

Abstract
We report the development of a multicatalytic, one-pot, asymmetric Michael/Stetter reaction between salicylaldehydes and electron-deficient alkynes. The cascade proceeds via amine-mediated Michael addition followed by an N-heterocyclic carbene-promoted intramolecular Stetter reaction. A variety of salicylaldehydes, doubly activated alkynes, and terminal, electrophilic allenes participate in a one-step or two-step protocol to give a variety of benzofuranone products in moderate to good yields and good to excellent enantioselectivities. The origin of enantioselectivity in the reaction is also explored; E/Z geometry of the reaction intermediate as well as the presence of catalytic amounts of catechol additive are found to influence reaction enantioselectivity.
Keywords: catalysis, organic synthesis, tandem catalysis
Cascade catalysis has garnered significant recent attention from the synthetic community as a means to swiftly assemble complex molecules from simple starting materials with minimal time, waste, and manipulation of reaction intermediates (1–11). Especially powerful in its application to total synthesis, asymmetric tandem catalysis has enabled rapid access to enantioenriched products with high levels of selectivity (1, 4–8, 10, 11). Although most examples exploit a single catalyst to promote multiple, sequential transformations (12–19), systems relying on two or more catalysts have been reported (2, 3, 11, 20). Inherent in any multiple catalyst system is the challenge of compatibility. Avoidance of mutual interference often obliges stepwise addition of catalysts or reagents and variation of reaction conditions over time (21–26). Nevertheless, cascades triggered by a single operation have been accomplished (20, 27, 28).
Very recently, we reported the development of a one-step, asymmetric Michael/benzoin reaction of β-ketoesters 1 and α,β-unsaturated aldehydes 2 mediated by compatible amine 3 and N-heterocyclic carbene (NHC) catalysts (Scheme 1) (29). Importantly, the one-pot procedure was found to give elaborated cyclopentanone products 5 in higher yield and enantioselectivity than a stepwise protocol, wherein intermediate aldehyde 6 is isolated and subjected to the benzoin reaction in a subsequent step (Scheme 1). This observation testifies to the power of cascade catalysis: By quickly relaying intermediates from one reaction to the next, catalysts can work synergistically to discourage undesired pathways.
Scheme 1.

Multicatalytic Michael/Benzoin cascade.
Encouraged by the discovery that NHCs could participate in cascade catalysis, we were inspired to use NHCs to mediate the cascade assembly of benzofuranone products 9 asymmetrically (30). A range of biological activities associated with 3(2H)-benzofuranones including antifungal (31, 32), antipsychotic (33), and anticancer (34–36) properties make these products attractive synthetic targets. Among natural products containing a 2,2′-disubstituted benzofuranone core, rocaglamide demonstrates appreciable cytotoxicity in mice and human cells lines (37), vinigrol displays antihypertensive properties (38), and Sch202596 shows promise in Alzheimer’s therapy (39).
Although a number of methods for the racemic assembly of benzofuranones containing C2 quaternary centers have been reported, many proceed from relatively advanced starting materials (40, 41) or suffer from competitive reaction pathways (42, 43). More rare are enantioselective preparations of 2,2′-disubstituted benzofuranones. In 2008, Jørgensen and coworkers reported that 2-tert-butoxy carbonyl benzofuranone could be alkylated asymmetrically with tetraethyl ethylidene-bisphosphonate to give the corresponding 2,2′-disubstituted product in excellent enantioselectivity (44). In a different approach, we have shown that chiral triazolinylidene carbenes mediate the cyclization of aldehyde-tethered, β,β-disubstituted Michael acceptors related to 12 to give benzofuranone products in excellent enantioselectivities (Scheme 2) (45–47).
Scheme 2.

Envisioned multicatalytic Michael/Stetter cascade.
Although the strategies described provide benzofuranone products in good yield and exceptional selectivities, both make use of substrates that require multiple steps to prepare (47). We imagined that we could expedite the synthesis of benzofuranone products 9 by assembling intermediate aldehydes 12 in situ via a base-catalyzed conjugate addition reaction of salicylaldehydes 7 and electrophilic alkynes 8 (Scheme 2). Fan et al. have shown that 1,4-diazabicyclo[2.2.2]octane (DABCO) (10) efficiently mediates the addition of amine and oxygen nucleophiles to dimethyl acetylenedicarboxylate (DMAD) (8a) and alkyl propiolates (48, 49). In our envisioned sequence, a tertiary amine such as quinuclidine (11) or DABCO (10) activates alkyne 8 toward nucleophilic attack to give intermediate aldehyde 12 (Scheme 2). Subsequent chiral carbene-promoted Stetter reaction sets a quaternary stereocenter and yields product 9 asymmetrically.
Crucial to the success of any catalytic cascade is a compatible catalyst system. For many Stetter systems, tertiary amines perform as optimal bases for carbene generation (47, 50, 51). For this reason, we were encouraged that DABCO or quinuclidine would not only serve as nucleophilic “triggers” (48) to promote our imagined conjugate addition reaction but would also prove suitable bases to deprotonate triazolium salt precatalyst 4b and generate the active carbene species.
Results and Discussion
We first examined whether our envisioned cascade could be performed in a one-pot, stepwise fashion. Carbenes have been shown capable of nucleophilic addition into DMAD and other activated alkynes (52, 53). To circumvent this undesired reaction pathway, a mixture of salicylaldehyde (7a) and DMAD (8a) was treated first with quinuclidine 11 and then with triazolium salt 4b. Benzofuranone product 9aa was isolated from this one-pot, two-step sequence in good overall yield and enantioselectivity (Table 1, entry 1). In a brief solvent screen, dichloromethane and 9∶1 toluene/t-amyl alcohol provided product 9aa in similarly high yields (Table 1, entries 1 and 4), whereas toluene gave the highest level of enantioselectivity (Table 1, entries 1–5). Lowering temperatures of the Stetter reaction improves enantioselectivity slightly but results in longer reaction times and lower product yields (Table 1, entries 6–8). No productive reaction is observed when the conjugate addition reaction is conducted at low temperatures (Table 1, entry 9).
Table 1.
Solvent and temperature screen 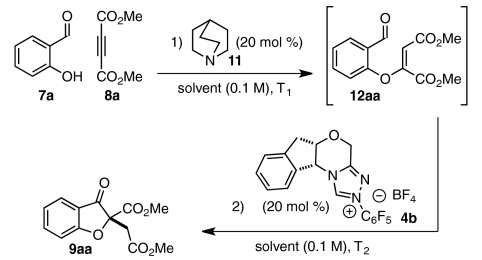
| Entry | Solvent | T1, °C | T2, °C | Yield, % | ee, % |
| 1 | CH2CI2 | 23 | 0 | 85 | 81 |
| 2 | THF | 23 | 0 | 76 | 83 |
| 3 | t-amyl alcohol | 23 | 0 | 63 | 78 |
| 4 | PhMe/t-amyl alcohol (9∶1) | 23 | 0 | 87 | 86 |
| 5 | PhMe | 23 | 0 | 82 | 89 |
| 6 | PhMe | 23 | 23 | 75 | 86 |
| 7 | PhMe | 23 | −10 | 60 | 90 |
| 8 | PhMe | 23 | −40 | 64 | 92 |
| 9 | PhMe | −40 | NA | NR | NA |
NA, not applicable; NR, no reaction.
Although we had developed conditions to mediate two bond-forming events in one reaction vessel with high levels of asymmetric induction, we hoped to reduce the number of required synthetic manipulations to a single operation. To this end, we treated a mixture of 7a, 8a, and 4b with quinuclidine (11) at 0 °C in toluene. To our delight, the cascade proceeds smoothly to give 9aa in undiminished yield and enantioselectivity (Eq. 1). The one-step protocol was found to be scalable: On a 1 g scale, product 9aa (1.48 g, 79% yield) is obtained in 88% enantiomeric excess (ee). When the one-step reaction is performed in dichloromethane, however, only starting material and decomposition products are recovered; under these conditions, nucleophilic addition of 4b-derived carbene into DMAD may interfere with the desired conjugate addition reaction (Eq. 1).  1
1
A series of control experiments were performed to probe the mechanism of the conjugate addition reaction. We had envisioned that Michael addition proceeds through nucleophilic activation of DMAD via intermediate I (Scheme 2). However, an alternative pathway could be imagined in which quinuclidine deprotonates salicylaldehyde, which adds conjugately to DMAD. To examine the viability of a base-catalyzed pathway, we exchanged quinuclidine for diisopropylethylamine. Treatment of salicylaldehyde and DMAD with this less competent nucleophile but similarly strong base resulted in complete recovery of starting material, suggesting that the proposed nucleophilic pathway is likely at work in our developed conditions (Eq. 2). Moreover, exposure of salicylaldehyde and DMAD to the free carbene derived from azolium salt 4b gave no observable product by 1H NMR spectroscopy (Eq. 3). It is highly probable that quinuclidine (11) rather than carbene-4b participates as the nucleophilic activator in our system.  2
2  3
3
With a productive one-step protocol in hand, we investigated the scope of the reaction with respect to salicylaldehyde 7. Indeed, both electron-rich and electron-deficient salicylaldehydes with various substitution patterns participate in the Michael/Stetter sequence to give products 9 in good yields and moderate to excellent enantioselectivites (Table 2). Electron-deficient salicylaldehydes give generally higher enantioselectivities but poorer yields than electron-rich salicylaldehydes. The lower yields observed for these substrates are attributed to the competitive formation of chromene side products 13 derived from intramolecular aldol of intermediate enolate III (Eq. 4). Ortho-substituted salicylaldehydes give the lowest observed yields; steric bulk surrounding the phenoxide apparently impedes nucleophilic addition into DMAD (Table 2, entries 9–10). Indeed, when the 3-substituent is sufficiently large, no conjugate addition is observed (Table 2, entry 10). Absolute configuration of products 9 was assigned by X-ray crystal structure of iodide 9da. The others were assigned by anology.  4
4
Table 2.
Salicylaldehyde scope 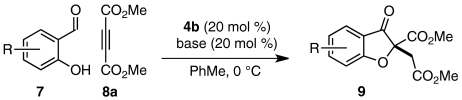
| Entry | 7 | Base | Product (9) | Yield, % | ee, % |
| 1 | 7a | 11 | 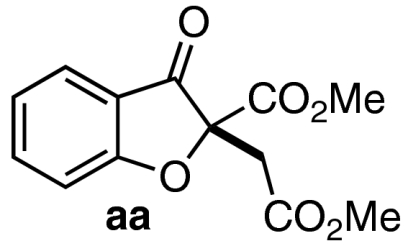 |
78 | 89 |
| 2 | 7b | 10 | 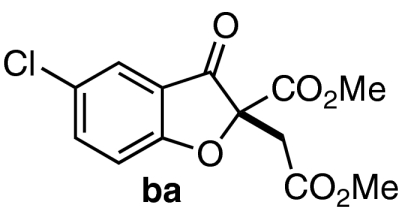 |
65 | 89 |
| 3 | 7c | 10 | 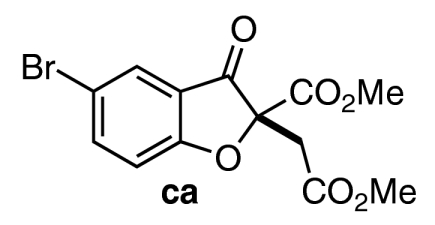 |
64 | 94 |
| 4 | 7d | 11 | 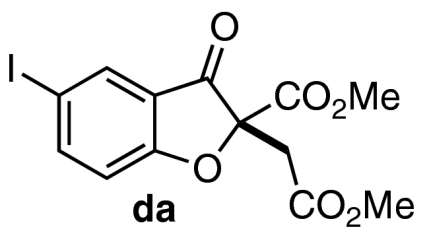 |
68 | 94 |
| 5 | 7e | 11 | 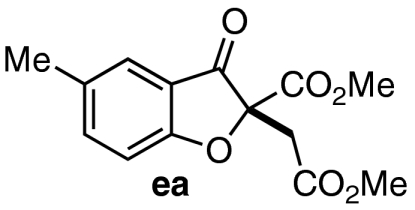 |
77 | 86 |
| 6 | 7f | 11 | 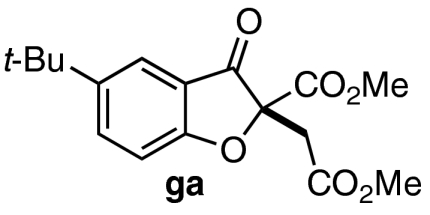 |
80 | 85 |
| 7 | 7g | 10 | 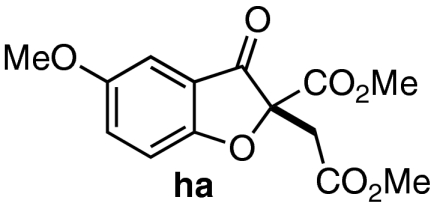 |
66 | 86 |
| 8 | 7h | 11 | 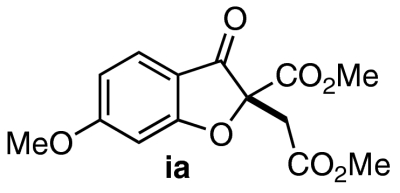 |
68 | 85 |
| 9 | 7i | 10 | 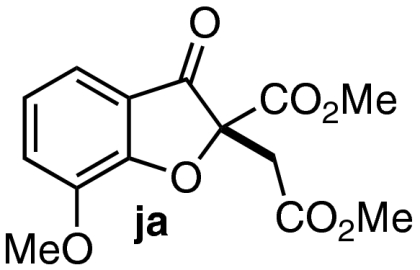 |
62* | 92 |
| 10 | 7j | 11 | 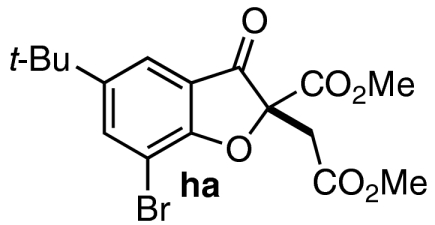 |
NR | NA |
NA, not applicable; NR, no reaction.
*9ja could not be separated from a minor impurity which coeluted in a variety of solvent systems.
We were intrigued by the variation of enantioselectivity across products 9 which appeared to be independent of steric or electronic factors. For example, 4- and 5-methoxy substrates 7h and 7g afford identical ees in spite of their differing electronic impact on both aldehyde and tethered alkene (Table 2, entry 7 vs. 8). Furthermore, sterically similar substrates 7d and 7e give products with widely disparate ees (Table 2, entry 4 vs. 5, 94% and 86% ee, respectively, corresponding to ∼0.5 kcal/mol energy difference). To probe the origin of ee variation, we performed a two-pot Michael/Stetter protocol wherein intermediate aldehyde 12 was isolated and subjected to Stetter conditions. Treatment of DMAD and salicylaldehydes 7f, 7a, and 7c with base gives the corresponding intermediate aldehydes 12fa, 12aa, and 12ca in good yields (Scheme 3). When intermediate aldehydes 12fa, 12aa, and 12ca are exposed to precatalyst 4b and base in the usual manner, however, products 9fa, 9aa, and 9ca are obtained in appreciably lower and more uniform enantioselectivities than those observed in the one-pot procedure (Scheme 3). We speculated that a trace impurity present in or side-product derived from certain salicylaldehydes 7 might be crucial for the high enantioselectivities obtained in the one-pot protocol; removal of the species during isolation of intermediate 12 would result in a drop in enantioselectivity of the subsequent Stetter reaction. Indeed, when the Stetter reactions of intermediate aldehydes 12ca and 12fa are conducted in the presence of an equivalent of exogenous salicylaldehyde 7c, the high enantioselectivities of products 9ca and 9fa are recovered (Table 3, entries 1 and 2).
Scheme 3.

Two-pot Michael/Stetter reaction.
Table 3.
Additive effect on Stetter enantioselectivity 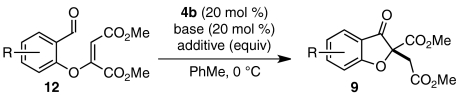
| ee, % |
|||||||
| Entry | 12 | Base | Additive | Equiv | Yield, % | W/o additive | W/additive |
| 1 | 12ca | 10 | 7c | 1.05 | 78 | 86 | 93 |
| 2 | 12fa | 11 | 7c | 1.05 | 63 | 83 | 89 |
| 3 | 12ca | 10 | 14a | 0.1 | 90 | 86 | 94 |
| 4 | 12fa | 11 | 14a | 0.1 | 84 | 83 | 92 |
We aimed to identify the species present in exogenous salicylaldehydes 7 that might promote elevation of enantioselectivity. Strong H-bond donors such as catechols have been shown to improve yields and enantioselectivities of enamine-promoted Michael additions of aldehydes into enones (54–56). We hypothesized that trace catechol derived from Dakin-oxidation (57, 58) of salicylaldehyde might contribute to Stetter enantioselectivity. Addition of 10 mol % catechol 14a to the Stetter reaction of intermediate aldehydes 12ca and 12fa improved product enantioselectivities to excellent 94% ee and 92% ee, respectively (Table 3, entries 3 and 4). When catechol 14a is added with precatalyst 4b in the one-pot, two-step protocol, similar improvement in enantioselectivity is observed for a variety of substrates (Table 4, entries 1–3). Little change in selectivity is experienced in the one-pot, one-step procedure. Presumably, catechol 14a adds conjugately to DMAD (8a) (48). Finally, an observed match/mismatch effect provides convincing support for catechol participation in the selectivity-determining step of the Stetter reaction. When chiral, binapthyl-derived catechol 14b is used as an additive, enantioselectivity of product 9aa improves from 89% ee to 95% ee with (S,R)-precatalyst 4b but shows no change with (R,S) precatalyst ent-4b (Table 4, entries 5 and 6). When catechol 14a is substituted with acidic phenol 14c, no improvement in enantioselectivity is observed (Table 4, entry 6).
Table 4.
Catechol effect on enantioselectivity in the one-pot, two-step Michael/Stetter reaction 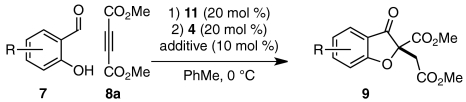
| ee, % |
||||||
| Entry | 7 | Additive | 4 | Yield, % | W/o additive | W/additive |
| 1 | 7a | 14a | 4b | 63 | 89 | 93 |
| 2 | 7f | 14a | 4b | 78 | 85 | 90 |
| 3 | 7h | 14a | 4b | 75 | 85 | 93 |
| 4 | 7a | 14b | 4b | 63 | 89 | 95 |
| 5 | 7a | 14b | ent-4b | 73 | −89 | −89 |
| 6 | 7a | 14c (1 equiv) | 4b | 60 | 89 | 89 |
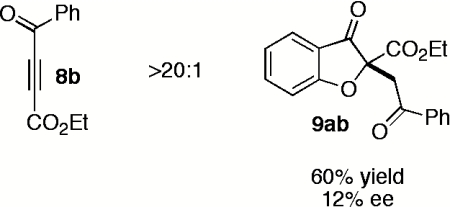
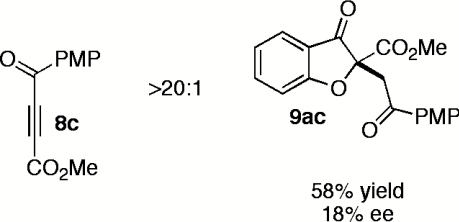
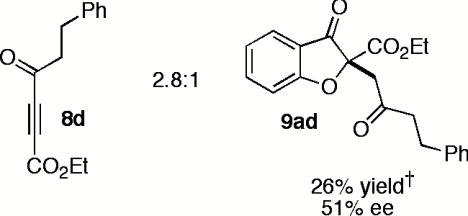
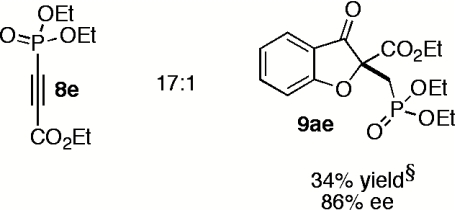
Having explored the scope and selectivity of our Michael/Stetter reaction between DMAD and a variety of salicylaldehydes, we focused on incorporation of unsymmetrical alkynes as Michael acceptors in this cascade. Ketoalkynoates 8b and 8c participate in the one-step reaction with salicylaldehyde (7a) to give moderate yields of products 9ab and 9ac regioselectively but with low enantioselectivity (Table 5, entries 1 and 2). Attenuation of alkyne electrophilicity by substitution of the aryl ketone with phenethyl ketone 8d improved the enantioselectivity of major product 9ad (Table 5, entry 3), but resulted in formation of a second regioisomer 15ad in ∼10% yield (see SI Text). Interestingly, minor regioisomer 15ad is obtained in high enantioselectivity (89%) relative to major product 9ad (Table 5, entry 3). Phosphonate ester 8e reacts in the one-pot, two-step protocol to give phosphonate 9ae in fair yield and good enantioselectivity (Table 5, entry 4).
Table 5.
Unsymmetrical alkyne scope 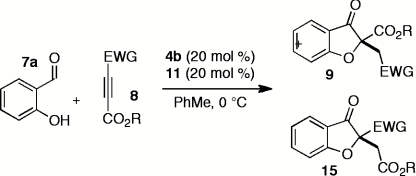
*Product ratios determined by 1H NMR of the unpurified reaction mixture.
†Yield of 9ad and 15ad determined by 1H NMR with reference to 2,6-Di-tert-butyl-4-methylphenol.
‡Phosphonate 9ae was prepared according to the one-pot, two-step protocol (see SI Text).
§Phosphonate 9ae could not be separated from a minor impurity which coeluted in multiple solvent systems.
Although we were pleased to find that a number of unsymmetrical alkynes were tolerated in our one-pot protocol, we were interested in identifying substrates that would participate with high levels of both regioselectivity and enantioselectivity. Intermediate aldehydes 16a containing a single electron-withdrawing substituent on the Michael acceptor have been shown to undergo the Stetter reaction with high enantioselectivity under conditions similar to these (Eq. 5) (47). Our initial attempt to access related intermediate aldehyde 16b via the necessarily regioselective Michael addition of salicylaldehyde (7a) into alkynoate 17 resulted only in isolation of starting material and decomposition products (Eq. 6) (59). Nevertheless, we were encouraged to try other modes of entry into 16b. Shi and Shi have shown that activated allenes behave as alkyne surrogates in a DABCO-catalyzed conjugate addition reaction with salicylaldehyde (59). Indeed, we were delighted to find that subjection of allenone 18a and allenoate 18b to our one-pot, two-step protocol in a variety of solvents gave benzofuranone products 19a and 19b in moderate yields and good to excellent enantioselectivities (Table 6).  5
5  6
6
Table 6.
One-pot, two-step Michael/Stetter reaction with activated allenes 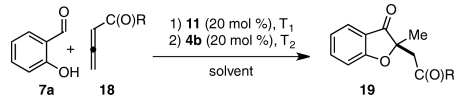
| Entry | 18 | Solvent | T1, °C | T2, °C | Product (19) | Yield, % | ee, % |
| 1 | 18a | THF | 23 | 0 | 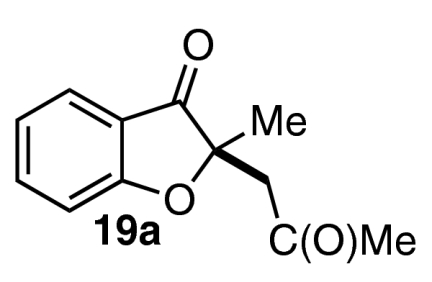 |
60 | 78 |
| 2 | 18b | PhMe | 23 | 23 | 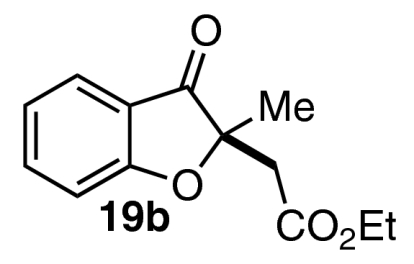 |
50 | 98 |
| 3 | 18b | PhMe | 110 | 110 | 19b | 50 | 89 |
| 4 | 18b | CH2CI2 | 23 | 23 | 19b | 59 | 96 |
We hoped to understand why certain substrates (DMAD, allenoate 18b) react with much greater enantioselectivity than others (ketoalkynoates 8b and 8c). A factor that has been shown to influence Stetter enantioselectivity is olefin geometry (46, 47). Although intermediate aldehydes 12aa and 16b derived from DMAD and allenoate 18b form with near perfect E-selectivity under our conditions (Eq. 7), intermediate aldehydes derived from ketoalkynoate substrates (8b-d) are observed as unselective mixtures of E/Z isomers (Eq. 8). For a number of Stetter scaffolds, the E-isomer has been shown to react in higher yield and with greater enantioselectivity than the corresponding Z-isomer (46, 47). To examine whether a relatively high Z/E ratio could contribute to the low enantioselectivities obtained for ketoalkynoate substrates 8a-c, we subjected a 6.5∶1 Z/E mixture (60) of intermediate aldehyde 12aa to our established Stetter conditions. Whereas E isomer 12aa reacts to afford product 9aa in 84% ee, Z-enriched 12aa gives 9aa in only 29% ee and in appreciably lower yield (Eq. 9). The disparity in enantioselectivity across batches of intermediate aldehyde 12aa reinforces previous observations that olefin geometry contributes significantly to Stetter selectivity.  7
7  8
8  9 Influence of olefin geometry on Stetter enantioselectivity (aE/Z ratios determined by 1H NMR).
9 Influence of olefin geometry on Stetter enantioselectivity (aE/Z ratios determined by 1H NMR).
In summary, we have described a unique and scalable one-pot procedure for the highly enantioselective preparation of benzofuranone products in moderate yields from simple starting materials. We have demonstrated that the one-pot Michael/Stetter protocol is superior to the two-step procedure with respect to enantioselectivity, and we have expanded on this observation to show that catechol additives improve enantioselectivity in the context of both two-pot and one-pot, two-step reactions. Moreover, we have identified olefin geometry as an important factor influencing Stetter enantioselectivity. Finally, we have illustrated that activated allenes behave as competent, E/Z-selective Michael acceptors in our one-pot, two-step reaction to provide access to alkyl-substituted benzofuranones 19 in moderate to excellent enantioselectivities. Investigations aimed at generalizing this concept are currently underway.
Materials and Methods
General methods, detailed experimental procedures, and characterization data for the compounds described in this article can be found in the SI Text, which is available free of charge on the PNAS web site.
General Procedure for the Multicatalytic Michael/Stetter Reaction.
A 1-dram vial was equipped with a magnetic stir bar under argon and charged sequentially with DMAD (8a) (21 mg, 0.15 mmol), salicylaldehdyde 7 (0.16 mmol), and triazolium salt 4b (14 mg, 0.030 mmol). Toluene (1.5 mL) was added, and the mixture was cooled to 0 °C. Quinuclidine (11) (3.0 mg, 0.030 mmol) or DABCO (10) (3.0 mg, 0.030 mmol) was added in one portion, and the reaction was monitored by TLC (hexanes/acetone). When the reaction was judged to be complete, the mixture was quenched with glacial acetic acid (1–2 drops), filtered through a plug of silica with Et2O (∼40 mL), and concentrated in vacuo. The resulting product 9 was purified via flash chromatography.
Procedure for the Preparation of 9aa on 7.0 mmol Scale.
A 250-mL, flame-dried, round-bottom flask was charged with triazolium salt 4b (164 mg, 0.350 mmol) and evacuated for 3 min, then filled with argon. After the evacuation procedure was repeated an additional two times, DMAD (8a) (1.01 g, 7.12 mmol), salicylaldehyde (7a) (933 mg, 7.64 mg), and toluene (72 mL) were added sequentially, and the reaction mixture was cooled to 0 °C. Quinuclidine (11) (156 mg, 1.40 mmol) was added portionwise to the reaction mixture. After it was allowed to stir at 0 °C for 9 h, the reaction was quenched with glacial acetic acid (150 μL) and poured directly onto a silica gel column (5∶1 - 1∶1 hexanes/EtOAc) to give 1.48 g (79% yield) of 9aa as a clear, amorphous solid.
Supplementary Material
Acknowledgments.
We thank Donald Gauthier (Merck) for a generous gift of aminoindanol, and Amgen for unrestricted support. We acknowledge Nick Emmendorfer (Colorado State University) for early experiments. We gratefully acknowledge the National Institute of General Medical Sciences (GM72586) and The Herman Frasch Foundation for support of this research.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. D.W.M. is a guest editor invited by the Editorial Board.
Data deposition: The coordinates for the crystal structure of 9da have been deposited in the Cambridge Structural Database, Cambridge Crystallographic Data Centre, www.ccdc.cam.ac.uk/products/csd/, Cambridge CB2 1EZ, United Kingdom (CSD reference no. CCDC 783384).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1002830107/-/DCSupplemental.
References
- 1.Ajamian A, Gleason JL. Two birds with one metallic stone: Single-pot catalysis of fundamentally different transformations. Angew Chem Int Edit. 2004;43(29):3754–3760. doi: 10.1002/anie.200301727. [DOI] [PubMed] [Google Scholar]
- 2.Lee JM, Na Y, Han H, Chang S. Cooperative multi-catalyst systems for one-pot organic transformations. Chem Soc Rev. 2004;33(5):302–312. doi: 10.1039/b309033g. [DOI] [PubMed] [Google Scholar]
- 3.Wasilke JC, Obrey SJ, Baker RT, Bazan GC. Concurrent tandem catalysis. Chem Rev. 2005;105(3):1001–1020. doi: 10.1021/cr020018n. [DOI] [PubMed] [Google Scholar]
- 4.Chapman CJ, Frost CG. Tandem and domino catalytic strategies for enantioselective synthesis. Synthesis. 2007;(1):1–21. [Google Scholar]
- 5.Enders D, Grondal C, Huttl MRM. Asymmetric organocatalytic domino reactions. Angew Chem Int Edit. 2007;46(10):1570–1581. doi: 10.1002/anie.200603129. [DOI] [PubMed] [Google Scholar]
- 6.Walji AM, MacMillan DWC. Strategies to bypass the taxol problem. Enantioselective cascade catalysis, a new approach for the efficient construction of molecular complexity. Synlett. 2007;(10):1477–1489. [Google Scholar]
- 7.Yu X, Wang W. Organocatalysis: Asymmetric cascade reactions catalysed by chiral secondary amines. Org Biomol Chem. 2008;6(12):2037–2046. doi: 10.1039/b800245m. [DOI] [PubMed] [Google Scholar]
- 8.Alba AN, Companyo X, Viciano M, Rios R. Organocatalytic domino reactions. Curr Org Chem. 2009;13(14):1432–1474. [Google Scholar]
- 9.Jiang H, et al. Achieving molecular complexity by organocatalytic one-pot strategies—A fast entry for synthesis of sphingoids, amino sugars, and polyhydroxylated α-amino acids. Angew Chem Int Edit. 2009;48(37):6844–6848. doi: 10.1002/anie.200901446. [DOI] [PubMed] [Google Scholar]
- 10.Bertelsen S, Jørgensen KA. Organocatalysis—after the gold rush. Chem Soc Rev. 2009;38(8):2178–2189. doi: 10.1039/b903816g. [DOI] [PubMed] [Google Scholar]
- 11.Zhou J. Recent advances in multicatalyst promoted asymmetric tandem reactions. Chem—Asian J. 2010;5(3):422–434. doi: 10.1002/asia.200900458. [DOI] [PubMed] [Google Scholar]
- 12.Enders D, Huttl MRM, Grondal C, Raabe G. Control of four stereocentres in a triple cascade organocatalytic reaction. Nature. 2006;441(7095):861–863. doi: 10.1038/nature04820. [DOI] [PubMed] [Google Scholar]
- 13.Marigo M, Bertelsen S, Landa A, Jørgensen KA. One-pot organocatalytic domino Michael-Aldol and intramolecular SN2 reactions. Asymmetric synthesis of highly functionalized epoxycyclohexanone derivatives. J Am Chem Soc. 2006;128(16):5475–5479. doi: 10.1021/ja058490o. [DOI] [PubMed] [Google Scholar]
- 14.Enders D, Narine AA, Benninghaus TR, Raabe G. Asymmetric organocatalytic domino reactions of γ-nitroketones and enals. Synlett. 2007;(11):1667–1670. [Google Scholar]
- 15.Wang J, et al. Organocatalytic enantioselective cascade Michael-Aldol condensation reactions: Efficient assembly of densely functionalized chiral cyclopentenes. Angew Chem Int Edit. 2007;46(47):9050–9053. doi: 10.1002/anie.200703163. [DOI] [PubMed] [Google Scholar]
- 16.Zu L, et al. Synthesis of highly functionalized chiral cyclopentanes by catalytic enantio- and diastereoselective double Michael addition reactions. Angew Chem Int Edit. 2007;46(20):3732–3734. doi: 10.1002/anie.200700485. [DOI] [PubMed] [Google Scholar]
- 17.Hayashi Y, Toyoshima M, Gotoh H, Ishikawa H. Diphenylprolinol silyl ether catalysis in an asymmetric formal carbo [3 + 3] cycloaddition reaction via a domino Michael/Knoevenagel condensation. Org Lett. 2008;11(1):45–48. doi: 10.1021/ol802330h. [DOI] [PubMed] [Google Scholar]
- 18.Ishikawa H, Suzuki T, Hayashi Y. High-yielding synthesis of the anti-influenza neuramidase inhibitor (-)-oseltamivir by three “one-pot” operations. Angew Chem Int Edit. 2009;48(7):1304–1307. doi: 10.1002/anie.200804883. [DOI] [PubMed] [Google Scholar]
- 19.Zu L, Zhang S, Xie H, Wang W. Catalytic asymmetric oxa-Michael‚ Michael cascade for facile construction of chiral chromans via an aminal intermediate. Org Lett. 2009;11(7):1627–1630. doi: 10.1021/ol9003433. [DOI] [PubMed] [Google Scholar]
- 20.Cernak TA, Lambert TH. Multicatalytic synthesis of α-pyrrolidinyl ketones via a tandem palladium(II)/indium(III)-catalyzed aminochlorocarbonylation/Friedel-Crafts acylation reaction. J Am Chem Soc. 2009;131(9):3124–3125. doi: 10.1021/ja809897f. [DOI] [PubMed] [Google Scholar]
- 21.Licandro E, et al. Michael addition of chiral Fischer aminocarbene complexes to nitroolefins: Study on the effect of the Michael acceptor structure on diastereoselectivity. Organometallics. 2001;20(3):485–496. [Google Scholar]
- 22.Dijk EW, et al. The asymmetric synthesis of (-)-pumiliotoxin C using tandem catalysis. Tetrahedron. 2004;60(43):9687–9693. [Google Scholar]
- 23.Huang Y, Walji AM, Larsen CH, MacMillan DWC. Enantioselective organo-cascade catalysis. J Am Chem Soc. 2005;127(43):15051–15053. doi: 10.1021/ja055545d. [DOI] [PubMed] [Google Scholar]
- 24.Shekhar S, Trantow B, Leitner A, Hartwig JF. Sequential catalytic isomerization and allylic substitution. Conversion of racemic branched allylic carbonates to enantioenriched allylic substitution products. J Am Chem Soc. 2006;128(36):11770–11771. doi: 10.1021/ja0644273. [DOI] [PubMed] [Google Scholar]
- 25.Simmons B, Walji AM, MacMillan DWC. Cycle-specific organocascade catalysis: Application to olefin hydroamination, hydro-oxidation, and amino-oxidation, and to natural product synthesis. Angew Chem Int Edit. 2009;48(24):4349–4353. doi: 10.1002/anie.200900220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang YG, Kumano T, Kano T, Maruoka K. Organocatalytic approach to enantioselective one-pot synthesis of pyrrolidine, hexahydropyrrolizine, and octahydroindolizine core structures. Org Lett. 2009;11(9):2027–2029. doi: 10.1021/ol900477x. [DOI] [PubMed] [Google Scholar]
- 27.Zhou J, List B. Organocatalytic asymmetric reaction cascade to substituted cyclohexylamines. J Am Chem Soc. 2007;129(24):7498–7499. doi: 10.1021/ja072134j. [DOI] [PubMed] [Google Scholar]
- 28.Scroggins ST, Chi Y, Fréchet JMJ. Polarity-directed one-pot asymmetric cascade reactions mediated by two catalysts in an aqueous buffer. Angew Chem Int Edit. 2010;49(13):2393–2396. doi: 10.1002/anie.200902945. [DOI] [PubMed] [Google Scholar]
- 29.Lathrop SP, Rovis T. Asymmetric synthesis of functionalized cyclopentanones via a multicatalytic secondary amine/n-heterocyclic carbene catalyzed cascade sequence. J Am Chem Soc. 2009;131(38):13628–13630. doi: 10.1021/ja905342e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Read de Alaniz J, Rovis T. The catalytic asymmetric intramolecular Stetter reaction. Synlett. 2009;(8):1189–1207. doi: 10.1055/s-0029-1216654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karthikeyan K, Perumal PT. Synthesis of highly functionalized spirobenzofuranols and spirobenzofuranones by Baylis-Hillman reaction. Synlett. 2009;(14):2366–2370. [Google Scholar]
- 32.Pirrung MC, Brown WL, Rege S, Laughton P. Total synthesis of (+)-griseofulvin. J Am Chem Soc. 1991;113(22):8561–8562. [Google Scholar]
- 33.Sheng R, et al. Design, synthesis and AChE inhibitory activity of indanone and aurone derivatives. Eur J Med Chem. 2009;44(1):7–17. doi: 10.1016/j.ejmech.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 34.Rønnest MH, et al. Synthesis and structure-activity relationship of griseofulvin analogues as inhibitors of centrosomal clustering in cancer cells. J Med Chem. 2009;52(10):3342–3347. doi: 10.1021/jm801517j. [DOI] [PubMed] [Google Scholar]
- 35.Awale S, et al. Constituents of Brazilian red propolis and their preferential cytotoxic activity against human pancreatic PANC-1 cancer cell line in nutrient-deprived condition. Bioorg Med Chem. 2008;16(1):181–189. doi: 10.1016/j.bmc.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 36.Charrier C, et al. Synthesis and modeling of new benzofuranone histone deacetylase inhibitors that stimulate tumor suppressor gene expression. J Med Chem. 2009;52(9):3112–3115. doi: 10.1021/jm9002439. [DOI] [PubMed] [Google Scholar]
- 37.Gerard B, Sangji S, O'Leary DJ, Porco JA. Enantioselective photocycloaddition mediated by chiral Brønsted acids: Asymmetric synthesis of the rocaglamides. J Am Chem Soc. 2006;128(24):7754–7755. doi: 10.1021/ja062621j. [DOI] [PubMed] [Google Scholar]
- 38.Morton JGM, Kwon LD, Freeman JD, Njardarson JT. An Adler-Becker oxidation approach to vinigrol. Tetrahedron Lett. 2009;50(15):1684–1686. [Google Scholar]
- 39.Katoh T, Ohmori O, Iwasaki K, Inoue M. Synthetic studies on Sch 202596, an antagonist of the galanin receptor subtype GalR1: An efficient synthesis of (+/−)-geodin, the spirocoumaranone part of Sch 202596. Tetrahedron. 2002;58(7):1289–1299. [Google Scholar]
- 40.Patonay-Péli E, Litkei G, Patonay T. Flavonoids, 42. Anomalous intramolecular conjugate addition: Synthesis of 2-amino-2-benzyl-3(2H)-benzofuranone and related compounds. Synthesis. 1990;(6):511–514. [Google Scholar]
- 41.He J, Zheng J, Liu J, She X, Pan X. N-heterocyclic carbene catalyzed nucleophilic substitution reaction for construction of benzopyrones and benzofuranones. Org Lett. 2006;8(20):4637–4640. doi: 10.1021/ol061924f. [DOI] [PubMed] [Google Scholar]
- 42.Kokubo K, Matsumasa K, Nishinaka Y, Miura M, Nomura M. Reaction of 2-hydroxybenzaldehydes with alkynes, alkenes, or allenes via cleavage of the aldehyde C-H bond using a rhodium catalyst system. Bull Chem Soc Jpn. 1999;72(2):303–311. [Google Scholar]
- 43.Kadnikov DV, Larock RC. Palladium-catalyzed carbonylative annulation of internal alkynes: Synthesis of 3,4-disubstituted coumarins. J Org Chem. 2003;68(24):9423–9432. doi: 10.1021/jo0350763. [DOI] [PubMed] [Google Scholar]
- 44.Capuzzi M, Perdicchia D, Jørgensen KA. Highly enantioselective approach to geminal bisphosphonates by organocatalyzed Michael-type addition of β-ketoesters. Chem—Eur J. 2008;14(1):128–135. doi: 10.1002/chem.200701317. [DOI] [PubMed] [Google Scholar]
- 45.Read de Alaniz J, Kerr MS, Moore JL, Rovis T. Scope of the asymmetric intramolecular Stetter reaction catalyzed by chiral nucleophilic triazolinylidene carbenes. J Org Chem. 2008;73(6):2033–2040. doi: 10.1021/jo702313f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kerr MS, Rovis T. Enantioselective synthesis of quaternary stereocenters via a catalytic asymmetric Stetter reaction. J Am Chem Soc. 2004;126(29):8876–8877. doi: 10.1021/ja047644h. [DOI] [PubMed] [Google Scholar]
- 47.Moore JL, Kerr MS, Rovis T. Enantioselective formation of quaternary stereocenters using the catalytic intramolecular Stetter reaction. Tetrahedron. 2006;62(49):11477–11482. [Google Scholar]
- 48.Fan MJ, Li GQ, Li LH, Yang SD, Liang YM. DABCO-catalyzed reaction of phenols or 1,2-diphenols with activated alkynes leading to the formation of alkenoic acid esters or 1,3-dioxole derivatives. Synthesis. 2006;(14):2286–2292. [Google Scholar]
- 49.Fan MJ, Li GQ, Liang YM. DABCO catalyzed reaction of various nucleophiles with activated alkynes leading to the formation of alkenoic acid esters, 1,4-dioxane, morpholine, and piperazinone derivatives. Tetrahedron. 2006;62(29):6782–6791. [Google Scholar]
- 50.DiRocco DA, Oberg KM, Dalton DM, Rovis T. Catalytic asymmetric intermolecular Stetter reaction of heterocyclic aldehydes with nitroalkenes: Backbone fluorination improves selectivity. J Am Chem Soc. 2009;131(31):10872–10874. doi: 10.1021/ja904375q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu Q, Perreault S, Rovis T. Catalytic asymmetric intermolecular Stetter reaction of glyoxamides with alkylidenemalonates. J Am Chem Soc. 2008;130(43):14066–14067. doi: 10.1021/ja805680z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nair V, Bindu S, Sreekumar V, Rath NP. Unprecedented reactivity of N-heterocyclic carbenes toward DMAD and aldehydes leading to novel multicomponent reactions. Org Lett. 2003;5(5):665–667. doi: 10.1021/ol0273856. [DOI] [PubMed] [Google Scholar]
- 53.Nair V, Sreekumar V, Bindu S, Suresh E. Two unprecedented multicomponent reactions involving N-heterocyclic carbenes, activated acetylenes, and aldehydes. Org Lett. 2005;7(12):2297–2300. doi: 10.1021/ol0502782. [DOI] [PubMed] [Google Scholar]
- 54.Campbell MJ, Johnson JS. Asymmetric synthesis of (+)-polyanthellin A. J Am Chem Soc. 2009;131(30):10370–10371. doi: 10.1021/ja904136q. [DOI] [PubMed] [Google Scholar]
- 55.Chen K, Baran PS. Total synthesis of eudesmane terpenes by site-selective C-H oxidations. Nature. 2009;459(7248):824–828. doi: 10.1038/nature08043. [DOI] [PubMed] [Google Scholar]
- 56.Peelen TJ, Chi Y, Gellman SH. Enantioselective organocatalytic Michael additions of aldehydes to enones with imidazolidinones: Cocatalyst effects and evidence for an enamine intermediate. J Am Chem Soc. 2005;127(33):11598–11599. doi: 10.1021/ja0532584. [DOI] [PubMed] [Google Scholar]
- 57.Dakin HD. The oxidation of hydroxy derivatives of benzaldehyde, acetophenone and related substances. Am Chem J. 1909;42(6):477–498. [Google Scholar]
- 58.Dakin HD. Oxidation of hydroxy derivatives of benzaldehyde and acetophenone. Proc Chem Soc. 1910;25:194. [Google Scholar]
- 59.Shi YL, Shi M. DABCO-catalyzed reaction of allenic esters and ketones with salicyl N-tosylimines: Synthesis of highly functionalized chromenes. Org Lett. 2005;7(14):3057–3060. doi: 10.1021/ol051044l. [DOI] [PubMed] [Google Scholar]
- 60.Gupta RK, George MV. Reactions of dimethyl acetylenedicarboxylate-X. Reaction with salicylaldehyde, ortho hydroxyacetophenone, 2-hydroxychalcones and 2,2′-dihydroxychalcones. Tetrahedron. 1975;31(10):1263–1275. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.