

Abstract
A simple organocatalytic one-pot protocol for the construction of optically active allylic alcohols and amines using readily available reactants and catalyst is presented. The described reaction is enabled by an enantioselective enone epoxidation/aziridination-Wharton-reaction sequence affording two highly privileged and synthetically important classes of compounds in an easy and benign way. The advantages of the described sequence include easy generation of stereogenic allylic centers, also including quaternary stereocenters, with excellent enantio- and diastereomeric-control and high product diversity. Furthermore, using monosubstituted enones as substrates, having moderate enantiomeric excess, the one-pot reaction sequence proceeds with an enantioenrichment of the products and high diastereoselectivity was achieved.
Keywords: asymmetric catalysis, allylic amines, enantioselectivity
Allylic alcohols and amines represent two of the most abundant and fundamental building blocks in contemporary organic synthesis. The possible transformations of these important compounds are numerous and proceed often with excellent stereoinduction (1–3). Consequently, optically active allylic alcohols and amines have appeared innumerable times as key intermediates in asymmetric total syntheses, showing the need for efficient and benign methods of their formation.
One of the traditional synthetic approaches to optically active allylic alcohols is the catalytic kinetic resolution applying enzymes (4). In addition, Sharpless and coworkers have successfully established an asymmetric titanium-catalyzed resolution of racemic secondary allylic alcohols via the Sharpless-Katsuki epoxidation (5), resulting in a mixture of epoxy-alcohols and optically active allylic alcohols (Scheme 1, expression 1). However, the fact that the resolution method is restricted to a theoretical yield of 50%, makes other direct routes to optically active allylic alcohols highly desirable. With the dynamic kinetic resolution (6), a procedure that allows full conversion of the starting compound to one desired product with excellent stereocontrol was established. In this process, a fast and efficient in situ racemization reaction of the undesired enantiomer of the starting material is the key to overcome the 50%-yield limit of the traditional resolution method. However, the scope of this reaction is limited to mainly acyclic substrates and the combined use of transition metal and enzyme is necessary (Scheme 1, expression 2).
Scheme 1.

Different methods for synthesis of allylic alcohols and amines; X = O, NH.
As an alternative to the resolution method, several different enantioselective reduction protocols of enones have been developed. Examples are the Corey-Bakshi-Shibata reduction (7), applying a borane-prolinol complex, and the catalytic enantioselective hydrogenation, applying BINAP [2,2'-Bis(diphenylphosphino)-1,1'-binaphthyl]-diamine ruthenium complexes developed by Noyori and coworkers (8) (Scheme 1, expression 3). The needs of bulky functional groups, different substitution patterns at the enone functionality, and inert reaction conditions for obtaining high selectivities are some of the drawbacks of these methods. Another protocol for the enantioselective synthesis of allylic alcohols is the 1,2-addition of vinylic metal species, generated either by transmetallation of boronates or by rhodium- or iridium-catalyzed reductive coupling of acetylenes (9) (Scheme 1, expression 4). While the transmetallation strategy requires the preparation of a “primary organometallic reagent,” the reductive coupling of acetylenes is usually performed under high pressure of hydrogen. Foreshadowed by the pioneering work of Trost et al., metal-catalyzed π-allyl substitutions of racemic allylic carbonates or esters have been extensively investigated in the last years (Scheme 1, expression 5). These methods provide straightforward and widely applied synthetic routes to nonterminal allylic alcohols (10–15). Nevertheless, despite the wide applicability, significant challenges remain yet to be solved. Among them is the restriction of π-allyl substitutions to symmetrically disubstituted π-allyl systems, which excludes certain substitution patterns in the allylic products. Moreover, the formation of quaternary allylic centers is, to the best of our knowledge, not possible using these methods.
Optically active allylic amines have so far been synthesized by almost similar protocols. While the metal-catalyzed π-allyl substitution for the synthesis of allylic amines (Scheme 1, expression 5) is restricted by the same issues mentioned above (16, 17), the 1,2-addition of vinylic metal species to imines (Scheme 1, expression 4) often applies an additional chiral auxiliary attached to the nitrogen atom in order to obtain high stereocontrol (9). An efficient transformation with complete stereoinversion, is the Mitsunobu reaction (Scheme 1, expression 6) which has been applied numerous times for the synthesis of optically active allylic amines (18). However, this process requires optically active allylic alcohols as starting materials and thus provides an indirect route to this important class of compounds.
Despite the achievements published so far for the synthesis of optically active allylic alcohols and amines, there is still need for developments of general, robust, and easy synthetic approaches to these classes of compounds. Incorporation of organocatalytic functionalizations in complex one-pot reactions has recently emerged as an efficient strategy for the formation of many important optically active compounds, otherwise difficult to obtain (19–25). The present work shows the development of an organocatalytic synthetic process, making use of a single amino-catalyst (26–31) in combination with cheap and readily available starting materials, for the formation of allylic alcohols and amines in moderate to good yields and excellent enantioselectivities (Scheme 2). The described reactions are enabled by an enantioselective enone epoxidation/aziridination-Wharton-reaction sequence. While the organocatalytic epoxidation of cyclic and acylic enones has been recently described by List et al. (32, 33) and Deng et al. (34), the organocatalytic aziridination of enones has been mainly limited to acyclic substrates (35, 36). We envisioned that a merger of the catalytic enone functionalizations with the hydrazine mediated 1,3-transposition following the Wharton protocol (37) could allow simple access to a broad spectrum of optically active allylic products under metal-free conditions, and preferably in an one-pot process (Scheme 2).
Scheme 2.

An organocatalytic approach to allylic alcohols and amines.
Results and Discussion
To find the optimal reaction conditions, we started our investigations for the synthesis of optically active allylic alcohols by using cyclohexenone 1a as a model substrate and performing the envisaged sequence in two separate steps (Scheme 3). Both the epoxidation and Wharton transposition reactions could be achieved providing the desired allylic alcohol 4a with high enantioselectivity. Comparable results could be attained for 3-methyl cyclohexenone 1e allowing the formation of the tertiary allylic alcohol 4e, otherwise difficult to obtain, with excellent enantiomeric excess.
Scheme 3.

Enantioselective organocatalytic synthesis of allylic alcohols.
However, the volatility of the optically active epoxy ketones 3a, e and allylic alcohols 4a, e (Scheme 3) makes the development of an one-pot protocol, and therefore a minimization of purification steps, desirable. We have found that sequential addition of hydrazine, methanol, and acetic acid to the reaction mixture of the epoxidation process, after full conversion of the organocatalytic step has been achieved, resulted in higher isolated yields of the corresponding allylic alcohols and maintaining the high enantioselectivity.
A plethora of cyclic enones of various ring-size and substitution patterns were evaluated as substrates in this one-pot sequence and the results are outlined in Table 1. The obtained yields ranged from moderate to good, while the enantiomeric excess remained high in all cases (87–99% ee). Seven-membered allylic alcohols were formed in similar high enantioselectivity, while for the application of cyclopentenone a significant decrease in reactivity was observed, as expected in analogy to previous reports (32). Substitutions at 5-, 4-, and 3-position of the enone were all well tolerated. With respect to use of enones with substitutions at 3- and 4-positions it is noteworthy that they represent substitution patterns difficult to obtain using conventional methods, such as palladium-catalyzed π-allyl substitution or dynamic kinetic resolution. The formation of quaternary allylic stereogenic centers with good enantio-control by nonresolution methods is a highly challenging task (38). Using the present conditions, a variety of allylic alcohols carrying a quaternary allylic center are formed with enantioselectivities up to 99% ee. Finally, it is also demonstrated that acyclic enones (Table 1, entry 9) are valid substrates for this reaction. However, for the acyclic product 4h an E/Z ratio of 1∶1 is observed, which is in accordance with the mechanism of the Wharton rearrangement (39–41).
Table 1.
One-pot synthesis of allylic alcohols using an organocatalytic epoxidation-Wharton sequence 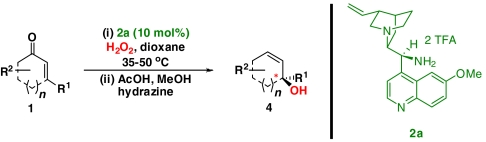
| Entry | Enone (1) | Product (4) | Yield (%)* | ee (%)† | ||
| 1 | 1a | 4a | 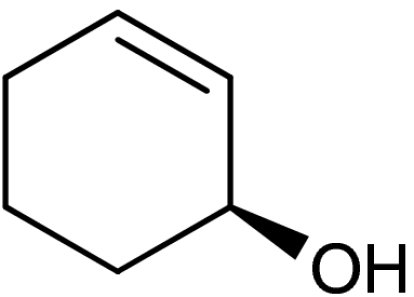 |
50 (32)§ | 93 (93)§ | |
| 2 | 1b | 4b | 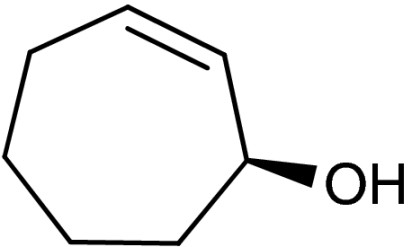 |
45 | 92 | |
| 3 | 1c | 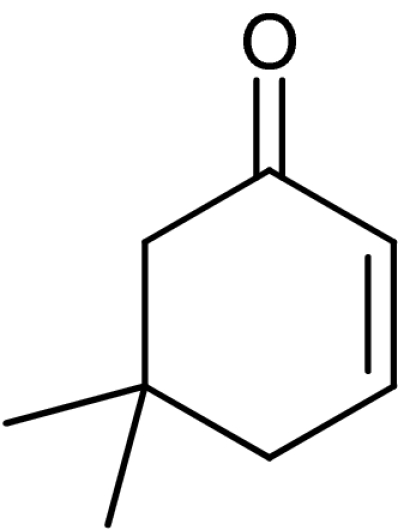 |
4c | 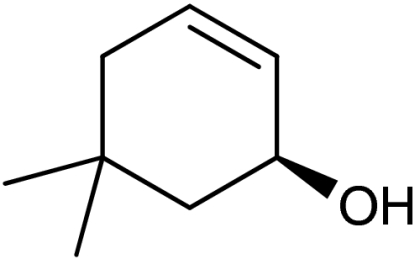 |
58 | 87 |
| 4 | 1d | 4d | 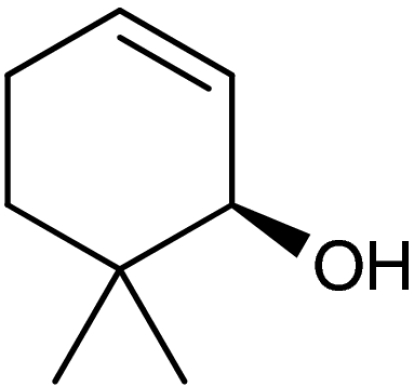 |
47 | 94 | |
| 5 | 1e | 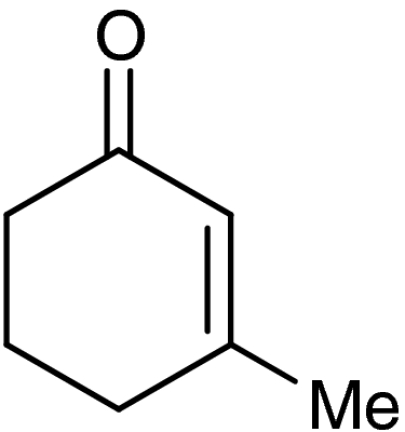 |
4e | 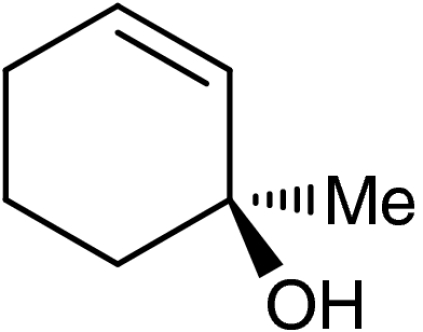 |
40 (35)§ | 94 (94)§ |
| 6 | 1f | 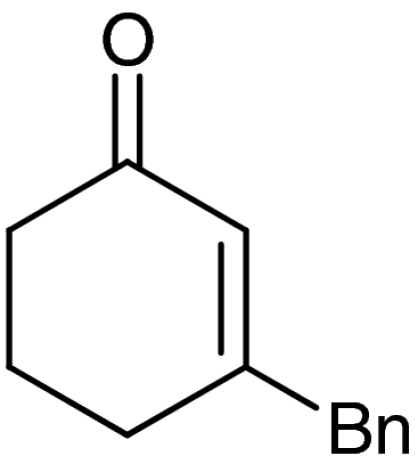 |
4f | 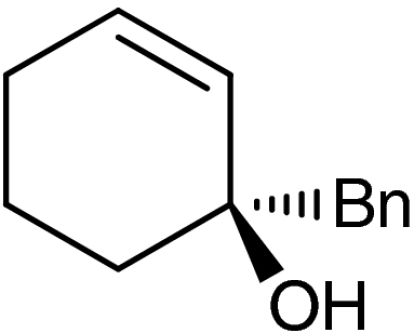 |
40 | 98 |
| 7 | 1g | 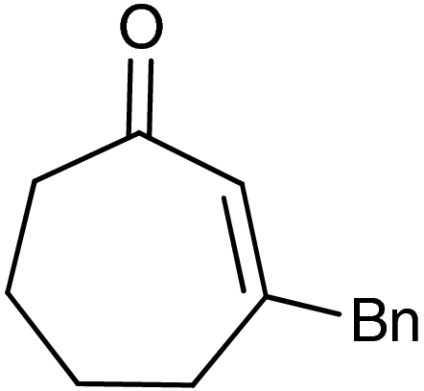 |
4g | 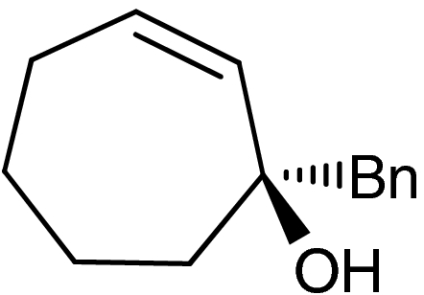 |
45 | 99 |
| 8‡ | 1g | 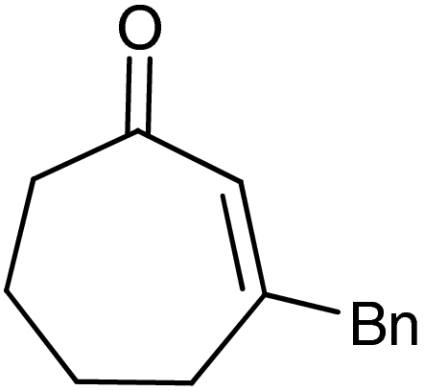 |
ent-4g | 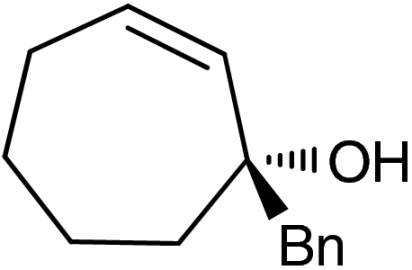 |
42 | 96 |
| 9 | 1h | 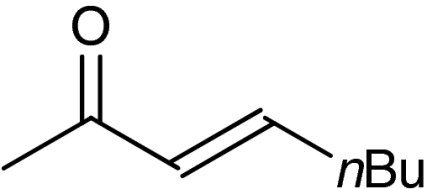 |
4h | 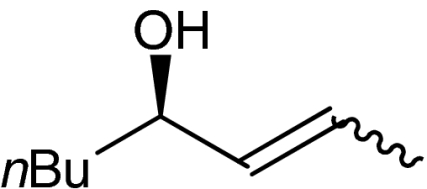 |
54 | 96 |
*Yields of isolated products.
†Determined by chiral stationary phase GC or HPLC.
‡The quasienantiomer of the catalyst is used (2b, see Scheme 5).
§Results for the sequence performed in two separate steps.
The traditional Wharton reaction is facilitated with a α-carbonyl leaving group, which typically is an epoxy motif as applied in the one-pot reaction sequence for the formation of the optically active allylic alcohols, by which the chirality is set during the first organocatalytic step. Intuitively, we propose that an electron-poor aziridine should be equally effective as the “chiral leaving group,” thereby facilitating the formation of the corresponding optically active allylic amines. To the best of our knowledge, such a reaction for this class of compounds has not yet been described. As mentioned previously, the organocatalytic aziridination of cyclic enones has only been rarely described, whereas acyclic enones have been studied more frequently (35, 36). Differently substituted hydroxylamine derivatives were tested as nucleophilic reagents, in which the hydroxy functionality serves as the leaving group and enables therefore the formation of the three-membered heterocycle after the nucleophilic 1,4-addition of the amine moiety. We chose 4-methyl-N-(tosyloxy)benzenesulfonamide 5 as a suitable nucleophile, because of its easy and simple access via ditosylation of hydroxylamine and the two-step sequence was initially performed separately (Scheme 4). We were pleased to observe, that the subjection of the synthesized and isolated aziridines to the Wharton transposition conditions led smoothly to the formation of optically active allylic N-tosyl amines in good yields and excellent enantioselectivities.
Scheme 4.

Enantioselective organocatalytic synthesis of allylic amines.
Although the products 7b, d proved to be nonvolatile, an one-pot sequence is highly desirable, reducing purification steps, material costs, and time. By increasing the amount of acetic acid used in the reductive elimination step, the allylic amines could be synthesized in an one-pot fashion in equally good or higher yields and without affecting the high enantioselectivity (Table 2).
Table 2.
One-pot synthesis of allylic amines using an organocatalytic aziridination-Wharton sequence 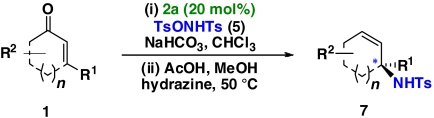
| Entry |
Enone (1) |
Product (7) |
Yield (%)* |
ee (%)† |
||
| 1 | 1a | 7a | 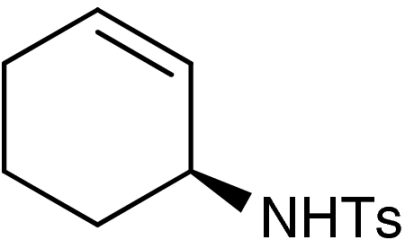 |
56 | 96 | |
| 2‡ | 1a | ent-7a | 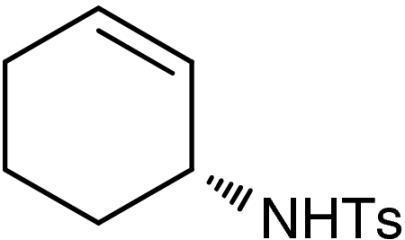 |
52 | 95 | |
| 3 | 1b | 7b |  |
48 (37)§ | 98 (98)§ | |
| 4 | 1d | 7d | 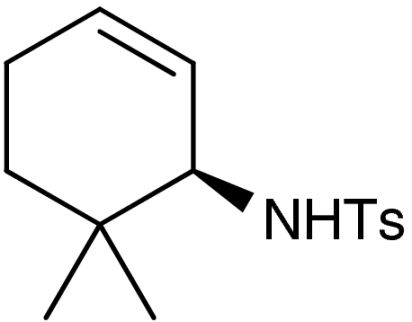 |
50 (50)§ | 97 (97)§ | |
| 5 | 1e | 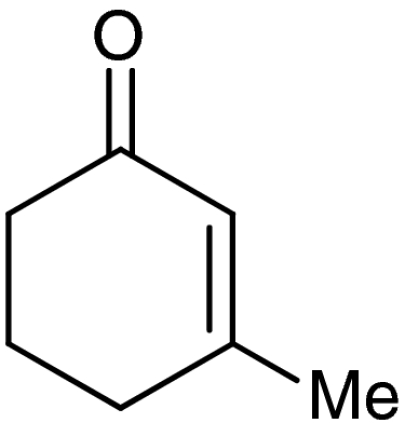 |
7e | 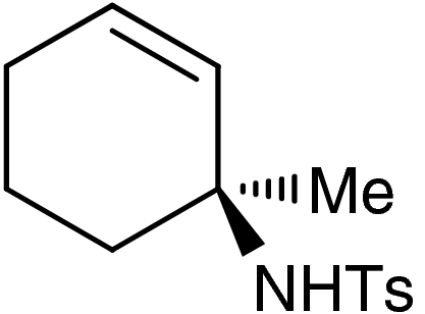 |
57 | 99 |
| 6 | 1f | 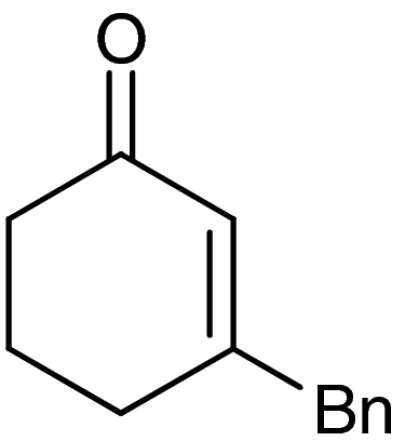 |
7f | 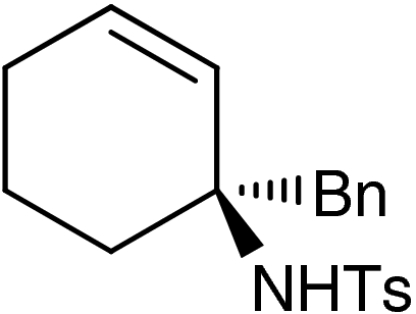 |
61 | 99 |
| 7‡ | 1f | 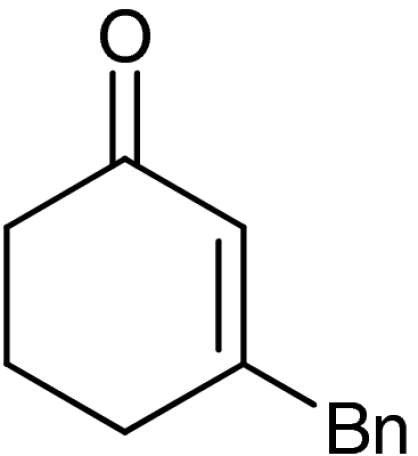 |
ent-7f | 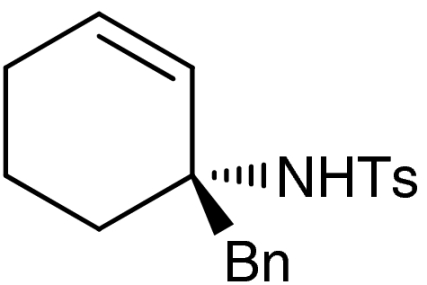 |
69 | 99 |
| 8 | 1g | 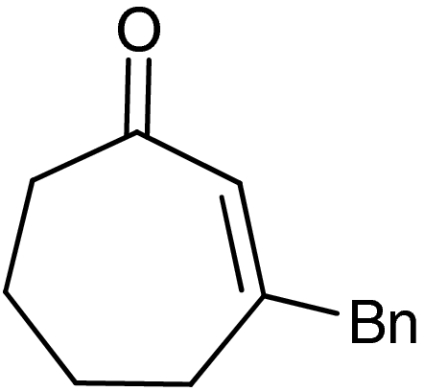 |
7g | 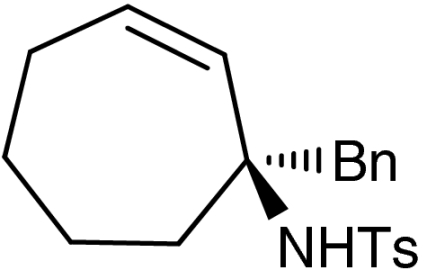 |
42 | 94 |
| 9 | 1h | 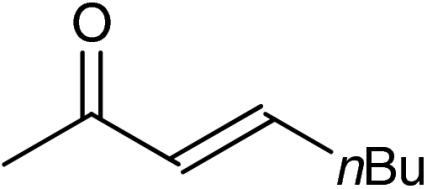 |
7h | 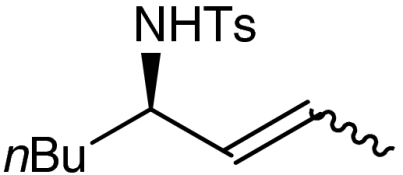 |
52 | 97 |
*Yields of isolated products.
†Determined by chiral stationary phase HPLC.
‡The quasi-enantiomer of the catalyst was used (2b, see Scheme 5).
§Results for the sequence performed in two separate steps.
Both six- and seven-membered enones participate in the reaction with satisfactory results and 95–99% ee. As expected, substitution at 4- and 3-position of the enone is tolerated. However, enones bearing substituents in 5-position resulted only in low conversion in the organocatalytic aziridination step. Although the aziridination of 3-substituted cyclic enones has been reported with only moderate enantioselectivity, we accomplished the generation of quaternary allylic centers (7e, f, g) with excellent enantio-control, using the nucleophile 5. The use of acyclic substrates (entry 9) furnished again the desired product 7h in high enantiomeric excess (97% ee); however also in this case a 1∶1 mixture of E/Z isomers was obtained.
To further challenge the robustness of the developed reaction sequence, we decided to evaluate the process applying the enantioenriched monosubstituted cyclohexenone 8 (42). As demonstrated in Scheme 5, the synthesis of optically active cyclic allylic alcohol and amine having two stereogenic centers was accomplished with good yields and diastereoselectivities. By employing catalyst 2b, the trans-substituted product 9a is formed in good yield and diastereomeric ratio, and an enantio-enrichment from 75% to 92% ee is achieved because of double stereoselection. However, by applying catalyst 2a, the cis-product is formed preferably, as demonstrated for compound 9b formed in 95% ee. A review of literature shows that methods for catalytic asymmetric synthesis of disubstituted cyclic allylic alcohols and amines are limited. Among the simplest and most applied methods is the palladium-catalyzed π-allyl substitution. However, stereo-selective π-allyl substitution is mostly carried out on cis-substituted substrates giving retention of the relative stereochemistry of the products, while the corresponding trans-product is obtained usually by Mitsunobu inversion (43). Consequently, we believe that the described diastereoselective reaction can serve as a complementary method to the palladium-catalyzed reactions for the formation of both cis and trans-substituted allylic products.
Scheme 5.

Diastereoselective synthesis of allylic alcohols and amines.
The mechanistic proposal of the reported one-pot reactions for the formation of optically active allylic alcohols and amines is outlined in Scheme 6. Condensation of the 9-amino-9-deoxyepiquinine ditrifluoroacetic acid salt catalyst 2a and enone 1 leads to the formation of an activated iminium species 10, which is subjected to nucleophilic attack of H2O2 or TsONHTs (4-Methyl-N-(tosyloxy)benzenesulfonamide) under formation of intermediate 11. Spontaneous ring closure and hydrolysis provided the optically active epoxy-ketone 3 and aziridine 6 intermediates and liberation of the amino-catalyst. Next, condensation of 3 or 6 with hydrazine under acidic conditions afforded the short-lived iminium species 12, which upon subsequent ring opening rearrange to intermediate 13. Final release of volatile N2 and protonation of the formed vinyl anions provided the desired optically active allylic alcohols 4 and amines 7. Despite the elevated temperatures and acidic conditions, no racemization was observed, furnishing the products in excellent enantioselectivities.
Scheme 6.

Mechanistic proposal of the formation of allylic alcohols and amines.
Conclusion
In conclusion, we have developed a simple and metal-free one-pot protocol for construction of optically active allylic alcohols and amines using readily available starting materials. The advantages of the described sequence include easy formation of quaternary allylic centers and expanded product diversity. It should therefore serve as practical complementary to presently available methods.
Materials and Methods
Unless otherwise stated, all commercially available reagents were purchased and used as received without further purification. The amino catalysts 2a, b, enones 1c, f, g and 8 were synthesized according to literature procedures (32, 42).
Typical Procedure for One-Pot Synthesis of Allylic Alcohols.
An ordinary glass vial equipped with a magnetic stirring bar was charged with the enone 1 (0.25 mmol, 1.0 equivalent), the catalyst 2 (0.025 mmol, 0.1 equivalent), and dioxane (1.0 mL). After 30 min of stirring at room temperature, H2O (50 wt% in H2O, 0.30 mmol, 1.2 equivalent) was added. The reaction was heated to 35–50 °C (see individual entries) and kept under vigorous stirring until complete conversion of the enone as monitored by TLC (usually 24–72 h). MeOH (5.0 mL), hydrazine monohydrate (1.25 mmol, 5.0 equivalent), and AcOH (0.63 mmol, 2.5 equivalent) were then added in the described sequence. After an additional 0.5–1 h of stirring at room temperature, the crude reaction mixture was diluted with NaHCO3 (sat.), extracted with CH2Cl2 (3 × 10 mL), dried over MgSO4, and most of the excess solvents were carefully removed in vacuo. The remaining volume (ca. 1–2 mL) was charged on silica gel and purified by FC (gradient: pentane to pentane/Et2 2∶1).
Typical Procedure for One-Pot Synthesis of Allylic Amines.
An ordinary glass vial equipped with a magnetic stirring bar was charged with TsONHTs 5 (0.38 mmol, 1.5 equivalent), the catalyst 2 (0.05 mmol, 0.2 equivalent), and CHCl3 (1.0 mL). After 10 min of stirring at room temperature, enone 1 (0.25 mmol, 1.0 equivalent) and NaHCO3 (0.5 mmol, 2 equivalent) were added sequentially. The reaction was kept under vigorous stirring until complete conversion of the enone as monitored by TLC (usually 24–72 h). MeOH (5.0 mL), hydrazine (1.25 mmol, 5.0 equivalent), and AcOH (1.13 mmol, 4.5 equivalent) were then added in the described sequence. After an additional 1 h of stirring at 50 °C, the crude reaction mixture was cooled to room temperature, diluted with NaHCO3 (sat.), extracted with CH2Cl2 (3 × 10 mL), dried over MgSO4, concentrated in vacuo, and purified by FC on silica gel (gradient: pentane to pentane/EtOAc 1∶1).
Supplementary Material
Acknowledgments.
This work was made possible by grants from the Danish National Research Foundation, the Carlsberg Foundation, Deutsche Forschungsgemeinschaft, and OChemSchool.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. D.W.C.M. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.0914523107/-/DCSupplemental.
References
- 1.Hoveyda AH, Evans DA, Fu GC. Substrate-directable chemical reactions. Chem Rev. 1993;93:1307–1370. [Google Scholar]
- 2.Cha JK, Kim N-S. Acyclic stereocontrol induced by allylic alkoxy groups. Synthetic applications of stereoselective dihydroxylation in natural product synthesis. Chem Rev. 1995;95:1761–1795. [Google Scholar]
- 3.Johansen M, Jørgensen KA. Allylic amination. Chem Rev. 1998;98:1689–1708. doi: 10.1021/cr970343o. [DOI] [PubMed] [Google Scholar]
- 4.Gais H-J, Theil F. In: Enzyme Catalysis in Organic Synthesis. Drauz K, Waldmann H, editors. Weinheim: Wiley-VCH; 2002. pp. 335–578. [Google Scholar]
- 5.Johnson RA, Sharpless KB. In: Catalytic Asymmetric Synthesis. Ojima I, editor. New York: Wiley-VCH; 1993. pp. 103–158. [Google Scholar]
- 6.Martin-Matute B, Bäckvall J-E. Asymmetric Organic Synthesis with Enzymes. Weinheim, Germany: Wiley-VCH; 2008. Dynamic kinetic resolutions; pp. 89–113. [Google Scholar]
- 7.Corey EJ, Helal CJ. Reduction of carbonyl compounds with chiral oxazaborolidine catalysts: A new paradigm for enantioselective catalysis and a powerful new synthetic method. Angew Chem Int Edit. 1998;37:1986–2012. doi: 10.1002/(SICI)1521-3773(19980817)37:15<1986::AID-ANIE1986>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 8.Noyori R, Okhuma T. Asymmetric catalysis by architectural and functional molecular engineering: Practical chemo- and stereoselective hydrogenation of ketones. Angew Chem Int Edit. 2001;40:40–73. [PubMed] [Google Scholar]
- 9.Skucas E, Ngai M-Y, Komanduri V, Krische MJ. Enantiomerically enriched allylic alcohols and allylic amines via C-C bond-forming hydrogenation: Asymmetric carbonyl and imine vinylation. Acc Chem Res. 2007;40:1394–1401. doi: 10.1021/ar7001123. [DOI] [PubMed] [Google Scholar]
- 10.Trost BM, Crawley ML. Asymmetric transition-metal-catalyzed allylic alkylations: Applications in total synthesis. Chem Rev. 2003;103:2921–2944. doi: 10.1021/cr020027w. [DOI] [PubMed] [Google Scholar]
- 11.Lu Z, Ma SM. Metal-catalyzed enantioselective allylation in asymmetric synthesis. Angew Chem Int Edit. 2008;47:258–297. doi: 10.1002/anie.200605113. [DOI] [PubMed] [Google Scholar]
- 12.Trost BM, Organ MG. Deracemization of cyclic allyl esters. J Am Chem Soc. 1994;116:10320–10321. [Google Scholar]
- 13.Trost BM, Tsui H-C, Toste FD. Deracemization of Baylis-Hillman adducts. J Am Chem Soc. 2000;122:3534–3535. [Google Scholar]
- 14.Lüssem BJ, Gais H-J. Palladium-catalyzed deracemization of allylic carbonates in water with formation of allylic alcohols: Hydrogen carbonate ion as nucleophile in the palladium-catalyzed allylic substitution and kinetic resolution. J Am Chem Soc. 2003;125:6066–6067. doi: 10.1021/ja034109t. [DOI] [PubMed] [Google Scholar]
- 15.Gais HJ, et al. Highly selective palladium catalyzed kinetic resolution and enantioselective substitution of racemic allylic carbonates with sulfur nucleophiles: Asymmetric synthesis of allylic sulfides, allylic sulfones, and allylic alcohols. Chem-Eur J. 2003;9:4202–4221. doi: 10.1002/chem.200204657. [DOI] [PubMed] [Google Scholar]
- 16.Trost BM, Bunt RC. Asymmetric induction in allylic alkylations of 3-(acyloxy)cycloalkenes. J Am Chem Soc. 1994;116:4089–4090. [Google Scholar]
- 17.Uozumi H, Tanaka K, Shibatomi K. Asymmetric allylic amination in water catalyzed by an amphiphilic resin-supported chiral palladium complex. Org Lett. 2004;6:281–283. doi: 10.1021/ol036264i. [DOI] [PubMed] [Google Scholar]
- 18.But TYS, Toy PH. The Mitsunobu reaction: Origin, mechanism, improvements and applications. Chem-Asian J. 2007;2:1340–1355. doi: 10.1002/asia.200700182. [DOI] [PubMed] [Google Scholar]
- 19.Jiang H, et al. Target-directed organocatalysis: A direct asymmetric catalytic approach to chiral propargylic and allylic fluorides. J Am Chem Soc. 2009;131:7153–7157. doi: 10.1021/ja901459z. [DOI] [PubMed] [Google Scholar]
- 20.Jiao P, Kawasaki M, Yamamoto H. A sequential O-nitrosoaldol and grignard addition process: An enantio- and diastereoselective entry to chiral 1,2-diols. Angew Chem Int Edit. 2009;48:3333–3336. doi: 10.1002/anie.200900682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Simmons B, Walji AM, MacMillan DWC. Cycle-specific organocascade catalysis: Application to olefin hydroamination, hydro-oxidation, and amino-oxidation, and to natural product synthesis. Angew Chem Int Edit. 2009;48:4349–4353. doi: 10.1002/anie.200900220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ishikawa H, Suzuki T, Hayashi Y. High-yielding synthesis of the anti-influenza neuramidase inhibitor (-)-oseltamivir by three “one-pot” operations. Angew Chem Int Edit. 2009;48:1304–1307. doi: 10.1002/anie.200804883. [DOI] [PubMed] [Google Scholar]
- 23.Enders D, Hüttl MRM, Runsink J, Raabe G, Wendt B. Organocatalytic one-pot asymmetric synthesis of functionalized tricyclic carbon frameworks from a triple-cascade/diels-alder sequence. Angew Chem Int Edit. 2007;46:467–469. doi: 10.1002/anie.200603434. [DOI] [PubMed] [Google Scholar]
- 24.Michrowska A, List B. Concise synthesis of ricciocarpin A and discovery of a more potent analogue. Nature Chemistry. 2009;1:225–228. doi: 10.1038/nchem.215. [DOI] [PubMed] [Google Scholar]
- 25.Lim SM, Hill N, Myers AG. A method for the preparation of differentiated trans-1,2-diol derivatives with enantio- and diastereocontrol. J Am Chem Soc. 2009;131:5763–5765. doi: 10.1021/ja901283q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Melchiorre P, Marigo M, Carlone A, Bartoli G. Asymmetric aminocatalysis—gold rush in organic chemistry. Angew Chem Int Edit. 2008;47:6138–6171. doi: 10.1002/anie.200705523. [DOI] [PubMed] [Google Scholar]
- 27.MacMillan DWC. The advent and development of organocatalysis. Nature. 2008;455:304–308. doi: 10.1038/nature07367. [DOI] [PubMed] [Google Scholar]
- 28.Dondoni A, Massi A. Asymmetric organocatalysis: From infancy to adolescence. Angew Chem Int Ediy. 2008;47:4638–4660. doi: 10.1002/anie.200704684. [DOI] [PubMed] [Google Scholar]
- 29.Barbas CF., III Organocatalysis lost: Modern chemistry, ancient chemistry, and an unseen biosynthetic apparatus. Angew Chem Int Edit. 2007;47:42–47. doi: 10.1002/anie.200702210. [DOI] [PubMed] [Google Scholar]
- 30.Erkkila A, Majander I, Pihko PM. Iminium catalysis. Chem Rev. 2007;107:5416–5470. doi: 10.1021/cr068388p. [DOI] [PubMed] [Google Scholar]
- 31.Enders D, Grondal C, Hüttl MRM. Asymmetric organocatalytic domino reactions. Angew Chem Int Ediy. 2007;46:1570–1581. doi: 10.1002/anie.200603129. [DOI] [PubMed] [Google Scholar]
- 32.Wang X-W, Reisinger CM, List B. Catalytic asymmetric epoxidation of cyclic enones. J Am Chem Soc. 2008;130:6070–6071. doi: 10.1021/ja801181u. [DOI] [PubMed] [Google Scholar]
- 33.Reisinger CM, Wang X-W, List B. Catalytic asymmetric hydroperoxidation of α,β-unsaturated ketones: An approach to enantiopure peroxyhemiketals, epoxides, and aldols. Angew Chem Int Edit. 2008;47:8112–8115. doi: 10.1002/anie.200803238. [DOI] [PubMed] [Google Scholar]
- 34.Lu X-J, Liu Y, Sun B-F, Cindric B, Deng Li. Catalytic enantioselective peroxidation of α,β-unsaturated ketones. J Am Chem Soc. 2008;130:8134–8135. doi: 10.1021/ja802982h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pesciaioli F, et al. Organocatalytic asymmetric aziridination of enones. Angew Chem Int Edit. 2008;47:8703–8706. doi: 10.1002/anie.200803647. [DOI] [PubMed] [Google Scholar]
- 36.Armstrong S, Baxter CA, Lamont SG, Pape AR, Wincewicz R. Amine-promoted, organocatalytic aziridination of enones. Org Lett. 2007;9:351–353. doi: 10.1021/ol062852v. [DOI] [PubMed] [Google Scholar]
- 37.Wharton P, Bohlen DH. Hydrazine reduction of α,β-epoxy ketones to allylic alcohols. J Org Chem. 1961;26:3615–3616. [Google Scholar]
- 38.Siewert J, Sandmann R, von Zezschwitz P. Rhodium-catalyzed enantioselective 1,2-addition of aluminum organyl compounds to cyclic enones. Angew Chem Int Edit. 2007;46:7122–7124. doi: 10.1002/anie.200701087. [DOI] [PubMed] [Google Scholar]
- 39.Stork G, Williard PG. Five- and six-membered-ring formation from olefinic α,β-epoxy ketones and hydrazine. J Am Chem Soc. 1977;99:7067–7068. [Google Scholar]
- 40.Ohloff G, Uhde G. Uncommon cyclization in the reaction of (+)-epoxy-alfa-dihydroionone and hydrazine hydrate. Helv Chim Acta. 1970;53:531–541. [Google Scholar]
- 41.Stork G, Baine NH. Cyclization of vinyl radicals: A versatile method for the construction of five- and six-membered rings. J Am Chem Soc. 1982;104:2321–2323. [Google Scholar]
- 42.Carlone A, Marigo M, North C, Landa A, Jørgensen KA. A simple asymmetric organocatalytic approach to optically active cyclohexenones. Chem Commun. 2006;47:4928–4930. doi: 10.1039/b611366d. [DOI] [PubMed] [Google Scholar]
- 43.Skull BK, Sakai T, Nichols JB, Koreeda M. Mitsunobu reaction of unbiased cyclic allylic alcohols. J Org Chem. 1997;62:8294–8303. doi: 10.1021/jo9615155. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.