: preparation of the C1-N15 fragment. Tetrahedron Lett. 2007;48:1827–1830. doi: 10.1016/j.tetlet.2007.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu S, Cui Y-M, Nan F-J. Total synthesis of the originally proposed and revised structures of scleritodermin A. Org Lett. 2008;10:3765–3768. doi: 10.1021/ol801419m. [DOI] [PubMed] [Google Scholar]
- 19.Sellanes D, Campot F, Nunez I, Lin G, Esposito P, Dematteis S, Saldana J, Dominguez L, Manta E, Serra G. Preparation and biological evaluation of key fragments and open analogs of scleritodermin A. Tetrahedron. 2010;66:5384–5395. [Google Scholar]
- 20.Garcia-Reynaga P, VanNieuwenhze MS. A new total synthesis of patellamide A. Org Lett. 2008;10:4621–4623. doi: 10.1021/ol801895y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ying Y, Taori K, Kim H, Hong J, Luesch H. Total Synthesis and Molecular Target of Largazole, a Histone Deacetylase Inhibitor. J Am Chem Soc. 2008;130:8455–8459. doi: 10.1021/ja8013727. [DOI] [PubMed] [Google Scholar]
- 22.Nasveschuk CG, Ungermannova D, Liu X, Phillips AJ. A Concise Total Synthesis of Largazole, Solution Structure, and Some Preliminary Structure Activity Relationships. Org Lett. 2008;10:3595–3598. doi: 10.1021/ol8013478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seiser T, Kamena F, Cramer N. Synthesis and biological activity of largazole and derivatives. Angew Chem Int Ed. 2008;47:6483–6485. doi: 10.1002/anie.200802043. [DOI] [PubMed] [Google Scholar]
- 24.Bowers A, West N, Taunton J, Schreiber SL, Bradner JE, Williams RM. Total synthesis and biological mode of action of largazole: A potent class I histone deacetylase inhibitor. J Am Chem Soc. 2008;130:11219–11222. doi: 10.1021/ja8033763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghosh AK, Kulkarni S. Enantioselective total synthesis of (+)-largazole, a potent inhibitor of histone deacetylase. Org Lett. 2008;10:3907–3909. doi: 10.1021/ol8014623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ying Y, Liu Y, Byeon SR, Kim H, Luesch H, Hong J. Synthesis and activity of largazole analogues with linker and macrocycle modification. Org Lett. 2008;10:4021–4024. doi: 10.1021/ol801532s. [DOI] [PubMed] [Google Scholar]
- 27.Ren Q, Dai L, Zhang H, Tan W, Xu Z, Ye T. Total synthesis of largazole. Synlett. 2008:2379–2383. [Google Scholar]
- 28.Numajiri Y, Takahashi T, Takagi M, Shin-ya K, Doi T. Total synthesis of largazole and its biological evaluation. Synlett. 2008:2483–2486. [Google Scholar]
- 29.Bowers AA, Greshock TJ, West N, Estiu G, Schreiber SL, Wiest O, Williams RM, Bradner JE. Synthesis and conformation-activity relationships of the peptide isosteres of FK228 and largazole. J Am Chem Soc. 2009;131:2900–2905. doi: 10.1021/ja807772w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bowers AA, West N, Newkirk TL, Troutman-Youngman AE, Schreiber SL, Wiest O, Bradner JE, Williams RM. Synthesis and histone deacetylase inhibitory activity of largazole analogs: alteration of the zinc-binding domain and macrocyclic scaffold. Org Lett. 2009;11:1301–1304. doi: 10.1021/ol900078k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seiser T, Cramer N. Syntheses and biological activity of the HDAC class I inhibitor largazole. Chimia. 2009;63:19–22. [Google Scholar]
- 32.Chen F, Gao A-H, Li J, Nan F-J. Synthesis and biological evaluation of C7-demethyl largazole analogues. ChemMedChem. 2009;4:1269–1272. doi: 10.1002/cmdc.200900125. [DOI] [PubMed] [Google Scholar]
- 33.Yan W, O’Doherty GA. Total synthesis of (+)-largazole, a histone deacetylase inhibitor. Chemtracts. 2009;22:50–58. [Google Scholar]
- 34.Wang B, Forsyth CJ. Total synthesis of largazole-devolution of a novel synthetic strategy. Syntesis. 2009:2873–2880. [Google Scholar]
- 35.Zeng X, Yin B, Hu Z, Liao C, Liu J, Li S, Li Z, Nicklaus MC, Zhou G, Jiang S. Total synthesis and biological evaluation of largazole and derivatives with promising selectivity for cancers cells. Org Lett. 2010;12:1368–1371. doi: 10.1021/ol100308a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Souto JA, Vaz E, Lepore I, Poppler A-C, Franci G, Alvarez R, Altucci L, de Lera AR. Synthesis and biological characterization of the histone deacetylase inhibitor largazole and C7-modified analogues. J Med Chem. 2010;53:4654–4667. doi: 10.1021/jm100244y. [DOI] [PubMed] [Google Scholar]
- 37.Li D, Yang HS, Cui Q, Mao SJ, Xu XH. Synthesis of bacillamide 3 and its analogue. Chin Chem Lett. 2009;20:1195–1197. [Google Scholar]
- 38.Wang W, Joyner S, Khoury KAS, Doemling A. (−)-Bacillamide C: the convergent approach. Org Biom Chem. 2010;8:529–532. doi: 10.1039/b918214d. [DOI] [PubMed] [Google Scholar]
- 39.Beaumont S, Ilardi EA, Monroe LR, Zakarian A. Valence tautomerism in titanium enolates: catalytic radical haloalkylation and application in the total synthesis of neodysidenin. J Am Chem Soc. 2010;132:1482–1483. doi: 10.1021/ja910154f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Numajiri Y, Takahashi T, Doi T. Total synthesis of (−)-apratoxin A, 34-epimer, and its oxazoline analogue. Chem Asian J. 2009;4:111–125. doi: 10.1002/asia.200800365. [DOI] [PubMed] [Google Scholar]
- 41.Gao X, Liu Y, Kwong S, Xu Z, Ye T. Total synthesis and stereochemical reassignment of bisebromoamide. Org Lett. 2010;12:3018–3021. doi: 10.1021/ol101021v. [DOI] [PubMed] [Google Scholar]
- 42.Wright AE, Cook Botelho J, Guzmán E, Harmody D, Linley P, McCarthy PJ, Pitts TP, Pomponi SA, Reed JK. Neopeltolide, a macrolide from a lithistid sponge of the family Neopeltidae. J Nat Prod. 2007;70:412–416. doi: 10.1021/np060597h. [DOI] [PubMed] [Google Scholar]
- 43.Oku N, Adachi K, Matsuda S, Kasai H, Takatsuki A, Shizuri Y. Ariakemicins A and B, novel polyketide-peptide Antibiotics from a marine gliding bacterium of the genus. Rapidithrix Org Lett. 2008;10:2481–2484. doi: 10.1021/ol8007292. [DOI] [PubMed] [Google Scholar]
- 44.Pettit GR, Hogan F, Xu JP, Tan R, Nogawa T, Cichacz Z, Pettit RK, Du J, Ye QH, Cragg GM, Herald CL, Hoard MS, Goswami A, Searcy J, Tackett L, Doubek DL, Williams L, Hooper JN, Schmidt JM, Chapuis JC, Tackett DN, Craciunescu F. Antineoplastic Agents. 536. New Sources of Naturally Occurring Cancer Cell Growth Inhibitors from Marine Organisms, Terrestrial Plants, and Microorganisms. J Nat Prod. 2008;71:438–444. doi: 10.1021/np700738k. [DOI] [PubMed] [Google Scholar]
- 45.Bishara A, Rudi A, Aknin M, Neumann D, Ben-Califa N, Kashman Y. Taumycins A and B, Two Bioactive Lipodepsipeptides from the Madagascar Sponge Fascaplysinopsis sp. Org Lett. 2008;10:4307–4309. doi: 10.1021/ol801750y. [DOI] [PubMed] [Google Scholar]
- 46.Bishara A, Rudi A, Aknin M, Neumann D, Ben-Califa N, Kashman Y. Salarin C, a new cytotoxic sponge-derived nitrogenous macrolide. Tetrahedron Lett. 2008;49:4355–4358. [Google Scholar]
- 47.Bishara A, Rudi A, Aknin M, Neumann D, Ben-Califa N, Kashman Y. Salarins D–J, seven new nitrogenous macrolides from the madagascar sponge Fascaplysinopsis sp. Tetrahedron. 2010;66:4339–4345. [Google Scholar]
- 48.Dalisay DS, Rogers EW, Edison AS, Molinski TF. Structure elucidation at the nanomole scale. 1. Trisoxazole macrolides and thiazole-containing cyclic peptides from the nudibranch Hexabranchus sanguineus . J Nat Prod. 2009;72:732–738. doi: 10.1021/np8007649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fernández R, Martín MJ, Rodríguez-Acebes R, Reyes F, Francesch A, Cuevas C. Diazonamides C–E, new cytotoxic metabolites from the ascidian Diazona sp. Tetrahedron Lett. 2008;49:2283–2285. [Google Scholar]
- 50.Oku N, Takada K, Fuller RW, Wilson JA, Peach ML, Pannell LK, McMahon JB, Gustafson KR. Isolation, Structural Elucidation, and Absolute Stereochemistry of Enigmazole A, a Cytotoxic Phosphomacrolide from the Papua New Guinea Marine Sponge Cinachyrella enigmatica. J Am Chem Soc. 2010;132:10278–10285. doi: 10.1021/ja1016766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Youngsaye W, Lowe JT, Pohlki F, Ralifo P, Panek JS. Total Synthesis and Stereochemical Reassignment of (+)-Neopeltolide. Angew Chem Int Ed. 2007;46:9211–9214. doi: 10.1002/anie.200704122. [DOI] [PubMed] [Google Scholar]
- 52.Custar DW, Zabawa TP, Scheidt KA. Total Synthesis and Structural Revision of the Marine Macrolide Neopeltolide. J Am Chem Soc. 2008;130:804–805. doi: 10.1021/ja710080q. [DOI] [PubMed] [Google Scholar]
- 53.Custar DW, Zabawa TP, Hines J, Crews CM, Scheidt KA. Total synthesis and structure-activity investigation of the marine natural product neopeltolide. J Am Chem Soc. 2009;131:12406–12414. doi: 10.1021/ja904604x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Woo SK, Kwon MS, Lee E. Total synthesis of (+)-neopeltolide by a Prins macrocyclization. Angew Chem Int Ed. 2008;47:3242–3244. doi: 10.1002/anie.200800386. [DOI] [PubMed] [Google Scholar]
- 55.Fuwa H, Naito S, Goto T, Sasaki M. Total synthesis of (+)-neopeltolide. Angew Chem Int Ed. 2008;47:4737–4739. doi: 10.1002/anie.200801399. [DOI] [PubMed] [Google Scholar]
- 56.Paterson I, Miller NA. Total synthesis of the marine macrolide (+)-neopeltolide. Chem Comm. 2008;39:4708–4710. doi: 10.1039/b812914b. [DOI] [PubMed] [Google Scholar]
- 57.Vintonyak VV, Kunze B, Sasse F, Maier ME. Total synthesis and biological activity of neopeltolide and analogues. Chem Eur J. 2008;14:11132–11140. doi: 10.1002/chem.200801398. [DOI] [PubMed] [Google Scholar]
- 58.Kartika R, Gruffi TR, Taylor RE. Concise Enantioselective Total Synthesis of Neopeltolide Macrolactone Highlighted by Ether Transfer. Org Lett. 2008;10:5047–5050. doi: 10.1021/ol802254z. [DOI] [PubMed] [Google Scholar]
- 59.Sasaki M, Fuwa H. Total synthesis of (+)-neopeltolide, antitumor marine macrolide. Farumashia. 2009;45:439–443. [Google Scholar]
- 60.Guinchard X, Roulland E. Total Synthesis of the Antiproliferative Macrolide (+)-Neopeltolide. Org Lett. 2009;11:4700–4703. doi: 10.1021/ol902047z. [DOI] [PubMed] [Google Scholar]
- 61.Fuwa H, Saito A, Naito S, Konoki K, Yotsu-Yamashita M, Sasaki M. Total Synthesis and Biological Evaluation of (+)-Neopeltolide and Its Analogues. Chem Eur J. 2009;15:12807–12818. doi: 10.1002/chem.200901675. [DOI] [PubMed] [Google Scholar]
- 62.Fuwa H, Saito A, Sasaki M. A Concise Total Synthesis of (+)-Neopeltolide. Angew Chem Int Ed. 2010;49:3041–3044. doi: 10.1002/anie.201000624. [DOI] [PubMed] [Google Scholar]
- 63.Cui Y, Tu W, Floreancig PE. Total synthesis of neopeltolide and analogs. Tetrahedron. 2010;66:4867–4873. doi: 10.1016/j.tet.2010.03.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yadav JS, Kumar GG. A concise stereoselective formal total synthesis of the cytotoxic macrolide (+)-Neopeltolide via Prins cyclization. Tetrahedron. 2010;66:480–487. [Google Scholar]
- 65.Skepper CK, Quach T, Molinski TF. Total Synthesis of Enigmazole A from Cinachyrella enigmatica. Bidirectional Bond Constructions with an Ambident 2,4-Disubstituted Oxazole Synthon. J Am Chem Soc. 2010;132:10286–10292. doi: 10.1021/ja1016975. [DOI] [PubMed] [Google Scholar]
- 66.Pattenden G, Ashweek NJ, Baker-Glenn CAG, Kempson J, Walker GM, Yee JGK. Total synthesis of (−)-ulapualide A, a novel tris-oxazole macrolide from marine nudibranchs, based on some biosynthesis speculation. Org Biomol Chem. 2008;6:1478–1497. doi: 10.1039/b801036f. [DOI] [PubMed] [Google Scholar]
- 67.Pattenden G, Ashweek NJ, Baker-Glenn CAG, Walker GM, Yee JGK. Total synthesis of (−)-ulapualide A: The danger of overdependence on NMR spectroscopy in assignment of stereochemistry. Angew Chem Int Ed. 2007;46:4359–4363. doi: 10.1002/anie.200700459. [DOI] [PubMed] [Google Scholar]
- 68.Smith TE, Kuo W, Bock VD, Roizen JL, Balskus EP, Theberge AB. Total Synthesis of (−)-Hennoxazole A. Org Lett. 2007;9:1153–1155. doi: 10.1021/ol070244p. [DOI] [PubMed] [Google Scholar]
- 69.Smith TE, Kuo W, Balskus EP, Bock VD, Roizen JL, Theberge AB, Carroll KA, Kurihara T, Wessler JD. Total Synthesis of (−)-Hennoxazole A. J Org Chem. 2008;73:142–150. doi: 10.1021/jo7018015. [DOI] [PubMed] [Google Scholar]
- 70.Nett M, Erol O, Kehraus S, Kock M, Krick A, Eguereva E, Neu E, König GM. Siphonazole, an Unusual Metabolite from Herpetosiphon sp. Angew Chem Int Ed. 2006;45:3863–3867. doi: 10.1002/anie.200504525. [DOI] [PubMed] [Google Scholar]
- 71.Linder J, Moody CJ. The total synthesis of siphonazole, a structurally unusual bis-oxazole natural product. Chem Commun. 2007:1508–1509. doi: 10.1039/b618160k. [DOI] [PubMed] [Google Scholar]
- 72.Linder J, Blake AJ, Moody CJ. Total synthesis of siphonazole and its O-methyl derivative, structurally unusual bis-oxazole natural products. Org Biomol Chem. 2008;6:3908–3916. doi: 10.1039/b810855b. [DOI] [PubMed] [Google Scholar]
- 73.Zhang J, Polishchuk EA, Chen J, Ciufolini MA. Development of an Oxazole Conjunctive Reagent and Application to the Total Synthesis of Siphonazoles. J Org Chem. 2009;74:9140–9151. doi: 10.1021/jo9018705. [DOI] [PubMed] [Google Scholar]
- 74.Chandrasekhar S, Sudhakar A. Total Synthesis of Bengazole A. Org Lett. 2010;12:236–238. doi: 10.1021/ol9024138. [DOI] [PubMed] [Google Scholar]
- 75.Enriquez-Garcia A, Ley SV. Total synthesis of the potent antifungal agents bengazole C and E Collect Czechoslovak. Chem Commun. 2009;74:887–900. [Google Scholar]
- 76.Bull JA, Balskus EP, Horan RAJ, Langner M, Ley SV. Total synthesis of potent antifungal marine bisoxazole natural products benzazoles A and B. Chem Eur J. 2007;13:5515–5538. doi: 10.1002/chem.200700033. [DOI] [PubMed] [Google Scholar]
- 77.Scarone L, Fajardo J, Saldana J, Dominguez L, Esposito P, Demastteis S, Wipf P, Manta E, Serra G. Synthesis and evaluation of anthelmintic and cytotoxic properties of [2,5′]bis-1,3-azole analogs of bengazoles. Lett Drug Des Discov. 2009;6:413–419. [Google Scholar]
- 78.Krauss J, Kalkbrenner S, Schuster A, Obainoke A, Bracher F. Synthesis of new bengazole analogues and their antimicrobial activity. Turk J Chem. 2008;32:125–130. [Google Scholar]
- 79.Mulder RJ, Shafer CM, Dalisay DS, Molinski TF. Synthesis and structure-activity relationships of bengazole A analogs. Bioorg Med Chem Lett. 2009;19:2928–2930. doi: 10.1016/j.bmcl.2009.04.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fresneda PM, Castaneda M, Blug M, Molina P. Iminophosphorane-based preparation of 2,5-disubstituted oxazole derivatives: synthesis of the marine alkaloid almazole C. Synlett. 2007;2:324–326. [Google Scholar]
- 81.N’Diaye I, Guella G, Mancini I, Pietra F. Almazole D, a New Type of Antibacterial 2,5-Disubstituted Oxazolic Dipeptide from a Red Alga of the Coast of Senegal. Tetrahedron Lett. 1996;37:3049–3050. [Google Scholar]
- 82.Miyake F, Hashimoto M, Tonsiengsom S, Yakushijin K, Horne DA. Synthesis of 5-(3-indolyl) oxazole natural products. Structure revision of Almazole D. Tetrahedron. 2010;66:4888–4893. [Google Scholar]
- 83.Lucas BS, Gopalsamuthiram V, Burke SD. Total synthesis of phorboxazole B. Angew Chem Int Ed. 2007;46:769–772. doi: 10.1002/anie.200603656. [DOI] [PubMed] [Google Scholar]
- 84.Smith AB, III, Razler TM, Meis RM, Pettit GR. Synthesis and Biological Evaluation of Phorboxazole Congeners Leading to the Discovery and Preparative Scale Synthesis of (+)-Chlorophorboxazole A Possessing Picomolar Human Solid Tumor Cell Growth Inhibitory Activity. J Org Chem. 2008;73:1201–1208. doi: 10.1021/jo701816h. [DOI] [PubMed] [Google Scholar]
- 85.Smith AB, III, Razler TM, Ciavarri JP, Hirose T, Ishikawa T, Meis RM. A Second-Generation Total Synthesis of (+)-Phorboxazole A. J Org Chem. 2008;73:1192–1200. doi: 10.1021/jo7018152. [DOI] [PubMed] [Google Scholar]
- 86.Haberhauer G, Drosdow E, Oeser T, Rominger F. Structural investigation of westiellamide analogs. Tetrahedron. 2008;64:1853–1859. [Google Scholar]
- 87.Comba P, Gahan LR, Haberhauer G, Hanson GR, Noble CJ, Seibold B, van den Brenk AL. Copper (II) coordination chemistry of westiellamide and its imidazole, oxazole, and thiazole analogues. Chem Eur J. 2008;14:4393–4403. doi: 10.1002/chem.200701778. [DOI] [PubMed] [Google Scholar]