

Abstract
In spite of the many catalytic methodologies available for the asymmetric functionalization of carbonyl compounds at their α and β positions, little progress has been achieved in the enantioselective carbon–carbon bond formation γ to a carbonyl group. Here, we show that primary amine catalysis provides an efficient way to address this synthetic issue, promoting vinylogous nucleophilicity upon selective activation of unmodified cyclic α,β-unsaturated ketones. Specifically, we document the development of the unprecedented direct and vinylogous Michael addition of β-substituted cyclohexenone derivatives to nitroalkenes proceeding under dienamine catalysis. Besides enforcing high levels of diastereo- and enantioselectivity, chiral primary amine catalysts derived from natural cinchona alkaloids ensure complete γ-site selectivity: The resulting, highly functionalized vinylogous Michael adducts, having two stereocenters at the γ and δ positions, are synthesized with very high fidelity. Finally, we describe the extension of the dienamine catalysis-induced vinylogous nucleophilicity to the asymmetric γ-amination of cyclohexene carbaldehyde.
Keywords: asymmetric synthesis, Michael reaction, organocatalysis
The stereoselective functionalization of carbonyl compounds, particularly while concomitantly forming a new carbon–carbon bond, represents one of the more efficient and potent chemical ways to produce valuable chiral molecules. This target has inspired generations of synthetic chemists to design unique chiral catalysts able to enantioselectively forge a stereocenter α or β to a carbonyl group (1–3). In this context, intense investigations into the aldol (4, 5) and Michael reactions (6, 7) have made them invaluable tools in modern organic chemistry. Recently, the classical organometallic-based approach has been enriched by the possibility of using chiral primary or secondary amines as efficient catalysts for the asymmetric functionalization of carbonyl compounds. This strategy is known as “asymmetric aminocatalysis” (8–11). Among the many advantages of this synthetic approach (12), one of its more attractive features is the ability to directly generate, in situ, the catalytically active intermediates from unmodified carbonyl compounds (13, 14).
Both metal- and organic-based approaches have achieved levels of reliability such that synthetic chemists can now address even the most daunting issues connected with the asymmetric catalytic functionalization of carbonyl compounds at their α and β positions. In contrast, little progress has been reported in the corresponding enantioselective carbon–carbon bond formation γ to a carbonyl group (15). Among the few useful approaches devised to date, the concept of vinylogous nucleophilicity is the most powerful (16–19). Formulated by Fuson in 1935 as the transmission of electronic effects through a conjugated π system (20), this principle accounts for the use of γ-enolizable α,β-unsaturated carbonyl compounds as precursors of nucleophilic dienolate equivalents, formally inverting the usual reactivity of this compound class. Vinylogous processes offer an efficient entry onto functionalized building blocks having high level of structural complexity; however, designing asymmetric catalytic versions is not simple. Indeed, every approach to vinylogous reactions overlays the challenge of site-selectivity onto the already present issue of stereo-selectivity. In general, the critical regiochemical issue can be addressed by judiciously preparing preformed, stable dienolate equivalents. This strategy has been successfully applied to asymmetric vinylogous aldol (17, 21), Mannich (18, 22), and Michael reactions (19, 23). Avoiding the stoichiometric preactivation of the vinylogous nucleophilic components would logically improve this approach, particularly from the standpoint of atom economy (24). However, examples of direct, catalytic, and asymmetric vinylogous reactions are rare: Recently, Trost and Hitce reported on the direct vinylogous Michael addition of 2(5H)-furanone to nitroalkenes under dinuclear zinc catalysis (25), whereas chiral Brønsted base catalysis proved successful to activate specific reactive alkenes, such as α,α-dicyano olefins, toward vinylogous nucleophilicity (26).
Within this context, we wondered whether asymmetric aminocatalysis could solve the challenge of a direct vinylogous nucleophilic addition of unmodified γ-enolizable carbonyl compounds. We were inspired by a recent report by Jørgensen and coworkers on the direct, enantioselective γ amination of α,β-unsaturated aldehydes using azodicarboxylates as the electrophilic nitrogen source (27). The process is based on a unique activation mode, dienamine catalysis, which exploits the condensation of a chiral secondary amine catalyst with β-aliphatic-substituted unsaturated compounds. This condensation leads to the formation of the expected electrophilic iminum ion intermediate, which is in equilibrium with an electron-rich dienamine intermediate. The dienamine, exploiting its γ-nucleophilic character, actually represents the catalytic active species. In principle, the ability to promote the in situ formation of a dienamine from γ-enolizable carbonyl compounds while enforcing γ-site selectivity during the nucleophilic path might offer a unique and potent way to design direct vinylogous processes (Fig. 1).
Fig. 1.

Dienamine catalysis and the concept of vinylogy; Elec, electrophile.
In spite of the potential to address important synthetic issues, such as the effective catalytic generation of a new carbon–carbon bond and a new stereocenter γ to a carbonyl group, dienamine catalysis has found limited application (28, 29). Accordingly, a recently published perspective on the advent of organocatalysis did not include dienamine catalysis among the generic modes of activation and induction (30). This lack was probably due to the fact that γ-amination of unsaturated aldehydes was originally proposed to follow a [4 + 2] cycloaddition path (27), instead of a more generalizable nucleophilic addition manifold. Moreover, some recent studies suggest that chiral secondary amines, such as proline (31) and its derivatives (32, 33), can activate γ-enolizable unsaturated aldehydes toward the formation of the dienamine intermediate, but generally promoting an α-site-selective alkylation step via enamine catalysis in the presence of suitable electrophiles. Nonetheless, a recent inspiring report by Christmann and coworkers has highlighted the potential of dienamine catalysis for enforcing a direct nucleophilic γ-addition path, albeit in an intramolecular way (34).
Here, we show that dienamine catalysis, induced by chiral primary amines, can efficiently promote the direct, intermolecular vinylogous Michael addition of unmodified β-substituted cyclohexenone derivatives to nitroalkenes, imparting high levels of diastereo- and enantioselectivity, and ensuring exclusive γ-site selectivity. Notably, the two stereocenters at the γ and δ positions of the carbonyl moiety are formed with very high fidelity (Scheme 1).
Scheme 1.

The direct vinylogous Michael addition developed in this study.
Results and Discussion
Background.
Our laboratory and others, independently, have recently introduced 9-amino(9-deoxy)epicinchona alkaloids (Fig. 2), chiral primary amines easily derived from natural sources, as general and effective catalysts for a wide variety of asymmetric α and β functionalizations of ketones (35, 36). We have further demonstrated the ability of catalyst A, derived from hydroquinine, to combine orthogonal aminocatalytic modes (iminium and enamine activations) into one mechanism, thus promoting cascade reactions with α,β-unsaturated ketones (37, 38) and even with the challenging α,β-disubstituted enals (39).
Fig. 2.

Primary amine catalysts used within this study.
Next, we decided to investigate the behavior of this versatile catalyst in the context of the elusive γ-site activation of unmodified enones, in order to design a dienamine-catalyzed direct, vinylogous Michael reaction. From the outset, we were fully aware of the inherent difficulties of our target, because the presence of multiple potential sites of enolization has greatly hampered the use of α,β-unsaturated ketones in even the stoichiometric version of vinylogous reactions (40). We decided to attack this problem by selecting β-substituted cyclohexenone derivatives as a model substrate. At first glance, this compound class seems highly challenging, because it further enhances the task of regioselectivity (Fig. 3). The condensation of a chiral primary amine catalyst with cyclic enones would lead to the formation of the iminium ion, which could equilibrate, upon selective α′-deprotonation or γ- and γ′-deprotonation, with the kinetic cross-conjugated dienamine intermediate I or the thermodynamic extended dienamines II and III, respectively. Although the former would open an avenue for α′-site alkylation (path a in Fig. 3), the extended dienamines could be alkylated at three different positions, namely α, γ, and even γ′, behaving as d2 or d4 reagents (paths b, d, and c, respectively).
Fig. 3.

Challenges arising from the site-selective formation of dienamines.
Despite the increased complexity posed by β-substituted cyclic enones, our choice was motivated by a variety of considerations. Our experience in designing of cascade reactions (37, 38) strongly indicated that, when reacted with acyclic, linear α,β-unsaturated ketones, catalyst A easily facilitates the equilibrium between the iminium ion and the nucleophilic cross-conjugated dienamine intermediate of type I, selectively directing the reaction manifold toward an α′-site alkylation (path a in Fig. 3). We thus considered using cyclohexenone derivatives to coax the regiocontrolled formation of extended dienamines within a more thermodynamically favored six-membered ring scaffold. This idea is based upon related enolization studies demonstrating that, under certain conditions, the selective formation of the thermodynamic exo-cyclic enolate of type III is strongly favored over both the endo-isomer and the kinetic cross-conjugated dienolate (41–45). This inherent behavior of β-substituted cyclohexenones has found a wonderful application in the conceptually unique, direct, vinylogous aldol reaction reported by Yamamoto and coworkers (46, 47): The complete γ-site selectivity induced by the nonchiral, bulky aluminum-based catalyst has been rationalized on the basis of catalyst steric effects (48, 49), which inhibit base deprotonation at the α′ position, whereas thermodynamic factors account for the formation of the reactive exo-dienolate isomer of the β-methyl 2-cyclohexen-1-one (46). On these grounds, we considered the unique ability of catalyst A, in combination with an acidic cocatalyst, to perturb the iminium-dienamine equilibrium, taken together with thermodynamic factors, that may govern the regioselective formation of the exo-cyclic dienamine intermediate. We wondered if this ability might be exploited to develop the challenging γ-site-selective, direct stereoselective addition of β-methyl 2-cyclohexen-1-one to nitrostyrene derivatives (following the nucleophilic path d in Fig. 3). Theoretical calculations accounting for a thermodynamically driven site-selective formation of a dienamine of type III are reported in the SI Appendix.
Organocatalytic Vinylogous Michael Addition.
The vinylogous Michael addition under dienamine catalysis was first evaluated by mixing 2 equivalents of β-methyl 2-cyclohexen-1-one 1 and nitrostyrene 2a in toluene (1 M, 48 h, 40 °C). In accordance with our mechanistic postulate, the chiral primary amine A (20 mol %), in combination with 30 mol % of 2-fluorobenzoic acid as the cocatalyst, directed the reaction manifold toward a γ-site-selective addition, leading to compound 3a as the unique product of the process and with a good level of enantiocontrol (Table 1, entry 1). Examination of the reaction media revealed that the catalytic process was greatly influenced by polarity, with solvents with a high dielectric constant strongly affecting both reactivity and enantioselectivity (Table 1, entries 1–5). The nature of the acidic cocatalysts was also a crucial parameter, with stronger acids leading to worse results (Table 1, entries 1, 6, and 7). This evidence suggests that fine tuning the carboxylic acid cocatalyst is essential to modulate the perturbation of the delicate equilibrium between iminium ion, cross-conjugated and extended dienamine intermediates during the reaction.
Table 1.
Optimization studies of the vinylogous Michael addition under dienamine catalysis
| Entry | Amine | Acid | Solvent | Conversion, %* | ee, %† |
| 1 | A | 2F-C6H4CO2H | Toluene | > 95 | 82 |
| 2 | A | 2F-C6H4CO2H | CHCl3 | 52 | 79 |
| 3 | A | 2F-C6H4CO2H | THF | 30 | 82 |
| 4 | A | 2F-C6H4CO2H | MeOH | < 5 | — |
| 5 | A | 2F-C6H4CO2H | MeCN | 12 | — |
| 6 | A | TFA | Toluene | < 5 | — |
| 7 | A | 2NO2-C6H4CO2H | Toluene | 45 | 80 |
| 8 | B | 2F-C6H4CO2H | Toluene | > 95 | 98 |
| 9 | B | 2F-C6H4CO2H | Toluene | > 95 (77)‡ | 98§ |
TFA, trifluoroacetic acid; reactions carried out using two equivalents of enone 1
*Conversion determined by 1H NMR analysis of the crude mixture; only product 3a, derived from a γ-site-selective alkylation step, has always been detected.
†The enantiomeric excess (ee) was determined by HPLC analysis on chiral stationary phases.
‡Number in parentheses refers to yield of the isolated compound 3a.
§Reaction performed in the presence of 10 mol % of B and 20 mol % of 2F-C6H4CO2H and using [2]0 = 0.2 M
These results further consolidate A as a general, highly versatile catalyst for the activation of keto compounds, even toward vinylogous nucleophilicity. However, we were not satisfied with the level of stereocontrol in the present chemistry. We speculated that using a bifunctional catalyst capable of simultaneously activating both the electrophilic and nucleophilic components might lead to higher catalytic activity and, more importantly, to better stereocontrol (50). To this end, we tested the potential of 6′-hydroxy-9-amino-9-deoxyepiquinine B to synergistically and productively bind the two reaction partners of the vinylogous Michael addition. B has recently been introduced by Chen et al. as an efficient bifunctional catalyst of the asymmetric 1,3-dipolar cycloaddition of cyclic enones (51). Catalyst B greatly improved the enantioselectivity as well as the reaction rate of the vinylogous addition of 1 to 2a while maintaining a complete γ-selectivity (Table 1, entry 8). The enhanced catalytic activity allowed us to lower the catalyst loading to 10 mol % (Table 1, entry 9), delineating a more practical synthetic protocol (extensive optimization studies can be found within the SI Appendix).
The best result was obtained with the catalytic salt made by combining B (10 mol %) and 2-fluorobenzoic acid (20 mol %) in toluene (0.2 M). These conditions were selected to examine the scope of the vinylogous Michael addition by evaluating a variety of nitroalkenes (Table 2). Different substituents at the aromatic moiety of β-nitrostyrene derivatives were well-tolerated, regardless of their electronic properties, because the corresponding adducts 3 were obtained in good to high yield and almost perfect stereocontrol (enantiomeric excesses ranging from 95% to 98%). The pseudoenantiomeric catalyst C, derived from quinidine, accounted for the possibility of accessing both antipodes of the products (Table 2, entries 2 and 10).
Table 2.
Vinylogous Michael addition: Scope of the nitrostyrene derivatives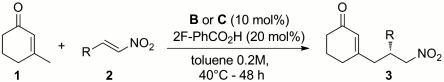
| Entry | Catalyst | R | 3 | Yield, %* | ee, %† |
| 1 | B | Ph | a | 77 | 98 |
| 2 | C | Ph | a | 55 | 95‡ |
| 3 | B | 4-MeO-Ph | b | 70 | 97 |
| 4 | B | 4-Me-Ph | c | 70 | 98 |
| 5 | B | 4-Br-Ph | d | 73 | 97 |
| 6 | B | 2-Cl-Ph | e | 84 | 96 |
| 7 | B | 2-F-Ph | f | 83 | 97 |
| 8 | B | 2-thiophenyl | g | 68 | 98 |
| 9 | B | 2-OBn-Ph | h | 87 | 97 |
| 10 | C | 2-OBn-Ph | h | 68 | 96‡ |
Bn, benzyl; reactions carried out using two equivalents of enone 1. 1H NMR analysis of the crude mixture indicated a highly γ-site-selective alkylation pathway, other products arising from different reaction manifolds (e.g., α′-alkylation under enamine catalysis, see ref. 52) being sporadically detected in negligible amounts.
*Yield of the isolated compounds 3.
†The enantiomeric excess (ee) was determined by HPLC analysis on chiral stationary phases.
‡The opposite (S) enantiomer has been obtained using catalyst C.
As limitations of the direct vinylogous Michael reactions, aliphatic nitroalkenes did not react under the described conditions. Moreover, modifying the cyclic scaffold of the nucleophilic component (i.e., 3-methyl 2-cyclopenten-1-one) resulted in a complete loss of reactivity, a result that highlights how strongly the cyclic scaffold geometry influences (and drives) the selective formation of the thermodynamic, extended dienamine intermediate.
The absolute configuration of the stereogenic center was unambiguously determined to be R by anomalous dispersion X-ray crystallographic analysis of the bromide derivative 3d. The ability of the catalyst to communicate its inherent stereochemical information while forging the new stereocenter at the δ position, several atoms apart from the catalyst binding point within the covalent dienamine intermediate, seemed noteworthy to us. It is also intriguing to consider how primary amine catalysis can impart unique mechanistic pathways, thus complementing and enriching the established reactivity profile of secondary amine catalysis. Within this context, a recent report has demonstrated that a chiral secondary amine behaves in a completely different way when exposed to the very same reagents combination, namely β-methyl cyclohexenone and nitrostyrene, catalyzing a Diels-Alder reaction between 1 and 2a via the selective formation of cross-conjugated dienamine of type I (52).
Our vinylogous protocol could be successfully extended to a β,β-disubstituted nitrostyrene, leading to the stereocontrolled generation of compound 4 having an all-carbon quaternary (53) stereocenter (Scheme 2). Moreover, a different class of Michael acceptor has proven to be a viable component of the dienamine-catalyzed direct vinylogous addition. Mixing β-methyl 2-cyclohexen-1-one with trans-α-cyanocinnamate under the catalysis of A furnished the corresponding vinylogous adduct with two stereogenic centers. As reported in Scheme 3, to avoid the epimerization event, we designed a one-pot vinylogous Michael addition/amination tandem sequence, directly leading to compound 5 with complete control over the relative stereochemistry and high enantioselectivity (54).
Schemes 2 and 3.

Next, we studied the possibility of forging two contiguous stereogenic centers at the γ and δ positions, examining the vinylogous Michael addition of differently β-substituted cyclohexanones to a variety of nitrostyrene derivatives under the catalysis of the salt made by combining 20 mol % of the chiral primary amine B with 30 mol % of 2-fluorobenzoic acid. In line with previous observations, the nature of the carboxylic acid cocatalyst strongly influenced both the reactivity and the stereochemical outcome of the process: Carrying out the reaction in the presence of 40 mol % of salicylic acid afforded higher enantiocontrol and an increased reaction rate, albeit with slightly lower diastereoselectivity (Table 3, entries 1 and 2). Thus, both catalyst salt combinations, where B is mixed with 2-fluoro- or 2-hydroxybenzoic acids, have been evaluated. Selected results are reported in Table 3. In general, a variety of substrate combinations can be realized, leading to products 6, readily amenable to further functionalizations, with high levels of γ-site, diastereo- and enantioselectivity.
Table 3.
Stereoselective creation of vicinal stereocenters under dienamine catalysis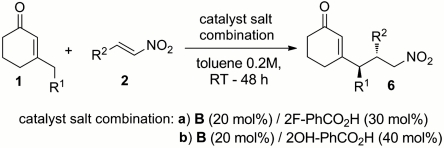
| Entry | Catalyst salt combination | R1 | R2 | 6 | Yield, %* | dr | ee, %† |
| 1 | a | Me | Ph | a | 72 | 9∶1 | 92 |
| 2 | b | Me | Ph | a | 80 | 6∶1 | 94§ |
| 3 | a | Me | 4-MeO-Ph | b | 78 | 10∶1 | 94 |
| 4 | a | Me | 4-Me-Ph | c | 76 | 10∶1 | 92 |
| 5 | b | Me | 4-Br-Ph | d | 81 | 5∶1 | 93 |
| 6 | b | Me | 4-NO2-Ph | e | 65 | 3∶1 | 95 |
| 7 | b | Me | 2-F-Ph | f | 80 | 6∶1 | 90 |
| 8 | b | Me | 2-thiophenyl | g | 90‡ | 3.3∶1 | 94 |
| 9 | b | Bn | Ph | h | 65‡ | 7∶1 | 85 |
| 10 | b | allyl | Ph | i | 44 | 11.5∶1 | 94§ |
| 11 | b | propyl | Ph | j | 86‡ | 11.5∶1 | 95 |
| 12 | b | propyl | 4-MeO-Ph | k | 72 | 13.5∶1 | 91§ |
| 13 | b | propyl | 4-Br-Ph | l | 50 | 10∶1 | 94§ |
| 14 | b | Ph | Ph | m | 86‡ | 2∶1 | 90§ |
Bn, benzyl; reactions carried out using two equivalents of enones 1. 1H NMR analysis of the crude mixture indicated a highly γ-site-selective alkylation pathway, other products arising from different reaction manifolds (e.g., α′-alkylation under enamine catalysis, see ref. 52) being sporadically detected in negligible amounts.
*Refers to the isolated single, major diastereoisomer.
†The enantiomeric excess (ee) was determined by HPLC analysis on chiral stationary phases.
‡Refers to the isolated mixture of diastereoisomers.
§Enantiomerically pure products obtained after a single crystallization.
Crystals from bromide 6l and from compound 6i were suitable for X-ray analysis, which established the absolute configuration of the vinylogous reaction as well as its anti-stereochemical outcome. Interestingly, the observed sense of relative stereoinduction is not common in the corresponding enamine-catalyzed Michael addition of carbonyls to nitroalkenes, which generally leads to a syn-relationship (55, 56).
Finally, to explore the potential of the chiral primary amine-induced vinylogous nucleophilicity, we wondered whether this unique reactivity concept may be translated to an aldehyde derivative adorned with a six-membered ring scaffold, reminiscent of the β-substituted cyclohexanone framework. Although the vinylogous Michael addition of 1-cyclohexene-1-carboxaldehyde 7 to nitrostyrene 2a did not proceed at all, the combination with tert-butylazodicarboxylate under the catalysis of A furnished the γ-amination product 8 with perfect regio- and enantioselectivity (Scheme 4).
Scheme 4.

Concluding Remarks
The direct, γ-site-selective addition of β-substituted cyclohexenone derivatives to nitroalkenes represents one of the few examples of catalytic, asymmetric vinylogous Michael reactions of unmodified carbonyl compounds. This unprecedented chemical transformation affords highly functionalized compounds, having two stereocenters at the γ and δ positions, with high enantiomeric purity. In addition to its synthetic interest, this study confirms the ability of chiral primary amine catalysis to impart unique reactivity profiles, thus expanding the potential of asymmetric aminocatalysis. Specifically, dienamine catalysis has been exploited to promote vinylogous nucleophilicity within addition reaction manifolds. We believe this reactivity may be further extended to a variety of vinylogous donors and acceptors as well as to nucleophilic substitution reactions.
Methods and Materials
All the vinylogous adducts were fully characterized: Structural proofs and spectral data for all compounds are provided in the SI Appendix.
All the reactions were carried out in undistilled solvent without any precautions to exclude moisture. To a solution of 9-amino(9-deoxy)epicinchona alkaloids A–C (0.08 mmol) in 0.8 mL of toluene, ortho-fluorobenzoic acid (0.16 mmol, 22.4 mg) was added at room temperature under stirring. After 10 min, the reaction was started with the addition of β-substituted cyclohexenone derivative 1 (0.8 mmol, 2.0 equiv) immediately followed by the nitroalkene 2 (0.4 mmol, 1.0 equiv), and the mixture was allowed to reach 40 °C. Stirring was continued until complete conversion of the starting material (24–48 h, checked by thin layer chromatography). The reaction mixture was then directly purified by flash column chromatography (SiO2, 20–30% ethyl acetate in hexane) to yield the vinylogous adducts 3 or 6.
Supplementary Material
Acknowledgments.
We thank F. Pesciaioli for fruitful discussions during the early stage of the research project. G. Bergonzini is gratefully acknowledged for her experimental support. This work was supported by Bologna University, the Catalan Institution for Research and Advanced Studies, and the Institute of Chemical Research of Catalonia Foundation.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. D.W.C.M. is a guest editor invited by the Editorial Board.
Data deposition: The atomic coordinates have been deposited in the Cambridge Structural Database, Cambridge Crystallographic Data Centre, Cambridge CB2 1EZ, United Kingdom (CSD reference nos. 763174, 763175, and 763176).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1001150107/-/DCSupplemental.
References
- 1.Noyori R. Synthesizing our future. Nat Chem. 2009;1:5–6. doi: 10.1038/nchem.143. [DOI] [PubMed] [Google Scholar]
- 2.Jacobsen EN, Pfaltz A, Yamamoto H, editors. Comprehensive Asymmetric Catalysis. Vol. 3. Berlin: Springer; 1999. pp. 997–1473. [Google Scholar]
- 3.Enders D, Narine AA. Lessons from nature: Biomimetic organocatalytic carbon-carbon bond formations. J Org Chem. 2008;73:7857–7870. doi: 10.1021/jo801374j. [DOI] [PubMed] [Google Scholar]
- 4.Mahrwald R, editor. Modern Aldol Reactions. Vol. 1. New York: Wiley; 2004. pp. 1–326. [Google Scholar]
- 5.Carreira EM, Fettes A, Marti C. Catalytic enantioselective aldol addition reactions. Org Reactions. 2006;67:1–216. [Google Scholar]
- 6.Christoffers J, Koripelly G, Rosiak A, Rössle M. Recent advances in metal-catalyzed asymmetric conjugate additions. Synthesis. 2007;(9):1279–1300. [Google Scholar]
- 7.Tsogoeva SB. Recent advances in asymmetric organocatalytic 1,4-conjugate additions. Eur J Org Chem. 2007;(11):1701–1716. [Google Scholar]
- 8.List B. Emil Knoevenagel and the roots of aminocatalysis. Angew Chem Int Edit. 2010;49:1730–1734. doi: 10.1002/anie.200906900. [DOI] [PubMed] [Google Scholar]
- 9.Bertelsen S, Jørgensen KA. Organocatalysis—after the gold rush. Chem Soc Rev. 2009;38:2178–2189. doi: 10.1039/b903816g. [DOI] [PubMed] [Google Scholar]
- 10.Melchiorre P, Marigo M, Carlone A, Bartoli G. Asymmetric aminocatalysis—gold rush in organic chemistry. Angew Chem Int Edit. 2008;47:6138–6171. doi: 10.1002/anie.200705523. [DOI] [PubMed] [Google Scholar]
- 11.Barbas CF., III Organocatalysis lost: Modern chemistry, ancient chemistry, and an unseen biosynthetic apparatus. Angew Chem Int Edit. 2008;47:42–47. doi: 10.1002/anie.200702210. [DOI] [PubMed] [Google Scholar]
- 12.Dalko PI, editor. Enantioselective Organocatalysis Reactions and Experimental Procedures. Weinheim: Wiley-VCH; 2007. pp. 1–116. [Google Scholar]
- 13.List B. Enamine catalysis is a powerful strategy for the catalytic generation and use of carbanion equivalents. Acc Chem Res. 2004;37:548–557. doi: 10.1021/ar0300571. [DOI] [PubMed] [Google Scholar]
- 14.Saito S, Yamamoto H. Design of acid-base catalysis for the asymmetric direct aldol reaction. Acc Chem Res. 2004;37:570–579. doi: 10.1021/ar030064p. [DOI] [PubMed] [Google Scholar]
- 15.Smith SW, Fu GC. Asymmetric carbon-carbon formation γ to a carbonyl group: Phosphine-catalyzed addition of nitromethane to allenes. J Am Chem Soc. 2009;131:14231–14233. doi: 10.1021/ja9061823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Casiraghi G, Zanardi F, Appendino G, Rassu G. The vinylogous aldol reaction: A valuable, yet understated carbon-carbon bond-forming maneuver. Chem Rev. 2000;100:1929–1972. doi: 10.1021/cr990247i. [DOI] [PubMed] [Google Scholar]
- 17.Denmark SE, Heemstra JR, Jr, Beutner GL. Catalytic, enantioselective, vinylogous aldol reactions. Angew Chem Int Edit. 2005;44:4682–4698. doi: 10.1002/anie.200462338. [DOI] [PubMed] [Google Scholar]
- 18.Martin SF. Evolution of the vinylogous Mannich reaction as a key construction for alkaloid synthesis. Acc Chem Res. 2002;35:895–904. doi: 10.1021/ar950230w. [DOI] [PubMed] [Google Scholar]
- 19.Christoffers J. Catalysis of the Michael reaction and the vinylogous Michael reaction by ferric chloride hexahydrate. Synlett. 2001;(6):723–732. [Google Scholar]
- 20.Fuson RC. The principle of vinylogy. Chem Rev. 1935;16:1–27. [Google Scholar]
- 21.Denmark SE, Heemstra JR., Jr Lewis base activation of Lewis acids vinylogous aldol addition reactions of conjugated N,O-silyl ketene acetals to aldehydes. J Am Chem Soc. 2006;128:1038–1039. doi: 10.1021/ja056747c. [DOI] [PubMed] [Google Scholar]
- 22.Sickert M, Schneider C. The enantioselective, Brønsted acid catalyzed, vinylogous Mannich reaction. Angew Chem Int Edit. 2008;47:3631–3634. doi: 10.1002/anie.200800103. [DOI] [PubMed] [Google Scholar]
- 23.Brown SP, Goodwin NC, MacMillan DWC. The first enantioselective organocatalytic Mukaiyama-Michael reaction: A direct method for the synthesis of enantioenriched γ-butenolide architecture. J Am Chem Soc. 2003;125:1192–1194. doi: 10.1021/ja029095q. [DOI] [PubMed] [Google Scholar]
- 24.Trost BM. Atom Economy—a challenge for organic synthesis: Homogeneous catalysis leads the way. Angew Chem Int Edit. 1995;34:259–281. [Google Scholar]
- 25.Trost BM, Hitche J. Direct asymmetric Michael addition to nitroalkenes: Vinylogous nucleophilicity under dinuclear zinc catalysis. J Am Chem Soc. 2009;131:4572–4573. doi: 10.1021/ja809723u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cui H-L, Chen Y-C. α,α-Dicyanoalkenes: Versatile vinylogous nucleophiles for organic synthesis. Chem Commun. 2009;(30):4479–4486. doi: 10.1039/b906201g. [DOI] [PubMed] [Google Scholar]
- 27.Bertelsen S, Marigo M, Brandes S, Dinér P, Jørgensen KA. Dienamine catalysis: Organocatalytic asymmetric γ-amination of α,β-unsaturated aldehydes. J Am Chem Soc. 2006;128:12973–12980. doi: 10.1021/ja064637f. [DOI] [PubMed] [Google Scholar]
- 28.Hong B-C, et al. Organocatalytic asymmetric Robinson annulation of α,β-unsaturated aldehydes: Applications to the total synthesis of (+)-palitantin. J Org Chem. 2007;72:8459–8471. doi: 10.1021/jo701477v. [DOI] [PubMed] [Google Scholar]
- 29.Liu K, Chougnet A, Woggon W-D. A short route to α-tocopherol. Angew Chem Int Edit. 2008;47:5827–5829. doi: 10.1002/anie.200801765. [DOI] [PubMed] [Google Scholar]
- 30.MacMillan DWC. The advent and development of asymmetric organocatalysis. Nature. 2008;455:304–308. doi: 10.1038/nature07367. [DOI] [PubMed] [Google Scholar]
- 31.Utsumi N, Zhang H, Tanaka F, Barbas CF., III A way to highly enantiomerically enriched aza-Morita-Baylis-Hillman-type products. Angew Chem Int Edit. 2007;46:1878–1880. doi: 10.1002/anie.200603973. [DOI] [PubMed] [Google Scholar]
- 32.Marqués-López E, et al. Crossed intramolecular Rauhut-Currier-type reactions via dienamine activation. Org Lett. 2009;11:4116–4119. doi: 10.1021/ol901614t. [DOI] [PubMed] [Google Scholar]
- 33.Han B, Xiao Y-C, He Z-Q, Chen Y-C. Asymmetric Michael addition of γ,γ-disubstituted α,β-unsaturated aldehydes to nitroolefins via dienamine catalysis. Org Lett. 2009;11:4660–4663. doi: 10.1021/ol901939b. [DOI] [PubMed] [Google Scholar]
- 34.de Figueiredo RM, Fröhlich R, Christmann M. Amine-catalyzed cyclizations of tethered α,β-unsaturated carbonyl compounds. Angew Chem Int Edit. 2008;47:1450–1453. doi: 10.1002/anie.200704688. [DOI] [PubMed] [Google Scholar]
- 35.Bartoli G, Melchiorre P. A novel organocatalytic tool for the iminium activation of α,β-unsaturated ketones. Synlett. 2008;(12):1759–1771. [Google Scholar]
- 36.Chen Y-C. The development of asymmetric primary amine catalysts based on cinchona alkaloids. Synlett. 2008;(13):1919–1930. [Google Scholar]
- 37.Wu L-Y, et al. Organocascade reactions of enones catalyzed by a chiral primary amine. Angew Chem Int Edit. 2009;48:7196–7199. doi: 10.1002/anie.200903280. [DOI] [PubMed] [Google Scholar]
- 38.Bencivenni G, et al. Targeting structural and stereochemical complexity by organocascade catalysis: Construction of spirocyclic oxindoles having multiple stereocentres. Angew Chem Int Edit. 2009;48:7200–7203. doi: 10.1002/anie.200903192. [DOI] [PubMed] [Google Scholar]
- 39.Galzerano P, Pesciaioli F, Mazzanti A, Bartoli G, Melchiorre P. Asymmetric organocatalytic cascade reactions with α-substituted α,β-unsaturated aldehydes. Angew Chem Int Edit. 2009;48:7892–7894. doi: 10.1002/anie.200903803. [DOI] [PubMed] [Google Scholar]
- 40.Denmark SE, Heemstra JR., Jr Lewis base activation of Lewis acids: Catalytic enantioselective vinylogous aldol addition reactions. J Org Chem. 2007;72:5668–5688. doi: 10.1021/jo070638u. [DOI] [PubMed] [Google Scholar]
- 41.Krafft ME, Holton RA. The Kharasch reagent regioselective generation of dienol ethers from enones. J Am Chem Soc. 1984;106:7619–7621. [Google Scholar]
- 42.Stork G, Benaim J. Monoalkylation of α,β-unsaturated ketones via metalloenamines. J Am Chem Soc. 1971;93:5938–5939. [Google Scholar]
- 43.Meinwald J, Hendry L. The deconjugation of isophorone. J Org Chem. 1971;36:1446–1447. [Google Scholar]
- 44.Luo S, Mi X, Xu H, Wang PG, Cheng J-P. Efficient Baylis-Hillman reactions of cyclic enones in methanol as catalyzed by methoxide anion. J Org Chem. 2004;69:8413–8422. doi: 10.1021/jo0491760. [DOI] [PubMed] [Google Scholar]
- 45.Ceccarelli R, Insogna S, Bella M. Organocatalytic regioselective Michael additions of cyclic enones via asymmetric phase transfer catalysis. Org Biomol Chem. 2006;4:4281–4284. doi: 10.1039/b612606e. [DOI] [PubMed] [Google Scholar]
- 46.Saito S, Shiozawa M, Ito M, Yamamoto H. Conceptually new directed aldol condensation using aluminum tris(2,6-diphenylphenoxide) J Am Chem Soc. 1998;120:813–814. [Google Scholar]
- 47.Takikawa H, Ishihara K, Saito S, Yamamoto H. Asymmetric vinylogous direct aldol reaction using aluminum tris[2,6-bis(4-alkylphenyl)phenoxide] Synlett. 2004;(4):732–734. [Google Scholar]
- 48.Saito S, Shiozawa M, Nagahara T, Nakadai M, Yamamoto H. Molecular recognition of carbonyl compounds using aluminum tris(2,6-diphenylphenoxide) (ATPH): New regio- and stereoselective alkylation of α,β-unsaturated carbonyl compounds. J Am Chem Soc. 2000;122:7847–7848. doi: 10.1021/ja0205941. [DOI] [PubMed] [Google Scholar]
- 49.Saito S, Nagahara T, Shiozawa M, Nakadai M, Yamamoto H. Molecular recognition of α,β-unsaturated carbonyl compounds using aluminum tris(2,6-diphenylphenoxide) (ATPH): Structural and conformational analysis of ATPH complexes and application to the selective vinylogous aldol reaction. J Am Chem Soc. 2003;125:6200–6210. doi: 10.1021/ja0205941. [DOI] [PubMed] [Google Scholar]
- 50.Marcelli T, van Maarseveen JH, Hiemstra H. Cupreines and cupreidines: An emerging class of bifunctional cinchona organocatalysts. Angew Chem Int Edit. 2006;45:7496–7504. doi: 10.1002/anie.200602318. [DOI] [PubMed] [Google Scholar]
- 51.Chen W, et al. Enantioselective 1,3-dipolar cycloaddition of cyclic enones catalyzed by multifunctional primary amines: Beneficial effects of hydrogen bonding. Angew Chem Int Edit. 2007;46:7667–7670. doi: 10.1002/anie.200702618. [DOI] [PubMed] [Google Scholar]
- 52.Xu D-Q, et al. In situ enamine activation in aqueous salt solutions: Highly efficient asymmetric organocatalytic Diels-Alder reaction of cyclohexenones with nitroolefins. Angew Chem Int Edit. 2009;48:3821–3824. doi: 10.1002/anie.200900269. [DOI] [PubMed] [Google Scholar]
- 53.Christoffers J, Baro A, editors. Quaternary Stereocenters. Challenges and Solutions in Organic Synthesis. Weinheim: Wiley-VCH; pp. 1–359. [Google Scholar]
- 54.Saaby S, Bella M, Jørgensen KA. Asymmetric construction of quaternary stereocenters by direct organocatalytic amination of α-substituted α-cyanoacetates and β-dicarbonyl compounds. J Am Chem Soc. 2004;126:8120–8121. doi: 10.1021/ja047704j. [DOI] [PubMed] [Google Scholar]
- 55.McCooey SH, Connon SJ. Readily accessible 9-epi-amino cinchona alkaloid derivatives promote efficient, highly enantioselective additions of aldehydes and ketones to nitroolefins. Org Lett. 2007;9:599–602. doi: 10.1021/ol0628006. [DOI] [PubMed] [Google Scholar]
- 56.Uehara H, Barbas CF., III anti-Selective asymmetric Michael reactions of aldehydes and nitroolefins catalyzed by a primary amine/thiourea. Angew Chem Int Edit. 2009;48:9848–9852. doi: 10.1002/anie.200905313. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.