

Abstract
Complementary to enantioselective transformations of planar functionalities, catalytic desymmetrization of meso compounds is another fundamentally important strategy for asymmetric synthesis. However, experimentally established stereochemical models on how a chiral catalyst discriminates between two enantiotopic functional groups in the desymmetrization of a meso substrate are particularly lacking. This article describes our endeavor to elucidate the chemical mechanism and characterization of the active conformation of the cinchona alkaloid-derived catalyst for a desymmetrization of meso cyclic anhydrides via asymmetric alcoholysis. First, our kinetic studies indicate that the cinchona alkaloid-catalyzed alcoholysis proceeds by a general base catalysis mechanism. Furthermore, the active conformer of the cinchona alkaloid-derived catalyst DHQD-PHN was clarified by catalyst conformation studies with a designed, rigid cinchona alkaloid derivative as a probe. These key mechanistic insights enabled us to construct a stereochemical model to rationalize how DHQD-PHN differentiates the two enantiotopic carbonyl groups in the transition state of the asymmetric alcoholysis of meso cyclic anhydrides. This model not only is consistent with the sense of asymmetric induction of the asymmetric alcoholysis but also provides a rationale on how the catalyst tolerates a broad range of cyclic anhydrides. These mechanistic insights further guided us to develop a novel practical catalyst for the enantioselective alcoholysis of meso cyclic anhydrides.
Keywords: cinchona alkoloid, desymmetrization, organocatalysis, general base catalysis, hydrogen bonding
Complementary to enantioselective transformations of planar functionalities, catalytic desymmetrization of meso compounds is another fundamentally important strategy for asymmetric synthesis (1–5). However, our understanding of how a chiral catalyst discriminates between two enantiotopic functional groups in a meso substrate at the molecular level is particularly lacking. Our group reported a desymmetric alcoholysis of a wide range of meso cyclic anhydrides with modified cinchona alkaloids to generate highly enantiomerically enriched hemiesters (5–10). Thus, we have initiated mechanistic studies to investigate how the modified cinchona alkaloids are able to efficiently differentiate the two enantiotopic carbonyl groups while tolerating variations of the substituents of the anhydrides. Herein we describe the experimental results that have enabled us to construct a transition state model to answer these mechanistic questions and to develop a practical catalyst guided by insights gained from our mechanistic studies.
Results and Discussion
In order to shed light on the origin of the catalytic activity on the enantioselective alcoholysis of meso cyclic anhydrides, we carried out kinetic studies on the methanolysis of cis-2,3-dimethyl succinic anhydride (1a) (SI Appendix). Upon treatment with the mono cinchona alkaloid DHQD-PHN (3) and the bis cinchona alkaloid (DHQD)2AQN (4) in diethyl ether at room temperature, methanolysis of anhydride 1a furnished hemiester 2a in 91% and 93% enantiomeric excess (ee), respectively. This enantioselective alcoholysis was found to display a first-order dependence on anhydride 1a as well as a first-order dependence on either mono cinchona alkaloid DHQD-PHN (3) or bis cinchona alkaloid (DHQD)2AQN (4). The reaction first showed a first-order dependence on methanol, which turned to a zero-order dependence as an excess amount of methanol was employed. These kinetic results are consistent with a general base catalysis mechanism (Scheme 1) in which the cinchona alkaloid first forms an aminealcohol hydrogen-bonding complex (8). The alcohol, associated with and activated by the chiral amine, reacts selectively with one of the enantiotopic carbonyl groups of 1a. The same first-order dependence, similar reaction rate, and comparably high enantioselectivity displayed by mono cinchona alkaloid 3 and bis cinchona alkaloid 4, respectively, indicate that one dihydroquinidyl group is sufficient to activate methanol as a nucleophile for the enantioselective alcoholysis as a hydrogen-bonding acceptor.
Scheme 1.

General base catalysis mechanism
Next we turned our attention to the characterization of the active conformer of DHQD-PHN (3) in the transition state. This task was particularly challenging because cinchona alkaloids such as 3 possess considerable conformational flexibility resulting from rotation around both the C8-C9 and the C4′-C9 bonds. Extensive conformational studies on dihydroquinidine and its derivatives by Wynberg and coworkers identified four minimum energy conformers: 11 (app-closed), 12 (app-open), 13 (gauche-open), 14 (gauche-closed) (Fig. 2) (11–13). Our NMR studies indicate that DHQD-PHN (3) in toluene readily adopts all these low energy conformations (SI Appendix), although Sharpless and coworkers reported a solid state structure of 3 as determined by X-ray crystallography in which 3 was found to adopt a gauche-open conformation (14).
Fig. 2.

Four minimum energy conformations of DHQD and its derivatives.
Conformationally rigid cinchona alkaloid derivatives were shown by Corey et al. to be powerful probes for identifying the active conformation of cinchona alkaloids as chiral ligands for the Sharpless asymmetric dihydroxylations of olefins (15–18). Recently, we have also successfully applied β-isocuperidine as a probe to elucidate the active conformation of cinchona alkaloid-derived bifunctional organic catalysts for an asymmetric conjugate addition of β-ketoesters to nitroalkenes (19). Accordingly, we examined the methanolysis of 1a with a rigid cinchona alkaloid 6 (Fig. 1) (20), which was designed to mimic a cinchona alkaloid that is locked in a gauche conformation (open and closed). The 6-catalyzed methanolysis of 1a, compared to that catalyzed by DHQD-PHN (3), proceeded with a drastically reduced enantioselectivity (20% ee vs. 96% ee at -20 °C).
Fig. 1.

Structure of cinchona alkaloids.
This result prompted us to design and synthesize a previously undescribed cinchona alkaloid derivative 7 (Fig. 1) and to investigate its ability to promote the enantioselective alcoholysis. To our knowledge, 7 represents a unique mimicry for a cinchona alkaloid derivative locked in an app conformation. As shown in Scheme 2 our synthesis of 7 began with the preparation of QD-PHN (15) from quinidine. A three-step sequence of hydroboration-oxidation-hydrolysis transformed QD-PHN (15) to alcohol intermediate 16 in 77% overall yield. Treatment of 16 with NaSEt furnished the corresponding 6′-OH derivative, which was converted to the cinchona alkaloid-diborane complex 17. The complex 17 was subjected to sequential alkylation of the 6′-OH and allylation of primary alcohol to generate the diene precursor 19. Ring-closing metathesis of diene 19 with Grubbs (I) catalyst accomplished the key task of forming the 21-membered macrocyclic ring. Hydrolysis of the bisborane complex afforded the desired rigid catalyst 7. An extensive 1H NMR analysis of 7 confirmed that it provided a mimicry of a cinchona alkaloid locked in an app-closed conformation (SI Appendix). To our delight, under identical reaction conditions, methanolysis of 1a with 7 proceeded with similarly high enantioselectivity as that with DHQD-PHN (3) (93% ee for 7 vs. 96% ee for 3). This result demonstrates unambiguously that a cinchona alkaloid in the app-closed conformation is able to promote a highly enantioselective alcoholysis, which provides strong experimental evidence supporting that DHQD-PHN (3) adopts the app-closed conformation as the active conformation when mediating the highly enantioselective methanolysis of meso anhydride 1a. It is noteworthy that this conclusion emerging from our studies was implicated neither by X-ray crystallographic nor by NMR studies of ground state conformations of 3.
Scheme 2.

Synthesis of rigid catalyst 7
On the basis of this insight into the active conformer of DHQD-PHN (3) and results from our kinetic studies as described above, we were able to formulate a transition state model for 3-catalyzed desymmetric alcoholysis of 1a. As illustrated in Scheme 3, in the hydrogen-bonding complex between the alcohol and DHQD-PHN (3) adopting an app-closed conformation, the substituent of the alcohol is postulated to point away from the C6′-methoxy group of the quinoline ring of 3 in order to avoid a steric interaction between them (A vs. B). Thus, as shown in A, the activated alcohol is shielded on three sides by the quinoline ring, C6′-OMe group and the substituent (methyl) of the alcohol. When anhydride 1a approaches the activated alcohol via the open corner, the steric repulsion between 1a and the alcohol-amine complex is minimized when the two methyl groups of 1a are pointing away from the alcohol-amine complex as illustrated in T1. In this transition state (T1), the activated alcohol is able to attack the top carbonyl group (pro-S) in 1a via the stereoelectronically preferred Dunitz–Burgi angle (21). On the other hand, such an alignment between the activated alcohol and the bottom pro-R carbonyl group is not attainable. Thus the alcoholysis of 1a occurs preferentially with the pro-S carbonyl group. Alternative transition state assemblies (T2 and T3) could be conceived in which the anhydride 1a is rotated in various ways to allow the pro-R carbonyl group to be aligned with the activated alcohol for the nucleophilic attack via the Dunitz–Burgi angle. However, compared to T1, they are disfavored because the substituents of anhydride 1a engage in repulsive steric interactions with either the substituent of the alcohol (as shown in T2) or the C6′-OMe group of the cinchona alkaloid 3 (as shown in T3).
Scheme 3.

Transition state model for 3 catalyzed asymmetric alcoholysis
This stereochemical model that emerged from our mechanistic studies is consistent with the sense of asymmetric induction as determined by analysis of absolute configurations of the hemiesters obtained from the cinchona alkaloid-promoted alcoholysis (6). Moreover, the insensitivity of catalytic enantioselectivity toward variations of the substituents of the meso cyclic anhydrides can be easily rationalized, as in T1 the substituents of the anhydrides are located in an open space where variations of their steric and electronic properties are well-tolerated by the cinchona alkaloid catalyst 3.
Nonintuitive predictions can also be made according to this model: (i) The enantioselectivity of the alcoholysis is expected to decrease significantly if the C6′-OMe is removed from DHQD-PHN (3) [i.e., using DHCN-PHN (5) as the catalyst]. (ii) Due to steric hindrance, 3-catalyzed alcoholysis with a secondary alcohol would be dramatically slower than those with primary alcohols. Importantly, these hypotheses have been verified by experimental results (Table 1).
Table 1.
Asymmetric alcoholysis of succinic anhydride 1a 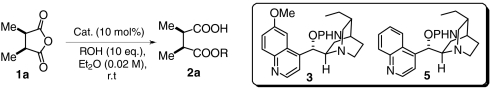
| entry | cat. | ROH | t/h | conv./% | ee/% |
| 1 | DHQD-PHN(3) | MeOH | 2 | 77 | 92 |
| 2 | DHCN-PHN(5) | MeOH | 2 | 49 | 70 |
| 3 | DHQD-PHN(3) | PriOH | 12 | < 5 | — |
This model also suggests that the presence of the sterically bulky phenanthryl group in DHQD-PHN may enforce an app conformation for 3; the steric interaction between the bulky C9-subsituent and the quinuclidinyl group is minimized when the cinchona alkaloid adopts the app-closed conformation (11), whereas it is significantly increased when the cinchona alkaloid adopts the gauche conformations (13 and 14). Following this line of analysis, we began to explore modified cinchona alkaloids bearing a bulky substituent at C9 with the goal of developing a more practical catalyst. Subsequently, we found that cinchona alkaloid QD-MN (21) that could be easily prepared in multigram quantity by a two-step route in 60% overall yield from cheap starting materials such as menthol, α-chloroacetyl chloride and quinidine (Scheme 4), matches the catalytic efficiency of DHQD-PHN (3) and (DHQD)2AQN (4) for the desymmetrization of a wide range of meso anhydrides (Table 2).
Scheme 4.

Preparation of QD-MN (21)
Table 2.
Asymmetric alcoholysis of cyclic anhydrides 1 with QD-MN and (DHQD)2AQN* 
| Entry | Substrate | Catalyst (mol%) | Alcohol (10 eq.) | Temp (°C) | Time | Yield † | ee ‡ |
| 1 | QD-MN (10%) | CF3CH2OH | RT | 5 h | 91% | 94% | |
| 2 | (DHQD)2AQN (5%) | CF3CH2OH | RT | 9 h | 90% | 90% | |
| 3 | QD-MN (10%) | MeOH | RT | 9 h | 90% | 90% | |
| 4 | (DHQD)2 AQN (5%) | MeOH | RT | 5 h | 89% | 94% | |
| 5 | 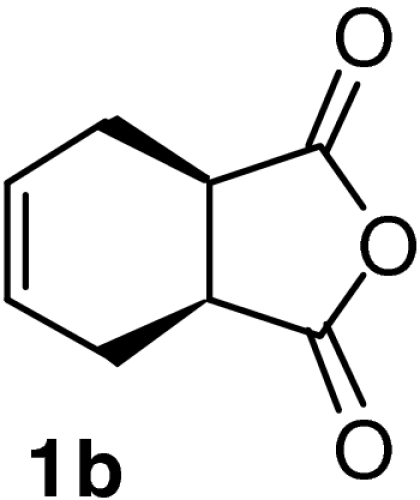 |
QD-MN (14%) | CF3CH2OH | RT | 6 h | 93% | 95% |
| 6 | (DHQD)2AQN (7%) | CF3CH2OH | RT | 5 h | 96% | 91% | |
| 7 | QD-MN (14%) | MeOH | RT | 8 h | 91% | 90% | |
| 8 | (DHQD)2AQN (7%) | MeOH | RT | 6 h | 92% | 94% | |
| 9 | 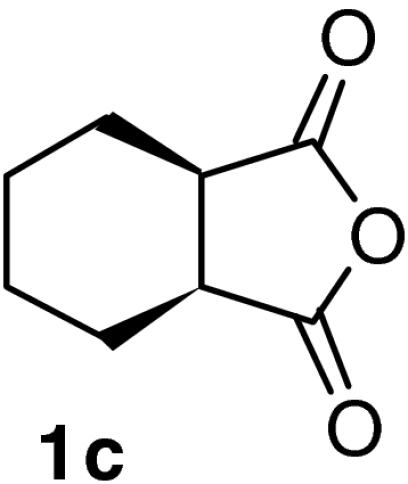 |
QD-MN (10%) | CF3CH2OH2 | RT | 7 h | 96% | 94% |
| 10 | (DHQD)2AQN (5%) | CF3CH2OH | RT | 7 h | 95% | 89% | |
| 11 | QD-MN (10%) | MeOH | RT | 8 h | 94% | 87% | |
| 12 | (DHQD)2AQN (5%) | MeOH | RT | 4 h | 95% | 93% | |
| 13 | 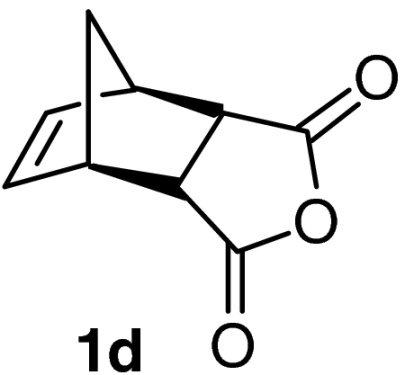 |
QD-MN (20%) | MeOH | RT | 36 h | 88% | 90% |
| 14 | (DHQD)2AQN (10%) | MeOH | RT | 12 h | 90% | 88% | |
| 15 | 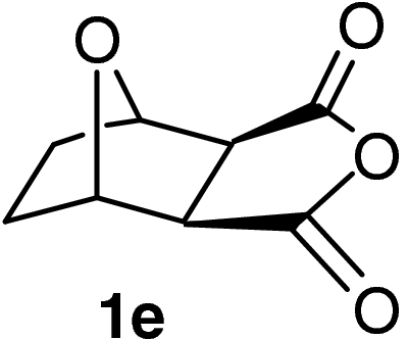 |
QD-MN (30%) | MeOH | −20 | 60 h | 91% | 95% |
| 16 | (DHQD)2AQN (15%) | MeOH | −20 | 120 h | 88% | 96% | |
| 17 | 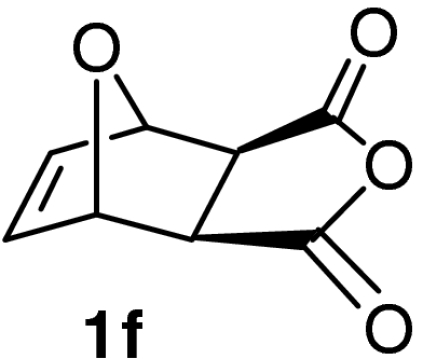 |
QD-MN (40%) | MeOH | −20 | 48 h | 87% | 92% |
| 18 | (DHQD)2AQN (20%) | MeOH | −20 | 96 h | 74% | 92% |
*The reaction was carried out with 1 (0.02 M in diethyl ether) and R’OH [10 equivalent (eq.)] in the presence of catalyst.
†Conversion was determined by 1H NMR.
‡See the SI Appendix for the determination of the ee value.
In summary, kinetic studies and catalyst conformational studies employing a designed rigid cinchona alkaloid derivative as a probe allowed us to determine the origin of catalytic activity and identify and the active conformer of the cinchona alkaloid catalyst for the highly enantioselective alcoholysis. These key mechanistic insights allowed us to formulate a transition state model that clarifies the molecular origin of the highly efficient recognition of enantiotopic carbonyl groups of the meso anhydride by the modified cinchona alkaloid. This model is consistent with the observed sense of asymmetric induction and provides a rationale for the broad scope of the asymmetric alcoholysis established from previous studies. These mechanistic insights also guided our development of a new modified cinchona alkaloid, QD-MN 21, as a more practical catalyst for the enantioselective alcoholysis of meso anhydrides.
Materials and Methods
Dry methanol or 2,2,2-trifluoroethanol (3.0 mmol) was added in one portion to a stirred solution of anhydride (0.3 mmol) and QD-MN (10–40 mol%) or (DHQD)2AQN (5–7 mol%) in diethyl ether (15.0 mL) at the temperature indicated in Table 2. The reaction mixture was stirred at that temperature and the conversion of the starting material was determined by 1H NMR analysis. The ee of each product was determined by GC analysis of the products or by HPLC analysis of a diastereoisomeric mixture of the corresponding amide-ester prepared from the hemiester.
Supplementary Material
Acknowledgments.
This work is dedicated to Professor Lixing Dai with deepest admiration and best wishes. We are grateful for financial support from the National Institutes of Health (GM-61591).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1004439107/-/DCSupplemental.
References
- 1.Wong C-H, Whitesides GM. In: Enzymes in synthetic organic chemistry. Baldwin JE, Magnus PD, editors. Oxford: Elsevier; 1994. [Google Scholar]
- 2.García-Urdiales E, Alfonso I, Gotor V. Enantioselective enzymatic desymmetrizations in organic synthesis. Chem Rev. 2005;105:313–354. doi: 10.1021/cr040640a. [DOI] [PubMed] [Google Scholar]
- 3.Willis MC. Enantioselective desymmetrization. J Chem Soc Perk T I. 1999:1765–1784. [Google Scholar]
- 4.Spivey AC, Andrews BI. Catalysis of the asymmetric desymmetrization of cyclic anhydrides by nucleophilic ring-opening with alcohols. Angew Chem Int Edit. 2001;40:3131–3134. doi: 10.1002/1521-3773(20010903)40:17<3131::AID-ANIE3131>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 5.Chen Y, McDaid P, Deng L. Asymmetric alcoholysis of cyclic anhydrides. Chem Rev. 2003;103:2965–2983. doi: 10.1021/cr020037x. [DOI] [PubMed] [Google Scholar]
- 6.Chen Y-G, Tian S-K, Deng L. A highly enantioselective catalytic desymmetrization of cyclic anhydrides with modified cinchona alkaloids. J Am Chem Soc. 2000;122:9542–9543. [Google Scholar]
- 7.Chen Y-G, Deng L. Parallel kinetic resolutions of monosubstituted succinic anhydrides catalyzed by a modified cinchona alkaloid. J Am Chem Soc. 2001;123:11302–11303. doi: 10.1021/ja011766h. [DOI] [PubMed] [Google Scholar]
- 8.Hang J-F, Tian S-K, Tang L, Deng L. Asymmetric synthesis of α-amino acids via cinchona alkaloid-catalyzed kinetic resolution of urethane-protected α-amino acid N-carboxyanhydrides. J Am Chem Soc. 2001;123:12696–12697. doi: 10.1021/ja011936q. [DOI] [PubMed] [Google Scholar]
- 9.Tang L, Deng L. Dynamic kinetic resolution via dual-function catalysis of modified cinchona alkaloids: Asymmetric synthesis of α-hydroxy carboxylic acids. J Am Chem Soc. 2002;124:2870–2871. doi: 10.1021/ja0255047. [DOI] [PubMed] [Google Scholar]
- 10.Hang J-F, Li H-M, Deng L. Development of a rapid, room-temperature dynamic kinetic resolution for efficient asymmetric synthesis of α-aryl amino acids. Org Lett. 2002;4:3321–3324. doi: 10.1021/ol026660l. [DOI] [PubMed] [Google Scholar]
- 11.Dijkstra GD, Kellogg RM, Wynberg H. Conformational analysis of some chiral catalysts of the cinchona and ephedra family. The alkaloid catalyzed addition of aromatic thiols to cyclic α,β -unsaturated ketones. Recl Trav Chim Pay-B. 1989;108:195–204. [Google Scholar]
- 12.Dijkstra GD, et al. Conformational study of cinchona alkaloids. A combined NMR, molecular mechanics and x-ray approach. J Am Chem Soc. 1989;111:8069–8076. [Google Scholar]
- 13.Dijkstra GD, Kellogg RM, Wynberg H. Conformational study of cinchona alkaloids: A combined NMR and molecular orbital approach. J Org Chem. 1990;55:6121–6131. [Google Scholar]
- 14.Amberg W, et al. Syntheses and crystal structures of the cinchona alkaloid derivatives used as ligands in the osmium-catalyzed asymmetric dihydroxylation of olefins. J Org Chem. 1993;58:844–849. [Google Scholar]
- 15.Corey EJ, Noe MC. Rigid and highly enantioselective catalyst for the dihydroxylation of olefins using osmium tetraoxide clarifies the origin of enantiospecificity. J Am Chem Soc. 1993;115:12579–12580. [Google Scholar]
- 16.Corey EJ, Noe MC, Sarshar S. X-ray crystallographic studies provide additional evidence that an enzyme-like binding pocket is crucial to the enantioselective dihydroxylation of olefins by OsO4-bis-cinchona alkaloid complexes. Tetrahedron Lett. 1994;35:2861–2864. [Google Scholar]
- 17.Corey EJ, Noe MC. Kinetic investigations provide additional evidence that an enzyme-like binding pocket is crucial for high enantioselectivity in the bis-cinchona alkaloid catalyzed asymmetric dihydroxylation of olefins. J Am Chem Soc. 1996;118:319–329. [Google Scholar]
- 18.Corey EJ, Noe MC. A critical analysis of the mechanistic basis of enantioselectivity in the bis-cinchona alkaloid catalyzed dihydroxylation of olefins. J Am Chem Soc. 1996;118:11038–11053. [Google Scholar]
- 19.Li H, et al. Stereocontrolled creation of adjacent quaternary and tertiary stereocenters via a catalytic diastereoselective and enantioselective conjugate addition. Angew Chem Int Edit. 2005;44:105–108. doi: 10.1002/anie.200461923. [DOI] [PubMed] [Google Scholar]
- 20.Bvaje W, Frackenpohl J, Langer P, Hoffmann HMR. Synthesis of oxazatwistanes and their homo- and bishomo-analogues from quinidine: Medium ring systems derived from cinchona alkaloids. Tetrahedron. 1998;54:3495–3512. [Google Scholar]
- 21.Bürgi HB, Dunitz JD, Shefter E. Geometrical reaction coordinates. II. Nucleophilic addition to a carbonyl group. J Am Chem Soc. 1973;95:5065–5067. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.