

Abstract
Given the significance of carbohydrates in life, medicine, and industry, the development of simple and efficient de novo methods to synthesize carbohydrates are highly desirable. Organocatalytic asymmetric assembly reactions are powerful tools to rapidly construct molecules with stereochemical complexity from simple precursors. Here, we present a simple and robust methodology for the asymmetric synthesis of pyranose derivatives with talo- and manno- configurations from simple achiral precursors through organocatalytic asymmetric intermolecular Michael–Henry reaction sequences. In this process, (tert-butyldimethylsilyloxy)acetaldehyde 1 was successfully utilized in two ways: as a donor in a highly selective anti-Michael reaction and as an acceptor in a consecutive Henry reaction. Varied nitroolefins served as Michael acceptors and varied aldehydes substituted for 1 as Henry acceptors providing for the construction of a wide range of carbohydrates with up to 5 stereocenters. In these reactions, a catalyst-controlled Michael reaction followed by a substrate-controlled Henry reaction provided 3,4-dideoxytalose derivatives 6 in a highly stereoselective manner. The Henry reaction was affected by addition of a simple base such as triethylamine: A complex chiral base was not necessary. 3,4-Dideoxymannose derivatives 7 were produced by simply changing the base from triethylamine to 1,8-diazabicyclo[5.4.0]undec-7-ene. Extension of this methodology to a syn-Michael initiated sequence was also successful. A mechanistic discussion is provided to explain the unusual substrate-induced stereoselectivity of the Henry reaction.
Keywords: amine-thiourea catalyst, asymmetric reaction, carbohydrates, Michael reaction, organocatalysis
Carbohydrates are one of the most important classes of organic molecules and play diverse and essential roles in life, medicine, and industry. Since Emil Fischer’s structural elucidation and synthesis of carbohydrates more than a century ago (1), carbohydrate synthesis has continued to challenge synthetic chemists. Robust, simple, direct, and highly stereoselective methods to carbohydrate synthesis remain largely elusive and the development of such methodologies is a driving force in synthetic chemistry. Indeed, our discovery of the proline catalyzed intermolecular aldol reaction (2, 3) and other related reactions (4, 5) were made possible by our development of antibody aldolases as synthetic tools for carbohydrate synthesis (6, 7). In the decade since this discovery, organocatalysis has emerged as a promising route to a wide range of chiral molecules (4, 5, 8–11). Our studies in organocatalysis prompted us (5, 12–14) and later others (15, 16) to develop cascade reactions and one-pot synthetic approaches toward the synthesis of complex molecules containing multiple stereocenters with the aim of producing robust and operationally simple approaches to the synthesis complex asymmetric molecules like carbohydrates. We have classified reactions of this type broadly as organocatalytic asymmetric assembly reactions because they provide for the asymmetric assembly of multiple substrates into higher order products with stereochemical complexity. The preparation of carbohydrates based on this type of organocatalytic approach has been a driving force in the field (14, 17–23).
Recently, we reported highly selective anti-Michael reactions of (tert-butyldimethylsilyloxy)acetaldehyde 1 to form γ-nitroaldehydes 4 catalyzed by primary amine-thiourea 3 (24). This type of catalyst provides for enamine-based activation of the aldehyde while enforcing configurational control of enamine geometry. Together with hydrogen bonding activation of β-nitroalkenes provided by the thiourea (25–29) functionality of the catalyst, this catalyst effectively merges two key functionalities in organocatalysis. The α-oxyaldehyde structure in the Michael product 4 suggested that successive Henry reactions of 4 to the parent aldehyde 1 could produce highly functionalized nitroalcohol 5, which might exist as its cyclized 3,4-dideoxypyranose form 6 as shown in Scheme 1. An asymmetric assembly reaction of this type would link three substrates through the formation of two new C-C bonds while installing four contiguous asymmetric centers. We were encouraged to explore this idea by development of several asymmetric Henry reactions (30), including intermolecular Michael-intramolecular Henry tandem reactions (31–33), an iminium mediated intermolecular Michael–Henry sequence (34) and Michael-aza-Henry reactions (35, 36). Here we demonstrate an organocatalytic intermolecular one-pot Michael–Henry reaction through enamine catalysis. As shown in Scheme 1, two stereocenters at C2 and C3 position in 6 were controlled with near perfection by the anti-Michael aldehyde reaction [up to 98∶2 diastereomeric ratio (dr) and 99% enantiomeric excess (ee)]. The challenge was to link this reaction with an intramolecular Henry reaction to produce a single product with defined C4 and C5 stereocenters. Herein we present our solution to this challenge and present a simple and robust methodology for synthesis of pyranose derivatives with talo- and manno- configurations through organocatalytic intermolecular Michael–Henry reaction sequences.
Scheme 1.

anti-Michael–Henry reaction sequence to construct carbohydrate structures.
Results and Discussion
We envisioned that if the Michael reaction of aldehyde 1 with β-nitrostyrene 2a was carried out in the presence of additional base, the Michael product 4a would react with remaining aldehyde 1. As a starting point, we used triethylamine as a second catalyst (Method A in Table 1, entry 1). The Michael reaction followed by the Henry reaction preceded stereoselectively to provide product 6a with the D-talo-configuration as a major product in good yield with only small amounts of the D-manno-isomer 7a. Only the α-anomers of 6a and 7a were observed in accord with the preference typical of manno- and talo-type carbohydrates. Small quantities of other diastereomers were removed by column chromatography. The closed form 6a and its open form 5a existed as an equilibrium mixture in CDCl3 (ca. 3∶1). This process provided stereoisomer 6a with five continuous stereocenters in good isolated yield; however, the enantiomeric excess was only 88%, which was lower than that of the original Michael adduct 4a as catalyzed by 3 (98% ee). Other chiral catalysts such as Takemoto catalyst 8, quinine 9, and quinidine 10 were tested as the second catalyst, but enantiomeric excess was not improved (entries 2–4). Because the enantioselectivity should be mainly induced at the irreversible Michael reaction step, we anticipated that these second catalysts promoted the Michael reaction nonselectively and that the competitive reaction resulted in a decreased ee. In fact, triethylamine and Takemoto catalyst 8 produced product 6 even in the absence of primary amine-thiourea 3 (entries 5 and 6).
Table 1.
Optimization of Michael–Henry reaction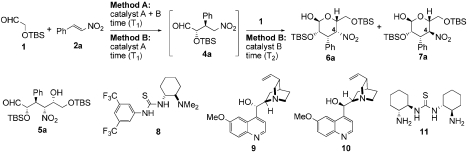
| Entry | Catalyst A | Catalyst B (mol%) | Time (T1 + T2) | Method * | Yield (%) † | dr ‡ (5a + 6a∶7a) | ee (%) § |
| 1 | 3 | Et3N (20) | 1 d | A | 62 | 7∶1 | 88 |
| 2 | 3 | 8 (20) | 1 d | A | 54 | 4∶1 | 89 |
| 3 | 3 | 9 (20) | 1 d | A | 62 | 6∶1 | 87 |
| 4 | 3 | 10 (20) | 1 d | A | 58 | 6∶1 | 87 |
| 5 | - | Et3N (20) | 1 d | A | 27 | 3∶1 | — |
| 6 | - | 8 (20) | 1 d | A | 17 | 3∶1 | -35 |
| 7 | 3 | Et3N (50) | 4 + 1 h | B | 68 | > 10∶1 | 98 |
| 8 | 3 | i Pr 2EtN (50) | 4 + 1 h | B | 68 | > 10∶1 | 98 |
| 9 | 3 | DABCO (25) | 4 + 1 h | B | 44 | 4∶1 | 98 |
| 10 | 3 | DMAP (50) | 4 + 1 h | B | 15 | 1∶1 | 98 |
| 11 | 3 | DBU (50) | 4 + 1 h | B | 51 | 0∶1 | 98 |
| 12 ¶ | 11 ∥ | Et3N (50) | 8 + 2 h | B | 38 | 8∶1 | 96 |
*Method A: catalyst A (20 mol%), catalyst B and 2a (0.1 or 0.2 mmol) were reacted with 1 (4 equiv.) in CH2Cl2 at rt for T1. Method B: catalyst A (20 mol%) and 2a (0.1 or 0.2 mmol) were reacted with 1 (4 equiv.) in CH2Cl2 at 30 °C for T1, then catalyst B was added (see text).
†Yield of isolated product.
‡Determined by 1H NMR analysis of purified product, (5a + 6a∶7a).
§Determined by chiral phase HPLC analysis of corresponding alcohol.
¶The reaction was carried out at rt.
∥10 mol% of 11 was used.
To overcome this problem, the second catalyst was added after the Michael reaction was complete (Method B). After insuring complete conversion of β-nitrostyrene 2a, 50 mol% of triethylamine was added to the reaction mixture, which was kept at 30 °C for 1 h (entry 7). An ee value of 98% was observed, comparable to that of the original Michael reaction. The reaction at 30 °C was more reproducible than the reaction at room temperature. We found that a short reaction time suppressed formation of C4-epimer 7a, preventing base-promoted epimerization at C4 position.
To identify the actual catalyst of the Henry reaction, the isolated Michael product 4a was treated with triethylamine without primary amine-thiourea catalyst 3 (Scheme 2). The product 6a was obtained in excellent yield and high ee. Thus, the actual catalyst is the tertiary amine (in this case, triethylamine). The Henry reaction provided predominantly one of the possible four isomers. This is a very rare example of stereoselective intermolecular Henry reaction controlled by the configuration of the nitroalkane (35, 36).
Scheme 2.

Henry reaction of isolated Michael product 4a.
Next, the effect of the second catalyst was studied. Reaction with sterically hindered diisopropylethylamine gave comparable results to those with triethylamine (Table 1, entry 8). Less hindered bases such as 1,4-diazabicyclo[2.2.2]octane (DABCO) and 4-dimethylaminopyridine (DMAP) were poorer catalysts (entries 9 and 10). When 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) was used, complete epimerization at C4 position was observed, affording 3,4-dideoxy-D-mannose derivative 7a with excellent ee. Derivative 7a existed only in the cyclized form in CDCl3. Dimeric catalyst 11 provided the product 6a in moderate yield with 10 mol% catalyst loading. The optimized one-pot conditions that provide for the synthesis of 3,4-dideoxy-D-talose derivative 6a with excellent ee and in good yield from two molecules of (tert-butyldimethylsilyloxy)acetaldehyde 1 and one β-nitrostyrene 2a via an anti-Michael-Henry reaction are listed in Table 1, entry 7. The C4-epimer 7a in the manno-configuration was produced by simply changing the second catalyst from triethylamine to DBU.
With these optimized conditions in hand, we surveyed scope of the reaction using triethylamine as the second catalyst (Table 2). Nitrostyrenes with both electron withdrawing groups and donating groups on the aromatic ring were good substrates for the reaction and the corresponding talose derivatives were obtained in good yield and excellent ee (entries 2–4). To suppress epimerization, substrates with electron withdrawing groups were subjected to short second reaction times (T2), whereas longer reaction times were necessary with electron rich substrates. Reaction of 2,6-dichloronitrostyrene 2e required 50 mol% of catalyst 3 and equimolar amounts of triethylamine to provide the product, 5e, with high ee (99% ee) in moderate yield (entry 5); note that 5e was present in the open form. Heteroaromatic substrates were also good candidates for this reaction: 2-(2-Nitrovinyl)thiophene 2f was converted to the product 6f in good yield and selectivity (entry 6). Enantiomeric excess of products 6 was comparable to that of Michael products 4 we reported previously (24). When nitrodiene 2g was used, alkenyl-substituted dideoxytalose derivative 6g formed with acceptable ee (entry 7). A second reaction time of 18 h was used to convert alkyl substituted nitroolefin 2h to 6h with good dr and ee (entry 8). Both 6g and 6h with smaller substituents existed in only in the cyclic form.
Table 2.
Michael–Henry reaction to 3,4-dideoxy-D-talose derivatives 6*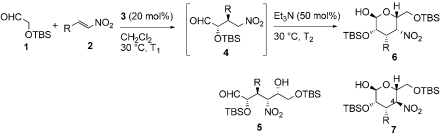
| Entry | Product | Time (T1, h) | Time (T2, h) | Yield (%) † | dr (6 + 5∶7) ‡ | 6∶5 ‡ | ee (%) § |
| 1 | Ph- | 4 | 1 | 68 | > 10∶1 | 3∶1 | 98 |
| 2 | 4 - BrC6H4- | 4 | 0.5 | 62 | > 10∶1 | 4∶1 | 98 |
| 3 | 4 - MeOC6H4- | 4 | 1.5 | 76 | > 10∶1 | 3∶1 | 97 |
| 4 | 3 - BrC6H4- | 5 | 0.5 | 68 | > 10∶1 | 6∶1 | 97 |
| 5 ¶∥ | 2,6 - Cl2C6H3- | 16 | 4 | 37 | 1∶0 | 0∶1 | 99 |
| 6** | 2 - Thiophenyl | 7 | 0.5 | 63 | 7∶1 | 13∶1 | 97 |
| 7¶ | (E) - PhCH = CH- | 6 | 0.3 | 43 | 6∶1 | 1∶0 | 93 |
| 8 ¶ | n - C7H15- | 5 | 18 | 44 | > 10∶1 | 1∶0 | 96 |
*3 (20 mol%) and 2 (0.2 mmol) were reacted with 1 (4 equiv.) in CH2Cl2 at 30 °C for T1, then Et3N (50 mol%) was added and reacted for T2.
†Yield of isolated product.
‡Determined by 1H NMR analysis of purified product in CDCl3.
§Determined by chiral phase HPLC analysis of corresponding alcohol.
¶3 (50 mol%) was used.
∥Et3N (100 mol%) was used.
**Et3N (30 mol%) was used.
Next, we investigated synthesis of 3,4-dideoxy-D-mannose derivatives 7 using DBU as the second catalyst (Table 3). The cyclized products were obtained in reasonably good yield via a three-step sequence: anti-Michael reaction, syn-Henry reaction, and C4-epimerization. We observed immediate consumption of Michael product 4 upon addition of DBU, followed by relatively slow but complete conversion of talo-type compound 6 to manno-configured 7. All tested nitroolefins with substituted phenyl, heteroaromatic, alkenyl, and alkyl groups were converted into their corresponding derivatives with excellent ee under these conditions, reflecting wide scope of this reaction. The exception was the sterically demanding 2,6-dichloro-β-nitrostyrene 2e; we could not isolate the corresponding manno-product in pure form probably due to poor selectivity of the Henry reaction and decomposition of product 5e during the prolonged reaction time.
Table 3.
Michael–Henry reaction to dideoxy-D-mannopyranose derivatives 7*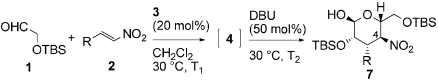
| Entry | Product | Time (T1, h) | Time (T2, h) | Yield (%) † | ee (%) ‡ |
| 1 | 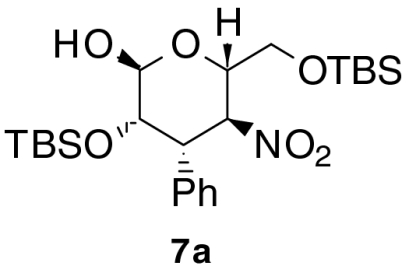 |
4 | 1 | 51 | 98 |
| 2 | 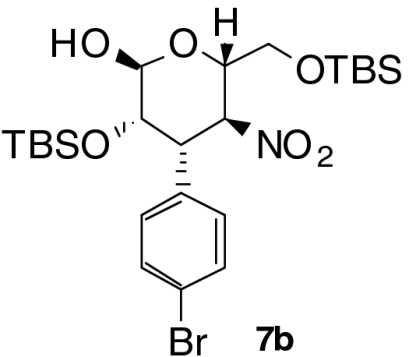 |
4 | 1 | 65 | 96 |
| 3 | 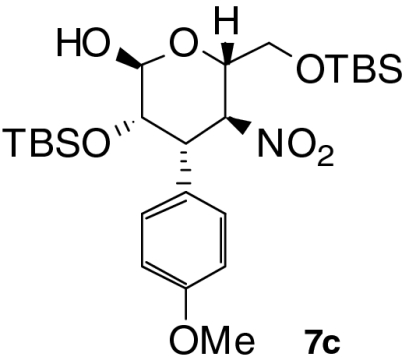 |
23 | 1 | 48 | 95 |
| 4 | 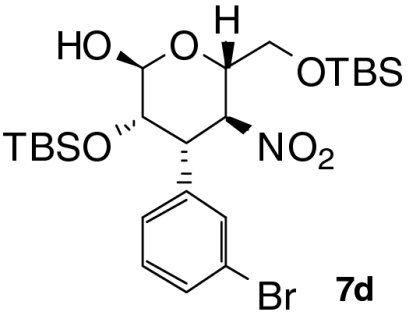 |
5 | 1 | 57 | 98 |
| 5 | 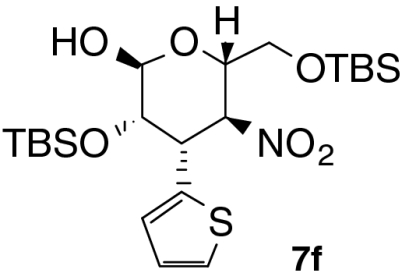 |
7 | 1 | 59 | 96 |
| 6 § | 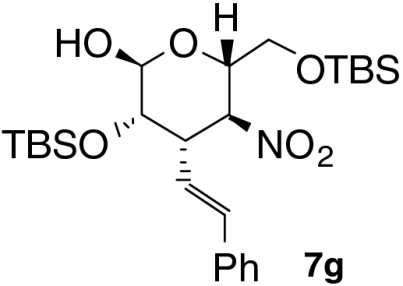 |
20 | 2 | 66 | 93 |
| 7 § | 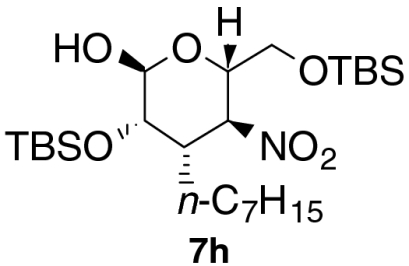 |
5 | 1 | 50 | 96 |
*3 (20 mol%) and 2 (0.2 mmol) were reacted with 1 (4 equiv.) in CH2Cl2 at 30 °C for T1, then DBU (50 mol%) was added and reacted for T2.
†Yield of isolated product.
‡Determined by chiral phase HPLC analysis.
§3 (50 mol%) was used.
Most typically, the Henry reaction proceeded in a stereo-specific manner with relatively slow C4-epimerization. Two pathways are possible for isomerization from the talo-configuration in 6 to the manno-type of 7: One is the direct epimerization of 6 by deprotonation at C4 and the other is the retro-Henry–Henry process (32). To clarify the mechanism, talo-product 6a was converted to acetyl pyranose 12a (Scheme 3). Both open form 5a and closed form 6a were converted to 12a and the minor manno-type product 13a could be separated at this stage. DBU treatment of 12a provided manno-product 13a, suggesting the epimerization occurred via direct epimerization at the C4 position. Moreover, both 12a and 13a were successfully converted into 4-amino-3,4-dideoxy-3-phenyl-D-talose derivative 14 and 4-amino-3,4-dideoxy-3-phenyl-D-mannose derivative 15, respectively. A 4-amino-substituted mannose scaffold related to 13a is found in the antitumor antibiotic spicamycin (37). The absolute and relative configurations of the 3,4-dideoxy-D-talose derivatives 6 were determined by X-ray crystallography of 12b (Fig. 1). A 1,3-diaxial interaction between the C2-alkoxy group and the C4-nitro group strained the tetrahydropyran ring and this is presumably the reason 6 exists as an equilibrium mixture between closed structure 6 and its open form 5 in most cases.
Scheme 3.

Epimerization experiment of 12a and conversion to 4-amino-3,4-dideoxy-3-phenyl-D-talose derivative 14 and 4-amino-3,4-dideoxy-3-phenyl-D-mannose derivative 15.
Fig. 1.

X-ray crystal structure of 12b.
A method for synthesis of both talo- and manno-type sugars has been established using (tert-butyldimethylsilyloxy)acetaldehyde 1 as both a donor in first Michael reaction and an acceptor in second Henry reaction. If other aldehydes could be used as acceptors in the second Henry reaction, the utility of the reaction would increase dramatically. Therefore, we evaluated other acceptors (Scheme 4). When ethyl glyoxylate 16 was added as the second acceptor in the presence of triethylamine, the talo-configured product 17 was obtained. In this case, the contaminating C4-epimer 18 was readily removed by column chromatography. By changing the second catalyst from triethylamine to DBU, the manno-type product 18 was obtained in good yield. In both cases, aldehyde 1 served as a donor only in the Michael reaction and products 17 and 18 were obtained in excellent ee. When aqueous formaldehyde was used as an acceptor in the second step (Scheme 4B), both the Henry reaction and epimerization occurred to give pentose derivative 19, which could serve as a precursor to human NK1 antagonists (38). Because α-oxyacetaldehyde 1 is a good acceptor in the Henry reaction, isolation of Michael adduct 4a and removal of unreacted 1 followed by addition and reaction with the second aldehyde provided better yields of products in cases where remaining 1 resulted in a competing Henry reaction in the one-pot format.
Scheme 4.

Michael–Henry reaction with other Henry acceptors. (A) Michael–Henry reaction with ethyl glyoxylate (16) as an acceptor. (B) Henry reaction with formaldehyde as an acceptor.
Although a large number of organocatalytic syn-Michael reactions with aldehyde nucleophiles have been reported since 2001 (39), there is no precedence for intermolecular Henry reactions of these Michael products. To evaluate this type of Michael–Henry reaction for syn-Michael adducts, we chose to study the Michael reaction of isovaleraldehyde 20 with diphenylprolinol silyl ether 21 (40) under our one-pot Henry reaction conditions (Scheme 5). The Michael–Henry product 24 formed in good yield with excellent enantiomeric excess in the presence of p-nitrobenzaldehyde 23 and triethylamine. It should be noted that isovaleraldehyde 20 did not act as the Henry acceptor under these conditions. The C4 and C5 stereocenters in the Henry product 24 possessed the same configurations as those in 6. Hence, the stereoselectivity of Henry reaction was controlled by the C3 stereocenter for both anti-Michael adduct 4 and syn-Michael product 22 regardless configuration of C2 stereocenter.
Scheme 5.

syn-Michael–Henry reaction.
In all of the reactions evaluated, only one isomer with four successive stereocenters was formed as the major product. It is clear that C2 and C3 stereochemistry was controlled in the anti-Michael reaction by primary amine-thiourea catalyst 3. Subsequent asymmetric induction at C4 and C5 positions occurred in the consecutive Henry reaction catalyzed by an achiral amine such as triethylamine. Substrate-controlled stereo induction in the Henry reaction is known for chiral aldehyde acceptors. However, diastereoselective Henry-type reactions of chiral nitroalkanes are rare (35, 36) and a general kinetic mechanism to explain their stereo induction is not reported.
To control C4 asymmetry in the Henry reaction, acceptor aldehyde 1 should approach from the si-face of the nitronate anion generated from Michael product 4a and base. Because both anti-Michael adducts 4 and syn-product 22 provide the same stereo-induction in the successive Henry reaction, asymmetric induction at this step is controlled by the chirality at C3 of the Michael products. It is known that allylic 1,3-strain restricts rotation of the σ-bond connected to enolates and nitronates (41). Therefore, the nitronate anion generated from 4a should exist predominantly in the conformation shown in Fig. 2A. The approach of aldehyde 1 should occur predominantly on the less crowded face. Fleming and Lewis have discussed diastereoselectivity of enolate alkylation, in which a Ph group was effectively smaller than the iPr group (42). The “effective radius” of phenyl group is smaller than that of a methyl group (43). Therefore, we rationalize that as Ph is smaller than the branched alkyl group (-CH(OTBS)CHO) on the C3 position in Fig. 2A, the electrophilic approach of the aldehyde should occur from the si-face of the nitronate. It is worth mentioning that the stereoselectivity at the α position to nitro group in other nitronate addition reactions, including Enders’ cascade reaction (16, 44), may be explained in a similar manner.
Fig. 2.

Proposed transition state of the Henry reaction. (A) Configuration of the nitronate is restricted by allylic 1,3-strain favoring si-face approach of the nitronate to control the developing C4 stereocenter. (B) Less favorable nitronate conformer. (C) Favored conformer that participates in the transition state (Left) and hyperconjugated structure of the conformer (Right). (D) Six-membered ring transition state structure to provide the syn-Henry product with C5 stereo-induction.
In addition, the stereoelectronic effect should enhance the facial selectivity. A theoretical study indicated that allylic bonds are staggered with respect to partially formed bonds in the transition states (45) and the two possible conformers are as shown in Fig. 2 B and C. In electrophilic attack on a π system such as a nitronate, a higher and more reactive highest occupied molecular orbital is obtained by mixing the π orbital and the σ orbital when the highest energy σ orbital is located perpendicular to the π system in the nucleophile. It is suggested that a homoallylic heteroatom (i.e., oxygen atom in Fig. 2C) raises the energy level of the σ orbital via lone pair participation (46). This hyperconjugative interaction, shown in Fig. 2C, makes a transition state through the conformer in Fig. 2C more likely.
On the other hand, C5 asymmetric induction could be explained by the syn-selective Henry reaction considering the six-membered ring transition state. In the proposed cyclic six-membered transition state shown in Fig. 2D, side chains of both nitronate and aldehyde occupy equatorial positions. This cyclic six-membered transition state was proposed for the highly syn-selective asymmetric Henry reactions catalyzed by transition metal complexes (47, 48). A chiral guanidine thiourea catalyst is also reported to provide the syn-Henry product (49). Thus, these two stereocontrolling factors, si-face approach to the nitronate anion and syn-selective Henry reaction, may control both C4 and C5 asymmetric induction and may result in the observed high selectivity of the Henry reaction described here.
Conclusions
The development of simple, robust and efficient methods for the de novo synthesis of carbohydrates is a significant challenge in organic synthesis. Our approach to this problem was based on the development of organocatalytic asymmetric assembly reactions. Here we describe asymmetric syntheses of pyranose derivatives with talo- and manno- configurations. 3,4-Dideoxytalose derivatives 6 were synthesized by combination of our anti-Michael reactions of (tert-butyldimethylsilyloxy)acetaldehyde 1 to form γ-nitroaldehydes 4 and a subsequent syn-Henry reaction with 1 as an acceptor. The extremely high selectivity of the anti-Michael reactions (up to 98∶2 dr and 99% ee) established the C3 stereocenter that controlled the stereochemistry at successive C4 and C5 stereocenters in the syn-Henry reaction resulting in the control of four contiguous asymmetric centers. These are the first reported examples of enamine catalysis of asymmetric intermolecular one-pot Michael–Henry reactions and are among rare examples of stereoselective intermolecular Henry reactions controlled by the configuration of the nitroalkanes and catalyzed by a simple base such as triethylamine. Use of DBU instead of triethylamine provided 3,4-dideoxymannose derivatives 7 via a three-step sequence: anti-Michael reaction, syn-Henry reaction, and C4-epimerization. Various products with aromatic, vinyl, and alkyl substituents at the C3 position were prepared demonstrating the wide scope and efficiency of this strategy.
In addition, the acceptor of the Henry reaction could be varied. When ethyl glyoxylate 16 was added as the second acceptor, both the talo-configured product 17 and the manno-type product 18 were obtained in the presence of triethylamine and DBU, respectively. Moreover, substitution of catalyst 21 for 3 allowed us to initiate the assembly reaction with a syn-Michael reaction to provide product 24 following the Henry reaction. Here, product 24 stereocenters at C4 and C5 possessed the same configurations as those in 6. This result implies that this type of diastereoselective Henry reaction is applicable to synthesis of a wide range of Michael products because the syn-Michael reaction works with a broad range of aldehydes and nitroolefins. As with the anti-Michael product, there is no previous literature precedence for the intermolecular Henry reaction with the syn-Michael products.
Stereoselectivity at C2 and C3 positions is explained by our anti-Michael reaction design as reported previously (24). Meanwhile, asymmetric induction at C4 and C5 positions occurred in the consecutive Henry reaction catalyzed by achiral bases. Stereocontrol at C4 position could be explained by si-face approach to the nitronate anion generated from Michael product 4a and base. Attack occurred from the side of the relatively small Ph group on the nitronate anion; rotation here might be restricted by allylic 1,3-strain. On the other hand, the C5 stereocenter could be induced by a syn-selective Henry reaction. In the proposed six-membered ring transition state shown in Fig. 2D, side chains of both nitronate and aldehyde occupy equatorial positions. These two stereo-controlling factors may result in the unusually high selectivity of the Henry reaction and consequent production of D-talo-configured product 6 with four contiguous stereocenters. In summary, we have demonstrated that catalytic asymmetric assembly reactions based on sequential Michael–Henry reactions allow rapid stereoselective assembly of varied aldehydes and nitroolefins into carbohydrates with very high levels of enantio- and diastereocontrol.
Materials and Methods
Typical experimental procedure for synthesis of 3,4-dideoxy-D-talose derivative 6a: (tert-butyldimethylsilyloxy)acetaldehyde 1 (152 μL, 0.8 mmol) was added to the solution of thiourea catalyst 3 (15.4 mg, 40 μmol) and β-nitrostyrene 2a (29.8 mg, 0.2 mmol) in CH2Cl2 (0.2 mL). The resulting solution was stirred at 30 °C for 4 h and then triethylamine (13.9 μL, 0.1 mmol) was added. After 1 h at 30 °C, Et2O and 1N HCl (0.2 mL) was added to the solution at rt. The aqueous layer was separated and extracted three times with Et2O. The combined organic layers were dried over MgSO4, concentrated, and purified by flash column chromatography to afford 3,4-dideoxy-D-talose derivative 6a (67.4 mg, 68%) as a colorless oil. 1H NMR (500 MHz, CDCl3) major (6a): δ 7.40 - 7.24 (m, 5H), 5.29 (brs, 1H), 4.79 (dd, J = 4.6, 2.5 Hz, 1H), 4.47 (ddd, J = 8.5, 6.2, 2.3 Hz, 1H), 3.90 (dd, J = 2.8, 1.4 Hz, 1H), 3.88 (dd, J = 9.9, 6.2 Hz, 1H), 3.84 (dd, J = 9.9, 8.6 Hz, 1H), 3.55 (dd, J = 4.7, 2.7 Hz, 1H), 3.03 (brs, 1H), 0.93 (s, 9H), 0.84 (s, 9H), 0.03 (s, 3H), 0.01 (s, 6H), -0.22 (s, 3H); 13C NMR (100 MHz, CDCl3) major (6a): δ 137.19, 129.95, 128.38, 128.11, 95.08, 81.65, 69.98, 68.85, 62.44, 43.47, 26.00, 25.95, 18.41, 18.29, -4.43, -4.98, -5.46, -5.55; high resolution mass spectrometry (HRMS) (m/z): [M + H]+ calcd for 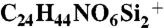 498.2702, found 498.2700. Enantiomeric excess: 98%, determined by HPLC after reduction to corresponding alcohol (Chiralpak IC, hexane/i-PrOH = 97∶3, flow rate 1.00 mL/ min, λ = 220 nm, rt): tR(major) = 13.6 min, tR(minor) = 15.2 min.
498.2702, found 498.2700. Enantiomeric excess: 98%, determined by HPLC after reduction to corresponding alcohol (Chiralpak IC, hexane/i-PrOH = 97∶3, flow rate 1.00 mL/ min, λ = 220 nm, rt): tR(major) = 13.6 min, tR(minor) = 15.2 min.
The procedure for synthesis of 3,4-dideoxy-D-mannose derivative 7a was similar to that for 6a, except for the use of DBU rather than triethylamine. 3,4-Dideoxy-D-mannose derivative 7a (50.6 mg, 51%) was obtained as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.33 - 7.21 (m, 5H), 5.47 (dd, J = 12.0, 9.8 Hz, 1H), 5.13 (d, J = 1.3 Hz, 1H), 4.47 (dt, J = 9.8, 3.5 Hz, 1H), 3.94 (dd, J = 12.0, 2.5 Hz, 1H), 3.82 - 3.77 (m, 2H), 3.74 (dd, J = 11.5, 3.9 Hz, 1H), 0.93 (s, 9H), 0.83 (s, 9H), 0.08 (s, 3H), 0.08 (s, 3H), -0.21 (s, 3H), -0.57 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 136.18, 129.08, 128.60, 127.92, 94.33, 82.45, 72.45, 71.32, 63.15, 45.66, 26.07, 25.95, 18.53, 18.13, -5.22, -5.24, -5.47, -5.73; HRMS (m/z): [M + H]+ calcd for 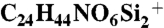 498.2702, found 498.2705. Enantiomeric excess: 98%, determined by HPLC (Chiralpak AD-H, hexane/i-PrOH = 99∶1, flow rate 1.00 mL/ min, λ = 220 nm, rt): tR(major) = 11.6 min, tR(minor) = 8.8 min.
498.2702, found 498.2705. Enantiomeric excess: 98%, determined by HPLC (Chiralpak AD-H, hexane/i-PrOH = 99∶1, flow rate 1.00 mL/ min, λ = 220 nm, rt): tR(major) = 11.6 min, tR(minor) = 8.8 min.
Supplementary Material
Acknowledgments.
We thank the Skaggs Institute for Chemical Biology for financial support. G.H.-T. thanks the Spanish Ministry of Education for a fellowship.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. D.W.M. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1003350107/-/DCSupplemental.
References
- 1.Freudenberg K. Emil Fischer and his contribution to carbohydrate chemistry. Adv Carbohydr Chem Biochem. 1966;21:1–38. doi: 10.1016/s0096-5332(08)60314-8. [DOI] [PubMed] [Google Scholar]
- 2.List B, Lerner RA, Barbas CF., III Proline-catalyzed direct asymmetric aldol reactions. J Am Chem Soc. 2000;122:2395–2396. [Google Scholar]
- 3.Sakthivel K, Notz W, Bui T, Barbas CF., III Amino acid catalyzed direct asymmetric aldol reactions: A bioorganic approach to catalytic asymmetric carbon–carbon bond-forming reactions. J Am Chem Soc. 2001;123:5260–5267. doi: 10.1021/ja010037z. [DOI] [PubMed] [Google Scholar]
- 4.Barbas CF., III Organocatalysis lost: Modern chemistry, ancient chemistry, and an unseen biosynthetic apparatus. Angew Chem Int Ed. 2008;47:42–47. doi: 10.1002/anie.200702210. [DOI] [PubMed] [Google Scholar]
- 5.Notz W, Tanaka F, Barbas CF., III Enamine-based organocatalysis with proline and diamines: The development of direct catalytic asymmetric aldol, Mannich, Michael, and Diels–Alder reactions. Acc Chem Res. 2004;37:580–591. doi: 10.1021/ar0300468. [DOI] [PubMed] [Google Scholar]
- 6.Tanaka F, Barbas CF., III . Antibody-catalyzed aldol reactions. In: Mahrwald R, editor. Modern Aldol Reactions, Volume 1: Enolates, Biocatalysis, and Natural Product Synthesis. New York: Wiley; 2004. pp. 273–310. [Google Scholar]
- 7.Wagner J, Lerner RA, Barbas CF., III Efficient aldolase catalytic antibodies that use the enamine mechanism of natural enzymes. Science. 1995;270:1797–1800. doi: 10.1126/science.270.5243.1797. [DOI] [PubMed] [Google Scholar]
- 8.Bertelsen S, Jørgensen KA. Organocatalysis—After the gold rush. Chem Soc Rev. 2009;38:2178–2189. doi: 10.1039/b903816g. [DOI] [PubMed] [Google Scholar]
- 9.Doyle AG, Jacobsen EN. Small-molecule H-bond donors in asymmetric catalysis. Chem Rev. 2007;107:5713–5743. doi: 10.1021/cr068373r. [DOI] [PubMed] [Google Scholar]
- 10.Takemoto Y. Recognition and activation by ureas and thioureas: stereoselective reactions using ureas and thioureas as hydrogen-bonding donors. Org Biomol Chem. 2005;3:4299–4306. doi: 10.1039/b511216h. [DOI] [PubMed] [Google Scholar]
- 11.Tian S, et al. Asymmetric organic catalysis with modified cinchona alkaloids. Acc Chem Res. 2004;37:621–631. doi: 10.1021/ar030048s. [DOI] [PubMed] [Google Scholar]
- 12.Bui T, Barbas CF., III A proline-catalyzed asymmetric Robinson annulation reaction. Tetrahedron Lett. 2000;41:6951–6954. [Google Scholar]
- 13.Chowdari NS, Ramachary DB, Barbas CF., III Organocatalytic asymmetric assembly reactions: One-pot synthesis of functionalized β-amino alcohols from aldehydes, ketones, and azodicarboxylates. Org Lett. 2003;5:1685–1688. doi: 10.1021/ol034333n. [DOI] [PubMed] [Google Scholar]
- 14.Chowdari NS, Ramachary DB, Córdova A, Barbas CF., III Proline-catalyzed asymmetric assembly reactions: enzyme-like assembly of carbohydrates and polyketides from three aldehyde substrates. Tetrahedron Lett. 2002;43:9591–9595. [Google Scholar]
- 15.Enders D, Grondal C, Hüttl MRM. Asymmetric organocatalytic domino reactions. Angew Chem Int Ed. 2007;46:1570–1581. doi: 10.1002/anie.200603129. [DOI] [PubMed] [Google Scholar]
- 16.Enders D, Hüttl MRM, Grondal C, Raabe G. Control of four stereocentres in a triple cascade organocatalytic reaction. Nature. 2006;441:861–863. doi: 10.1038/nature04820. [DOI] [PubMed] [Google Scholar]
- 17.Enders D, Grondal C. Direct organocatalytic de novo synthesis of carbohydrates. Angew Chem Int Ed. 2005;44:1210–1212. doi: 10.1002/anie.200462428. [DOI] [PubMed] [Google Scholar]
- 18.Jiang H, et al. Achieving molecular complexity by organocatalytic one-pot strategies—A fast entry for synthesis of sphingoids, amino sugars, and polyhydroxylated α-amino acids. Angew Chem Int Ed. 2009;48:6844–6848. doi: 10.1002/anie.200901446. [DOI] [PubMed] [Google Scholar]
- 19.Northrup AB, MacMillan DWC. Two-step synthesis of carbohydrates by selective aldol reactions. Science. 2004;305:1752–1755. doi: 10.1126/science.1101710. [DOI] [PubMed] [Google Scholar]
- 20.Ramasastry SSV, Albertshofer K, Utsumi N, Tanaka F, Barbas CF., III Mimicking fructose and rhamnulose aldolases: Organocatalytic syn-aldol reactions with unprotected dihydroxyacetone. Angew Chem Int Ed. 2007;46:5572–5575. doi: 10.1002/anie.200701269. [DOI] [PubMed] [Google Scholar]
- 21.Ramasastry SSV, Zhang H, Tanaka F, Barbas CF., III Direct catalytic asymmetric synthesis of anti-1,2-amino alcohols and syn-1,2-diols through organocatalytic anti-Mannich and syn-aldol reactions. J Am Chem Soc. 2007;129:288–289. doi: 10.1021/ja0677012. [DOI] [PubMed] [Google Scholar]
- 22.Suri JT, Ramachary DB, Barbas CF., III Mimicking dihydroxy acetone phosphate-utilizing aldolases through organocatalysis: A facile route to carbohydrates and aminosugars. Org Lett. 2005;7:1383–1385. doi: 10.1021/ol0502533. [DOI] [PubMed] [Google Scholar]
- 23.Zhang H, Ramasastry SSV, Tanaka F, Barbas CF., III Organocatalytic anti-Mannich reactions with dihydroxyacetone and acyclic dihydroxyacetone derivatives: A facile route to amino sugars. Adv Synth Catal. 2008;350:791–796. [Google Scholar]
- 24.Uehara H, Barbas CF., III anti-Selective asymmetric Michael reactions of aldehydes and nitroolefins catalyzed by a primary amine/thiourea. Angew Chem Int Ed. 2009;48:9848–9852. doi: 10.1002/anie.200905313. [DOI] [PubMed] [Google Scholar]
- 25.Lalonde MP, Chen Y, Jacobsen EN. A chiral primary amine thiourea catalyst for the highly enantioselective direct conjugate addition of α,α-disubstituted aldehydes to nitroalkenes. Angew Chem Int Ed. 2006;45:6366–6370. doi: 10.1002/anie.200602221. [DOI] [PubMed] [Google Scholar]
- 26.Liu Y, Sun B, Wang B, Wakem M, Deng L. Catalytic asymmetric conjugate addition of simple alkyl thiols to α,β-unsaturated N-acylated oxazolidin-2-ones with bifunctional catalysts. J Am Chem Soc. 2009;131:418–419. doi: 10.1021/ja8085092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mei K, et al. Simple cyclohexanediamine-derived primary amine thiourea catalyzed highly enantioselective conjugate addition of nitroalkanes to enones. Org Lett. 2009;11:2864–2867. doi: 10.1021/ol9010322. [DOI] [PubMed] [Google Scholar]
- 28.Okino T, Hoashi Y, Takemoto Y. Enantioselective michael reaction of malonates to nitroolefins catalyzed by bifunctional organocatalysts. J Am Chem Soc. 2003;125:12672–12673. doi: 10.1021/ja036972z. [DOI] [PubMed] [Google Scholar]
- 29.Wang J, Li H, Yu X, Zu L, Wang W. Chiral binaphthyl-derived amine-thiourea organocatalyst-promoted asymmetric Morita–Baylis–Hillman Reaction. Org Lett. 2005;7:4293–4296. doi: 10.1021/ol051822+. [DOI] [PubMed] [Google Scholar]
- 30.Palomo C, Oiarbide M, Laso A. Recent advances in the catalytic asymmetric nitroaldol (Henry) reaction. Eur J Org Chem. 2007:2561–2574. [Google Scholar]
- 31.Adibekian A, et al. Stereocontrolled synthesis of fully functionalized D-glucosamine monosaccharides via a domino nitro-Michael/Henry reaction. Chem Commun. 2008:3549–3551. doi: 10.1039/b805159c. [DOI] [PubMed] [Google Scholar]
- 32.Hayashi Y, Okano T, Aratake S, Hazelard D. Diphenylprolinol silyl ether as a catalyst in an enantioselective, catalytic, tandem Michael/Henry reaction for the control of four stereocenters. Angew Chem Int Ed. 2007;46:4922–4925. doi: 10.1002/anie.200700909. [DOI] [PubMed] [Google Scholar]
- 33.Xu D, et al. A novel enantioselective catalytic tandem oxa-Michael–Henry reaction: One-pot organocatalytic asymmetric synthesis of 3-Nitro-2H-chromenes. Adv Synth Catal. 2008;350:2610–2616. [Google Scholar]
- 34.García Ruano JL, Marcos V, Suanzes JA, Marzo L, Alemán J. One-pot synthesis of pentasubstituted cyclohexanes by a Michael addition followed by a tandem inter–intra double Henry reaction. Chem Eur J. 2009;15:6576–6580. doi: 10.1002/chem.200900894. [DOI] [PubMed] [Google Scholar]
- 35.Han B, Xiao Y, He Z, Chen Y. Asymmetric Michael addition of γ,γ-disubstituted α,β-unsaturated aldehydes to nitroolefins via dienamine catalysis. Org Lett. 2009;11:4660–4663. doi: 10.1021/ol901939b. [DOI] [PubMed] [Google Scholar]
- 36.Jakubec P, Helliwell M, Dixon DJ. Cyclic imine nitro-Mannich/lactamization cascades: a direct stereoselective synthesis of multicyclic piperidinone derivatives. Org Lett. 2008;10:4267–4270. doi: 10.1021/ol801666w. [DOI] [PubMed] [Google Scholar]
- 37.Sakai T, et al. Absolute configuration of spicamycin, an antitumor antibiotic produced by Streptomyces alanosinicus. J Antibiot. 1995;48:899–900. doi: 10.7164/antibiotics.48.899. [DOI] [PubMed] [Google Scholar]
- 38.Morriello GJ, et al. Fused bicyclic pyrrolizinones as new scaffolds for human NK1 antagonists. Bioorg Med Chem. 2008;16:2156–2170. doi: 10.1016/j.bmc.2007.11.081. [DOI] [PubMed] [Google Scholar]
- 39.Betancort JM, Barbas CF., III Catalytic direct asymmetric Michael reactions: Taming naked aldehyde donors. Org Lett. 2001;3:3737–3740. doi: 10.1021/ol0167006. [DOI] [PubMed] [Google Scholar]
- 40.Hayashi Y, Gotoh H, Hayashi T, Shoji M. Diphenylprolinol silyl ethers as efficient organocatalysts for the asymmetric Michael reaction of aldehydes and nitroalkenes. Angew Chem Int Ed. 2005;44:4212–4215. doi: 10.1002/anie.200500599. [DOI] [PubMed] [Google Scholar]
- 41.Hoffmann RW. Allylic 1,3-strain as a controlling factor in stereoselective transformations. Chem Rev. 1989;89:1841–1860. [Google Scholar]
- 42.Fleming I, Lewis JJ. A paradigm for diastereoselectivity in electrophilic attack on trigonal carbon adjacent to a chiral centre: The methylation and protonation of some open-chain enolates. J Chem Soc Chem Commun. 1985:149–151. [Google Scholar]
- 43.Bott G, Field LD, Sternhell S. Steric effects. A study of a rationally designed system. J Am Chem Soc. 1980;102:5618–5626. [Google Scholar]
- 44.Shinisha CB, Sunoj RB. Unraveling high precision stereocontrol in a triple cascade organocatalytic reaction. Org Biomol Chem. 2008;6:3921–3929. doi: 10.1039/b810901j. [DOI] [PubMed] [Google Scholar]
- 45.Paddon-Row MN, Rondan NG, Houk KN. Staggered models for asymmetric induction: attack trajectories and conformations of allylic bonds from ab initio transition structures of addition reactions. J Am Chem Soc. 1982;104:7162–7166. [Google Scholar]
- 46.McGarvey GJ, Williams JM. Stereoelectronic controlling features of allylic asymmetry. Application to ester enolate alkylations. J Am Chem Soc. 1985;107:1435–1437. [Google Scholar]
- 47.Arai T, Yokoyama N. Tandem catalytic asymmetric Friedel–Crafts/Henry reaction: Control of three contiguous acyclic stereocenters. Angew Chem Int Ed. 2008;47:4989–4992. doi: 10.1002/anie.200801373. [DOI] [PubMed] [Google Scholar]
- 48.Sasai H, et al. Efficient diastereoselective and enantioselective nitroaldol reactions from prochiral starting materials: Utilization of La–Li–6,6′-disubstituted BINOL complexes as asymmetric catalysts. J Org Chem. 1995;60:7388–7389. [Google Scholar]
- 49.Sohtome Y, et al. Organocatalytic asymmetric nitroaldol reaction: Cooperative effects of guanidine and thiourea functional groups. Chem Asian J. 2007;2:1150–1160. doi: 10.1002/asia.200700145. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.