

Abstract
The catalytic generation of chiral ester enolate equivalents from α,β-unsaturated aldehydes with chiral N-hetereocyclic carbene catalysts makes possible highly enantioselective hetero-Diels–Alder reactions. The reactions proceed under simple, mild conditions with both aliphatic and aromatic substituted enals as substrates. Previous attempts to employ these starting materials as enolate precursors gave structurally different products via catalytically generated homoenolate equivalents. Critical to the success of the enolate generation was the strength of the catalytic base used to generate the active N-heterocyclic carbene catalyst. To complement these studies, we have investigated the enolate structure using computational methods and find that it prefers conformations perpendicular to the triazolium core.
Keywords: asymmetric synthesis, catalysis, reaction mechanism
The catalytic generation of uniquely reactive chemical species from simple, inert starting materials makes possible synthetic access to complex, stereochemically rich organic molecules. This concept differs from the equally important and successful concept of substrate activation, in which a catalyst such as a Lewis acid or organocatalyst enhances the inherent reactivity of a substrate such as an aldehyde or electron-deficient olefin. Although great success in the generation of uniquely reactive species such as metallocarbenes or carbon nucleophiles is common in transition metal-mediated reactions, this mode of catalysis is less well established in the emerging area of organocatalysis.
An exception to this generalization is the rich chemistry of azolium-catalyzed reactions, exemplified by the long known benzoin dimerization of aldehydes promoted by the cofactor thiamine and its derivatives (1, 2). The catalysts derived from these azolium salts, typically categorized as N-heterocyclic carbenes (NHC) (3), undergo reactions with aldehydes that lead to catalytic generation of acyl anion equivalents. These species have long been known to act as nucleophiles in reactions with a range of electrophilic partners including aldehydes, electron-deficient olefins (Stetter reactions) (4), imines (5), and oxidants (6, 7). As we and Glorius first demonstrated in 2004 (8, 9), the use of α,β-unsaturated aldehydes and suitable catalysts makes possible the catalytic generation of homoenolate equivalents for the synthesis of γ-lactones, γ-lactams (10, 11), cyclopentenes (12, 13), and numerous catalytic cascades (14, 15) often with high levels of enantioselectivity.
Beyond acyl anion and homoenolate equivalents, the combination of N-heterocyclic carbenes and α-functionalized aldehydes also makes possible the catalytic generation of two other important species: acyl azoliums (16, 17), which serve as activated carboxylic acids, and ester enolate equivalents (18). Since the initial disclosures of this possibility in 2004 by our group (19), and that of Rovis (20), these concepts have been widely employed and developed into an impressive range of unique reactions, often with high levels of diastereoselectivity or enantioselectivity (21, 22) under mild, simple reaction conditions.
Among the most exciting of these discoveries has been the catalytic generation of chiral ester enolate equivalents for highly enantioselective carbon–carbon bond-forming reactions (Fig. 1). We have applied this approach to the generation of enolate equivalents from α-chloroaldehydes and their surrogates for highly enantioselective inverse-electron demand Diels–Alder reactions of enones to give enantiopure dihydropyranone products (23). We have likewise used the protonation of electron-deficient α,β-unsaturated aldehydes to generate such enolates and trapped these with N-sulfonyl, α,β-unsaturated imines to afford dihydropyridinone products (18). Similar enolates can also be generated from the corresponding stable ketenes, and the research group of Smith and Ye have documented several elegant applications of this chemistry (24–27). Most recently, Scheidt and co-workers have used enolates generated from α-aryloxy aldehydes for enantioselective Mannich reactions (28).
Fig. 1.

Starting materials for catalytically generated enolate equivalents using NHC catalysts.
All of these catalytic reactions proceed from starting materials that are either unstable or require several steps for their preparation. For example, the α-chloro aldehydes used in our own work have a modest shelf life, although they can be conveniently prepared and used as their corresponding bisulfite adducts (29). The use of isolated ketenes and α-aryloxy aldehydes adds several steps to the preparation of the starting materials. We therefore sought to use α,β-unsaturated aldehydes as substrates, as these compounds are widely employed in a variety of enantioselective organocatalytic reactions and they are increasingly commercially available or readily prepared by improved methods (30). Although this has proven possible in carefully designed intramolecular systems, such as those reported by Scheidt and co-workers (31), the use of simple α,β-unsaturated aldehydes as ester enolate precursors has not been achieved. Attempts to date have resulted in dramatically different reactivity as the homoenolate pathway dominates (Scheme 1) (12, 13). This can be exploited for highly enantioselective cascade reactions but not for the desirable intermolecular reactions of catalytically generated ester enolate equivalents.
Scheme 1.

In this paper, we document the successful use of α,β-unsaturated aldehydes as starting materials for the catalytic generation of chiral ester enolate equivalents and their use in inverse-electron demand Diels–Alder reaction with enones. Remarkably, a change in the cocatalytic amine base employed in these reactions can completely shift the reaction pathway to the hetero-Diels–Alder reaction, which proceeds via a catalytically generated enolate, to the complete exclusion of an alternative pathway that occurs via a fomal homoenolate equivalent (Scheme 1). The same chiral triazolium catalyst affords the hetero-Diels–Alder products in outstanding yield and enantioselectivity. These conditions are successful for a range of α,β-unsaturated aldehyde enolate precurors and various electron-deficient enones.
Results and Discussion
In 2009, we disclosed a highly enantioselective cascade reaction of α,β-unsaturated aldehydes and α′-hydroxyenone 2 to give cyclopentyl lactone 4 as a mixture of isomers (14, 15). During optimization of this reaction, we observed that weaker amine bases led to the formation of the hetero-Diels–Alder product 3. Although we had previously obtained such products using α-chloroaldehydes as starting materials, prior attempts to generate and trap enolate intermediates from simple enals were unsuccessful. Using enone 2 and cinnamaldehyde as substrates, we further investigated conditions that favored the formation of the Diels–Alder product (Table 1).
Table 1.
Conditions for catalytically generation of enolate equivalents from cinnamaldehyde for hetero-Diels–Alder reactions* 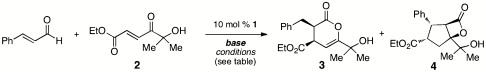
| Entry | Conditions | Base | Ratio 3∶4† |
| 1 | 0.1 M THF, RT | 25% DBU | 1∶1 |
| 2 | 0.1 M EtOH, RT | 25% DBU | No annulation products |
| 3 | 0.1 M DCE, RT | 25% DBU | 1∶1 |
| 0.1 M Tol, RT | 25% DBU | 1∶5 | |
| 8 | 25% DIPEA | 1∶0 (side products observed)‡ | |
| 9 | 25% DMAP | 1∶0 | |
| 10 | 1 equivalent DBU | 0∶1 | |
| 1.5 equivalent NEt3 | 1∶5 | ||
| 11 | 0.1 M Tol, 40 °C | 25% DBU | 1∶10 |
| 12 | 0.1 M CH2Cl2, 40 °C | 15% DMAP | 1∶0 (10∶1 d.r., 99% ee) |
| 13 | 15% NMM | 1∶0 (> 20∶1 d.r., 99% ee) |
*Reaction time: 18–24 h. DBU, 1,8-diazabicyclo[5.4.0]undec-7-ene; DMAP, N,N-4-dimethylaminopyridine; NMM, N-methylmorpholine; DIPEA. diisopropylethylamine RT, room temperature; d.r., diastereomeric ratio; ee, enantiomeric excess.
†Product ratios determined by 1H NMR of unpurified reaction mixtures. Minor cyclopentane-derived isomers also observed, see refs. 14 and 15 for details.
‡Side products include substrate decomposition or unidentified materials.
Three factors were found to be beneficial for favoring the desired Diels–Alder reaction. First, the use of weak amine bases was crucial; DMAP (conjugate acid pKa = 9.2) and N-methyl morpholine (NMM, conjugate acid pKa = 7.4) gave the best results. A stronger base such as DBU (conjugate acid pKa = 12) or excess of moderate bases such as NEt3 (conjugate acid pKa = 10.8) gave predominantly cyclopentane products (entries 1 and 2). Toluene and CH2Cl2 were the preferred solvents; selectivity decreased with THF or 1,2-dichloroethane. Finally, slightly elevated temperatures (40 °C) gave cleaner reactions. Although the reaction times and yields were similar with either DMAP or NMM (entries 12 and 13), better diastereoselectivities were observed with the less basic NMM. For reactions where epimerization is not an issue, either base may be used.
As the catalytic generation of the reactive enolate equivalent occurs via protonation of the β-position of the extended Breslow intermediate (32), we anticipated that this reaction might be limited to a narrow range of enals. This fear proved to be unfounded, and the conditions identified in Table 2 were applicable to electron-rich and electron-deficient cinnamaldehyde derivatives (Entries 1–5). Remarkably, the reaction tolerates an unprotected aromatic aldehyde without competing benzoin or oxidation reactions (entry 5). Acrolein is also a viable substrate (entry 6). Although not investigated in this series, simple aliphatic enals such as 2-hexenal are excellent substrates for the Diels–Alder reactions vide infra.
Table 2.
NHC-catalyzed reactions of alpha-hydroxy ketoenone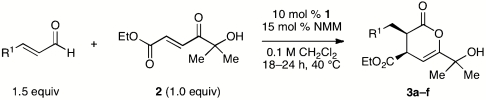
| Entry | R1 = | % yield* | d.r.† | % ee‡ |
| 1 | Ph | 93 | > 20∶1 | 99 |
| 2 | p-BrC6H4 | 93 | > 20∶1 | 99 |
| 3 | p-MeOC6H4 | 92 | > 20∶1 | 99 |
| 4 | 2-furyl | 85 | > 20∶1 | 99 |
| 5 | p-CHOC6H4 | 96 | > 20∶1 | 99 |
| 6 | H | 89 | > 20∶1 | 99 |
*Isolated yield following chromatography.
†Determined by 1H NMR of unpurified reaction mixtures.
‡Determined by SFC analysis on chiral stationary phases. SFC, supercritical fluid chromatography.
In addition to the use of synthetically versatile enone 2, we have also applied this catalyst and conditions to annulations with three other classes of enones. N-Cbz protected enone 5 is also an outstanding substrate and gave the expected products in excellent yield and enantiopurity using DMAP as the catalytic base (Table 3). In contrast to enone 2, the formation of cyclopentane derivatives was never observed as side products. It is not clear if this should be attributed to the reduced electrophilicity of 5, or the possibility that the amide N-H acts to protonate the conjugated Breslow intermediate, leading exclusively to the hetero-Diels–Alder pathway. When DBU was employed as the catalytic base, no cyclopentane products were observed, suggesting that these enones are less electrophilic than the corresponding alcohols.
Table 3.
NHC-catalyzed reactions of N-Cbz protected ketoenone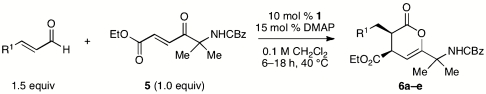
| Entry | R1 = | % yield* | d.r.† | % ee‡ |
| 1 | Ph | 98% | > 20∶1 | 99 |
| 2 | p-BrC6H4 | 98% | > 20∶1 | 99 |
| 3 | p-MeOC6H4 | 79% | > 20∶1 | 99 |
| 4 | 2-furyl | 98% | 20∶1 | 99 |
| 5 | n-C3H7 | 80% | > 20∶1 | 99 |
*Isolated yield following chromatography.
†Determined by 1H NMR of unpurified reaction mixtures.
‡Determined by SFC analysis on chiral stationary phases.
Our original work on NHC-catalyzed Diels–Alder reactions via catalytically generated enolates employed electron-deficient enones as substrates. With some limitations the optimized conditions from Table 1 were also applicable to these substrate classes. γ-Ketoenones bearing either electron-rich aromatic substituents (Table 4) or aliphatic (Table 5) gave products in good yields and outstanding stereoselectivies. Methyl-substituted enones required higher concentrations and longer reaction times to reach full conversion, but these changes did not compromise the high levels of stereoselectivity. With more electron-deficient variants, the formation of cyclopentane derivatives via the alternative annulation pathway became important (Eq. 1).  1
1
Table 4.
NHC-catalyzed reactions of para-methoxyphenyl ketoenone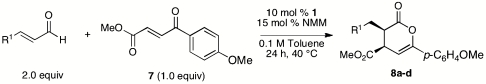
| Entry | R1 = | % yield* | d.r.† | % ee‡ |
| 1 | Ph | 62 | > 20∶1 | 99 |
| 2 | p-BrC6H4 | 70 | > 20∶1 | 99 |
| 3 | H | 54 | > 20∶1 | 99 |
| 4 | n-C3H7 | 60 | > 20∶1 | 99 |
*Isolated yield following chromatography.
†Determined by 1H NMR of unpurified reaction mixtures.
‡Determined by SFC analysis on chiral stationary phases.
Table 5.
NHC-catalyzed reactions of methyl ketoenone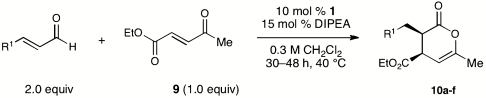
| Entry | R1 = | % yield* | d.r.† | % ee‡ |
| 1 | Ph | 94 | > 20∶1 | 99 |
| 2 | p-MeOC6H4 | 70 | > 20∶1 | 99 |
| 3 | 2-furyl | 54 | > 20∶1 | 99 |
| 4 | p-BrC6H4 | 88 | > 20∶1 | 99 |
| 5 | H | 63 | > 20∶1 | 99 |
| 6 | n-C3H7 | 95 | > 20∶1 | 94 |
*Isolated yield following chromatography.
†Determined by 1H NMR of unpurified reaction mixtures.
‡Determined by SFC analysis on chiral stationary phases.
In light of these findings, we were somewhat surprised to find that α-keto-β,γ-unsaturated esters were excellent substrates (Table 6). This makes possible the synthesis of synthetic challenging vicinal dialkyl stereocenters in excellent yield and with orthogonal synthetic handles for further elaboration. Although the use of acrolein in this reaction gave somewhat lower yields (entries 2 and 4), it provides access to important stereochemical arrays including erythro-dimethyl building blocks that have been the subject of recent efforts by several groups (33, 34). With α-keto-β-γ-enones bearing aromatic substituents, the reactions gave complex product mixtures under these conditions and further optimization has not yet been explored*.
Table 6.
NHC-catalyzed reactions of alpha-ketoenone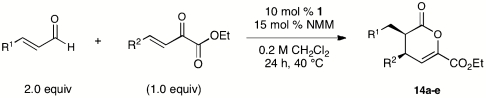
| Entry | R1 = | R2 = | % yield* | % ee†,‡ |
| 1 | Ph | Me | 85 | 98 |
| 2 | H | Me | 51 | 99 |
| 3 | n-C3H7 | cyclo-Hex | 79 | 98 |
| 4 | H | cyclo-Hex | 49 | 99 |
| 5 | Ph | iso-C3H7 | 91 | 96 |
*Isolated yield following chromatography.
†All products > 20∶1 dr as determined by 1H NMR of unpurified mixtures.
‡Determined by SFC analysis on chiral stationary phases.
In seeking to rationalize the high levels of enantioselectivity observed across several classes of enones and N-sulfonyl imines, we have revisited our initially proposed stereochemical model for the hetero-Diels–Alder cycloadditions. Our original analysis suggested that the intermediate azolium-enolate complex formed a conjugated, planar structure between the azolium and the enolate olefin. Upon conformational searching, we have found that the two lowest-lying enolate structures are ones in which the triazolium moiety and the enolate are nearly perpendicular. (For computational studies on the azolium catalyzed benzoin and Stetter reactions, see ref. 35.) These two conformers differ in the orientation of the enolate, with the lowest-lying structure positioning the enolate oxygen on the same face as the indene ring. The N-mesityl group blocks the approach of the oxodiene, providing a rationalization for the necessity of this substitutent in highly enantioselective annulations catalyzed by chiral N-heterocyclic carbenes. Approach of the oxodiene toward the open face of this enolate conformation via an endo transition state gives the observed absolute stereochemistry of the products. We therefore believe that an enolate with the structure shown in Fig. 2A is the operative nucleophile in these hetero-Diels–Alder reactions and in the several other unique reactions that invoke them as intermediates.
Fig. 2.

The two lowest energy conformers from geometry optimization with HF 6-31G*. Initial conformers were identified via Monte Carlo conformational searches using the Merck Force Field Model (MFFM). Four other low-lying conformational isomers were also optimized and found to be higher in energy (see SI Appendix).
Discussion
The catalytic generation of chiral enolates and their use in this hetero-Diels–Alder pathways highlights the impressive ability of a single catalyst and substrate combination to access multiple, nonobvious reactive species by subtle changes in the reaction conditions. As shown in Fig. 3, the combination of an α,β-unsaturated aldehyde and an N-heterocyclic carbene can lead to the formation of four discrete reactive intermediates: (i) acyl anion equivalents, (ii) homoenolate equivalents, (iii) ester enolate equivalents, and (iv) activated carboxylates. Both the structure of the azolium precatalyst and the nature of cocatalytic amine base determine the reaction pathway. Remarkable selectivity, often completely favoring one pathway to the exclusion of the other three, can be obtained in NHC-catalyzed reactions of aldehydes. In the present case, selectivity between the homoenolate and enolate pathways is determined by the fate of conjugated Breslow-intermediate i. If the conjugate acid of the catalytic base, generated by deprotonation of the azolium salt, is acidic enough to protonate the Breslow intermediate at the β-position, the formation of enolate equivalent iii occurs. Provided that the enone reactant is sufficiently electrophilic, annulation occurs to give the hetero-Diels–Alder products. The success of the reactions, and the product distribution, is therefore determined by the relative rates of C─C bond formation from the reactive intermediates and the two protonation steps.
Fig. 3.

Catalytically generated reactive species from α,β-unsaturated aldehydes with N-heterocyclic carbene catalysts.
Although not the focus of this study, the structure and electronics of the N-heterocyclic carbene catalyst also play a crucial role in determining the product outcome. We have previously demonstrated that more electronic-rich imidazolium-derived catalysts favor the homoenolate pathways, whereas triazolium-derived structures enhance protonation and lead to the enolate and activated carboxylates (36). The exceptions occur with very good electrophiles, such as enones 2 and 11, particularly when stronger amine bases, such as DBU, that give weaker conjugate acids are employed.
Conclusion
In summary, we have identified conditions for the selective generation of NHC-bound enolates from α,β-unsaturated aldehydes and their use in highly enantioselective hetero-Diels–Alder reactions with electron-deficient enones. These reactions occur under mild, simple conditions and afford synthetically valuable products in excellent yield with outstanding stereoselectivities using a single catalyst precursor. These studies reinforce the remarkable range of reactivities available to α,β-unsaturated aldehydes, which in this study serve as precursors to nucleophilic ester enolates rather than their more traditional roles as C1 or C3 electrophiles, and the versatility of N-heterocyclic carbenes in generating and controlling uniquely reactive intermediates. Further efforts to explore the utility of these catalytically generated intermediates and unique catalysts to enhance their reactivity are ongoing.
Supplementary Material
Acknowledgments.
We are grateful to National Institute of General Medical Sciences (National Institutes of Health, GM–079339), the National Science Foundation (CHE-0449587), Bristol–Myers Squibb, and Roche for support of this research. J.K. is a graduate fellow of the Royal Thai Government. Instrumentation support for computing was provided by the National Science Foundation Chemistry Research Instrumentation and Facilities program (CHE0131132).
Footnotes
The authors declare no conflict of interest.
*This probably reflects the higher electrophilicity of these substrates, leading to competing reactions. High yields of the identical Diels–Alder adducts can be obtained with these substrates with chiral enolates generated from α-halo aldehydes.
This article is a PNAS Direct Submission. D.W.C.M. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1007469107/-/DCSupplemental.
References
- 1.Enders D, Niemeier O, Henseler A. Organocatalysis by N-heterocyclic carbenes. Chem Rev. 2007;107:5606–5655. doi: 10.1021/cr068372z. [DOI] [PubMed] [Google Scholar]
- 2.Moore JL, Rovis T. Carbene catalysis. Top Curr Chem. 2009;291:77–144. doi: 10.1007/978-3-642-02815-1_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hermann WA. N-Heterocyclic carbenes: A new concept in organometallic catalysis. Angew Chem Int Edit. 2002;41:1290–1309. doi: 10.1002/1521-3773(20020415)41:8<1290::aid-anie1290>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 4.Stetter H, Kuhlmann H. Paquette LA, editor. The catalyzed nucleophilic addition of aldehydes to electrophilic double bonds. Org Reactions. 1991;40:407–496. [Google Scholar]
- 5.Murry JA, et al. Synthesis of α-amido ketones via organic catalysis: Thiazolium-catalyzed cross-coupings of aldehydes with acylimines. J Am Chem Soc. 2001;123:9696–9697. doi: 10.1021/ja0165943. [DOI] [PubMed] [Google Scholar]
- 6.Guin J, Desarkar S, Grimme S, Studer A. Biomimetic carbene-catalyzed oxidations of aldehydes using tempo. Angew Chem Int Edit. 2008;47:8727–8730. doi: 10.1002/anie.200802735. [DOI] [PubMed] [Google Scholar]
- 7.Sarkar SD, Grimme S, Studer A. NHC catalyzed oxidations of aldehydes to esters: Chemoselective acylation of alcohols in presence of amines. J Am Chem Soc. 2010;132:1190–1191. doi: 10.1021/ja910540j. [DOI] [PubMed] [Google Scholar]
- 8.Sohn SS, Rosen EL, Bode JW. N-heterocyclic carbene-catalyzed generation of homoenolates: γ-butyrolactones by direct annulations of enals and aldehydes. J Am Chem Soc. 2004;126:14370–14371. doi: 10.1021/ja044714b. [DOI] [PubMed] [Google Scholar]
- 9.Burstein C, Glorius F. Organocatalyzed conjugate umpolung of alpha,beta-unsaturated aldehydes for the synthesis of gamma-butyrolactones. Angew Chem Int Edit. 2004;43:6205–6208. doi: 10.1002/anie.200461572. [DOI] [PubMed] [Google Scholar]
- 10.He M, Bode JW. Catalytic synthesis of γ-lactams via direct annulations of enals and n-sulfonylimines. Org Lett. 2005;7:3131–3134. doi: 10.1021/ol051234w. [DOI] [PubMed] [Google Scholar]
- 11.Rommel M, Fukuzumi T, Bode JW. Cyclic ketimines as superior electrophiles for NHC-catalyzed homoenolate additions with broad scope and low catalyst loadings. J Am Chem Soc. 2008;130:17266–17267. doi: 10.1021/ja807937m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nair V, Vellalath S, Poonoth M, Suresh E. N-heterocyclic carbene-catalyzed reaction of chalcones and enals via homoenolate: An efficient synthesis of 1,3,4-trisubstituted cyclopentenes. J Am Chem Soc. 2006;128:8736–8737. doi: 10.1021/ja0625677. [DOI] [PubMed] [Google Scholar]
- 13.Chiang PC, Kaeobamrung J, Bode JW. Enantioselective, cyclopentene-forming annulations via NHC-catalyzed benzoin–oxy-Cope reactions. J Am Chem Soc. 2007;129:3520–3521. doi: 10.1021/ja0705543. [DOI] [PubMed] [Google Scholar]
- 14.He M, Bode JW. Enantioselective, NHC-catalyzed bicyclo-β-lactam formation via direct annulations of enals and unsaturated N-sulfonyl ketimines. J Am Chem Soc. 2008;130:17266–17267. doi: 10.1021/ja0778592. [DOI] [PubMed] [Google Scholar]
- 15.Kaeobamrung J, Bode JW. Stereodivergency of triazolium and imidazolium-derived NHCs for catalytic, enantioselective cyclopentane synthesis. Org Lett. 2009;11:677–680. doi: 10.1021/ol802739d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bruice TC, Kundu NG. Displacement reactions on 2-acetyl- 3,4-dimethylthiazolium iodide. J Am Chem Soc. 1966;88:4097–4098. [Google Scholar]
- 17.Lienhard G. Kinetics and mechanism of the hydrolysis of 2-acetyl-3,4-dimethylthiazolium ion. J Am Chem Soc. 1966;88:5642–5649. [Google Scholar]
- 18.He M, Struble JR, Bode JW. Highly enantioselective azadiene Diels-Alder reactions catalyzed by chiral N-heterocyclic carbenes. J Am Chem Soc. 2006;128:8418–8420. doi: 10.1021/ja062707c. [DOI] [PubMed] [Google Scholar]
- 19.Chow KYK, Bode JW. Catalytic generation of activated carboxylates: Direct, stereoselective synthesis of β-hydroxyesters from epoxyaldehydes. J Am Chem Soc. 2004;126:8126–8127. doi: 10.1021/ja047407e. [DOI] [PubMed] [Google Scholar]
- 20.Reynolds NT, Read de Alaniz J, Rovis T. Conversion of α-haloaldehydes into acylating agents by an internal redox reaction catalyzed by nucleophilic carbenes. J Am Chem Soc. 2004;126:9518–9519. doi: 10.1021/ja046991o. [DOI] [PubMed] [Google Scholar]
- 21.Reynolds NT, Rovis T. Enantioselective protonation of catalytically generated chiral enolates as an approach to the synthesis of α-chloroesters. J Am Chem Soc. 2005;127:16406–16407. doi: 10.1021/ja055918a. [DOI] [PubMed] [Google Scholar]
- 22.Vora HU, Rovis T. N-Heterocyclic carbene catalyzed asymmetric hydration: direct synthesis of α-protio and α-deuterio α-chloro and α-fluoro carboxylic acids. J Am Chem Soc. 2010;132:2860–2861. doi: 10.1021/ja910281s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He M, Uc GJ, Bode JW. Chiral, N-Heterocyclic carbene catalyzed oxodiene Diels-Alder reactions with low catalyst loadings. J Am Chem Soc. 2006;128:15088–15089. doi: 10.1021/ja066380r. [DOI] [PubMed] [Google Scholar]
- 24.Zhang YR, Lv H, Zhou D, Ye S. Chiral N-heterocyclic carbene-catalyzed formal [4 + 2] cycloaddition of ketenes with enones: Highly enantioselective synthesis of trans- and cis-delta-lactones. Chem Eur J. 2008;14:8473–8476. doi: 10.1002/chem.200801165. [DOI] [PubMed] [Google Scholar]
- 25.Wang XN, Lv H, Huang XL, Ye S. Asymmetric esterification of ketenes catalyzed by an N-heterocyclic carbene. Org Biomol Chem. 2009;7:346–350. doi: 10.1039/b815139c. [DOI] [PubMed] [Google Scholar]
- 26.Duguet N, Campbell CD, Slawin AMZ, Smith AM. N-heterocyclic carbene catalysed-lactam synthesis. Org Biomol Chem. 2008;6:1108–1113. doi: 10.1039/b800857b. [DOI] [PubMed] [Google Scholar]
- 27.He L, Zhang YR, Ye S. Formal cycloaddition of disubstituted ketenes with 2-oxoaldehydes catalyzed by chiral N-heterocyclic carbene. J Org Chem. 2008;73:8101–8103. doi: 10.1021/jo801494f. [DOI] [PubMed] [Google Scholar]
- 28.Kawanaka Y, Phillips EM, Scheidt KA. N-heterocyclic carbene-catalyzed enantioselective Mannich reactions with α-aryloxyacetaldehyde. J Am Chem Soc. 2009;131:18028–18029. doi: 10.1021/ja9094044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He M, Beahm BJ, Bode JW. Chiral NHC-catalyzed oxodiene Diels-Alder reactions with α-chloroaldehyde bisulfite salts. Org Lett. 2008;10:3817–3820. doi: 10.1021/ol801502h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Valenta P, Drucker NA, Bode JW, Walsh PJ. Simple one-pot conversion of aldehydes and ketones to enals. Org Lett. 2009;11:2117–2119. doi: 10.1021/ol9005757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Phillips EM, Wadamoto M, Chan A, Scheidt KA. Highly enantioselective intramolecular Michael additions catalyzed by N-heterocyclic carbenes. Angew Chem Int Edit. 2007;46:3107–3110. doi: 10.1002/anie.200605235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sohn SS, Bode JW. Catalytic generation of activated carboxylates from enals: A product determining role for the base. Org Lett. 2005;7:3873–3876. doi: 10.1021/ol051269w. [DOI] [PubMed] [Google Scholar]
- 33.Zhao J, Burgess K. Synthesis of vicinal dimethyl chirons by asymmetric hydrogenation of trisubstituted alkenes. J Am Chem Soc. 2009;131:13236–13237. doi: 10.1021/ja905458n. [DOI] [PubMed] [Google Scholar]
- 34.van Zijl AW, Szymanski W, López F, Minnaard AJ, Feringa BL. Catalytic enantioselective synthesis of vicinal dialkyl arrays. J Org Chem. 2008;73:6994–7002. doi: 10.1021/jo8010649. [DOI] [PubMed] [Google Scholar]
- 35.Dudding T, Houk KN. Computational predictions of stereochemistry in asymmetric thiazolium and triazolium catalyzed benzoin condensations. Proc Natl Acad Sci USA. 2004;101:5770–5775. doi: 10.1073/pnas.0307256101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Struble JR, Kaeobamrung J, Bode JW. Synthesis of an N-mesityl substituted chiral imidazolium salt for NHC-catalyzed reactions. Org Lett. 2008;10:957–960. doi: 10.1021/ol800006m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.