

Abstract
Despite the ubiquitous use of phosphoramidite chemistry in the synthesis of biophosphates, catalytic asymmetric phosphoramidite transfer remains largely unexplored for phosphate ester synthesis. We have discovered that a tetrazole-functionalized peptide, in the presence of 10-Å molecular sieves, functions as an enantioselective catalyst for phosphite transfer. This chemistry in turn has been used as the key step in a streamlined synthesis of myo-inositol-6-phosphate. Mechanistic insights implicate phosphate as a directing group for a highly selective kinetic resolution of a protected inositol monophosphate. This work represents a distinct and efficient method for the selective catalytic phosphorylation of natural products.
Keywords: kinetic resolution, organocatalysis
The inositol phosphates and their phosphoinositide relatives are hubs for an extraordinary amount of biochemistry, and there appears to be no wane in the pace of discovery in this field (1–3). Many early efforts have focused on the biochemistry of the inositol lipids, whereas the last decade has seen a revival of interest in the myriad of inositol phosphates and polyphosphates not associated with the lipid bilayer (1). Total syntheses of these compounds and their analogs have played a critical role in enabling high-precision studies of their biochemistry (4). Historically, the strategy for synthesis in this area has been dominated by protecting group manipulations and classical resolution (5). Our laboratory endeavored to take a step forward in 2001, introducing a class of catalysts that rely on catalytic enantioselective transfer of P(V) reagents (Fig. 1A) (6, 7). This work enabled access to a wide variety of myo-inositol phosphates (8), phosphatidyl-myo-inositol phosphates (9–11), and related analogs, as well as facilitating studies of their biochemistry (12–15).
Fig. 1.

Catalytic enantioselective P(V) group transfer. (A) Stereodifferentiation of the 1,3 hydroxyls. (B) Stereodifferentiation of the 4,6 hydroxyls.
Although catalytic enantioselective P(V) chemistry continues to prove useful, we have observed limitations. First, the nearly exclusive requirement of phenyl substituents on the chlorophosphate P(V) reagent necessitates additional protecting group manipulations in many syntheses (8–11) due to reagent stability and the incompatibility of phenyl phosphate hydrolysis conditions with synthetic schemes. Despite their utility in concert with peptide-based catalysts, chlorophosphates are generally regarded as less reactive and less versatile reagents compared to their phosphorous(III) analogues. In fact, these realities were an impetus for the development of P(III) reagents for complex phosphate and polynucleotide synthesis (16). Second, from the standpoint of asymmetric, catalytic syntheses of phosphoinositides, the P(V) methodology has thus far failed to effectively differentiate the respective 4- or 6-positions of myo-inositol (Fig. 1B) (17), even though it is highly selective for differentiation of the enantiotopic 1- and 3-positions of the myo-inositol ring.
We speculate that this discrepancy in 4/6 vs 1/3 desymmetrization is due to local structure of the reacting hydroxyl groups (Fig. 2). Whereas the 1- and 3-positions appear to be more differentiated from the local stereochemical perspective, with neighboring axial and equatiorial substituents, the enantiotopic 4- and 6-positions of myo-inositol are effectively equivalent in terms of their local environment, each flanked by two stereochemically identical equatorial positions.
Fig. 2.

Challenges posed by 4,6 desymmetrization. (A) Approach by electrophile for 1,3 desymmetrization. (B) Approach by electrophile for 4,6 desymmetrization.
In order to address this significant limitation in catalytic asymmetric phosphorylation methodology, we have developed a unique asymmetric catalytic system and applied it to a target-oriented total synthesis of a natural product. In doing so we have solved a long-standing challenge within our lab, 4- versus 6-positional differentiation in myo-inositol. We have approached this goal through the introduction of (i) a chiral peptide-based phosphitylation catalyst based on a nonproteinogenic amino acid, tetrazolylalanine (Atz) and (ii) its application to an efficient catalytic asymmetric synthesis of myo-inositol-6-phosphate. This report represents a distinct account, to our knowledge, of a synthetic strategy wherein catalytic enantioselective phosphoramidite-based bond construction can be implemented and applied to the synthesis of a naturally occurring, phosphorylated metabolite.
Results and Discussion
The fundamental concept we have applied for catalytic P(V) phosphate transfer chemistry has been nucleophilic catalysis employing a π-methylhistidine (Pmh) residue * (e.g., peptide 2 in Fig. 3A) (18, 19). Exploration of catalytic P(III)-based transfers necessitates the deployment of a different catalytic residue. Given the profound success of tetrazole as a promoter of phosphoramidite transfer (20), we set out to synthesize and explore Atz-based peptides (e.g., peptide 3) as chiral catalysts for this reaction.
Fig. 3.

(A) π-Methyl-histidine based peptide catalyst (2) and tetrazole based peptide catalyst (3). (B) Synthesis of tetrazole based peptides from commercially available amino acids.
We prepared Boc-L-tetrazolylalanine (21) from Boc-L-β-cyanoalanine and incorporated this residue into a small library of pentameric peptide catlaysts utilizing standard coupling procedures (Fig. 3B). To ensure a fully active catalyst, peptides were purified by reverse-phase chromatography with 0.1% trifluoroacetic acid in the mobile phase. Notably, we find that both the free tetrazole acid and the 1,8-Diazobicyclo[5.4.0]undec-7-ene (DBU) salt obtain directly from resin cleavage performed equally well for catalytic phosphitylation.
The transition from Pmh-based catalysis (Fig. 4A) (17) to Atz-based catalysis also requires the development of a fundamentally different catalytic cycle. The difference arises from the bifunctional activation of phosphoramidites by tetrazole; initial protonation of the phosphoramidite precedes tetrazole fixation on the phosphorous atom (Fig. 4B) (22, 23). Implicit in this catalytic cycle is the need to regenerate the free tetrazole acid; for each round of phosphoramidite activation one equivalent of dialkylamine is generated, effectively deactivating the catalyst as the dialkylammonium salt. As such, most procedures require an excess of tetrazole to achieve complete conversion (24).
Fig. 4.

(A) Pmh-based catalysis of P(V) transfer, versus (B) Atz-based catalysis of P(III)/phosphoramidite transfer.
To date, only two reports (25, 26) describe catalytic activation of phosphoramidites with efficient turnover of the catalytic species. It is generally accepted that catalyst deprotonation by the product dialkylamine is the main source of catalyst inactivation. As a result, we considered several amine scavengers, ultimately settling upon the method described by Hayakawa and Kataoka, utilizing 10-Å molecular sieves (M.S.) in conjunction with diethylamino phosphoramidites (25). Under optimized experimental conditions, we found that 10-Å molecular sieves are highly effective as a diethyl amine scavenger in CHCl3. Amine scavenging was evidenced by the complete disappearance of diethyl amino resonances as the reaction progressed when monitored by 1H—NMR. Moreover, 10-Å molecular sieves are inexpensive, require no special storage or preparation, and may be removed by filtration after the reaction is complete. They are also effective drying agents—a distinct advantage given that activated phosphoramidites are prone to hydrolysis.
We then set out to assess whether this new peptide-based, catalytic phosphitylation process could be rendered enantioselective. We thus turned our attention to the desymmetrization of 1,3,5-tri-O-PMB-myo-inositol 4 (Table 1). Rather than engage in a large-scale peptide screening exercise, we simply chose several peptide sequences that had previously proven useful in Pmh-based transfer of P(V) reagents, recognizing that the chiral environments presented by these catalysts would not necessarily be appropriate for the target reaction; † rather, we simply hoped to learn if even a small modicum of selectivity might be observed with this new phosphoramidite activation paradigm. We thus examined several peptide sequences, each terminated in Boc-L-Atz for the desymmetrization of 4. Most notably, three of the catalysts (entries 1, 4, and 5 in Table 1) exhibited enantiomer ratios (e.r.) of 70∶30 or better. Moreover, peptides 3 and 7 catalyzed the formation of opposite enantiomers of the product, despite the identical stereochemistry at the terminal residue (L-Atz). Also, as little as 5 mol % of the catalyst could be employed (entries 2 and 3) while maintaining excellent conversion to the product, confirming the efficiency of the molecular sieve strategy for catalyst turnover.
Table 1.
Selected data for phosphitylation catalyst screen
 | |||||||
| Entry* | Peptide† | Loading | i + 1 | i + 2 | i + 3 | i + 4 | e.r.‡ |
| 1 | 3 | 20% | Hyp(tBu) | hPhe | Phe | Phe | 26.0∶74.0 |
| 2 | 3 | 5% | Hyp(tBu) | hPhe | Phe | Phe | 24.0∶76.0 |
| 3 | 3 | 5% | Hyp(tBu) | hPhe | Phe | Phe | 15.0∶85.0 |
| 4 | 7 | 20% | Asn(Trt) | Aib | Ser(tBu) | (D)Phe | 69.5∶30.5 |
| 5 | 8 | 20% | Aib | Glu(tBu) | hPhe | Phe | 31.0∶69.0 |
*Entries 1, 2, 4, and 5 were performed at room temperature.
†All peptides were used as the C-terminal methyl ester. Each peptide was synthesized with Boc-Atz as the N-terminal amino acid that is designated as the i position.
‡Secondary kinetic resolution is presumed to partially contribute to the observed selectivities as was frequently observed with peptide 3. Enantiomer ratios of 5 were measured using chiral HPLC.
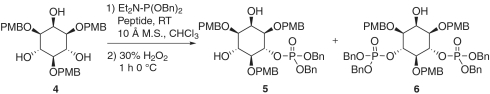
At 4 °C, on a 250-mg scale, the selectivity virtually doubles compared to the room temperature reaction (entry 3). This chemistry proceeds very cleanly with a 71% isolated yield of 5, 16% isolated yield of the bis-phosphorylated 6 and 6% recovered starting material. Furthermore, in monitoring the progress of the reaction by 31P and 1H we observed no other phosphorylated inositol products.
Interestingly, during the course of optimizing reactions we noticed a modest degree of secondary kinetic resolution of the product when the reaction was allowed to proceed to greater conversions [i.e., ultimately to the meso bis(phosphorylated) product 6] (28). Intrigued by the possibility that the in situ formed phosphite may offer a handle for substrate recognition, we were curious what effect a phosphate might have in the context of a kinetic resolution.‡ To this end, racemic monophosphate 5 was subjected to the same phosphitylation conditions used in the desymmetrization of 1,3,5-tri-O-PMB-myo-inositol 4 (Eq. 1). A dramatic kinetic resolution was observed, corresponding to a krel of 34.5 ± 3.7. This unprecedented, high degree of selectivity in a catalytic phosphoramidite transfer reaction establishes the notion of asymmetric catalysis with Atz-based peptides as a useful tool for chiral phosphate synthesis.
We suspected this high degree of selectivity could be attributed to the phosphate oxygen functioning as a hydrogen bond acceptor, forming a crucial contact with the catalyst for asymmetric induction. To test this hypothesis, racemic phosphorothioate 9 was subjected to the same kinetic resolution conditions (Eq. 2). Sulfur is known to be a weaker hydrogen bond acceptor than oxygen (29), yet phosphorothioates are frequently used as phosphate ester analogues (30), making 9 an appropriate probe for our hypothesis. Indeed, catalyst 3 exhibited considerably lower selectivity for racemic phosphorothioate 9 (krel < 10), pointing to the direct role that the phosphorous-oxygen bond serves in substrate recognition. This notion that a phosphate group may be an effective H-bond-based directing group during catalytic stereoselective phosphoramidite transfer reactions is now the basis of further study.


One could envision a total synthesis of myo-inositol-6-phosphate based upon the highly selective kinetic resolution of racemic 5. However, we found the preparation of racemic 5 to be significantly less efficient with achiral catalysts than with peptide catalyst 3 (Table 1). We observed that achiral catalysts lead to over conversion to the bis-phosphorylated product and small quantites of 2′-phosphorylated product with achiral promoters. Thus, by this route, the highest possible yield of optically pure monophosphate 5 would be less than 20% from 4.
Therefore, we proceeded with the total synthesis of myo-inositol-6-phosphate via two sequential asymmetric transformations from 1,3,5-tri-O-PMB-myo-inositol 4. On a 1-g scale of 1,3,5-tri-O-PMB-myo-inositol, the peptide catalyzed desymmetrization proceeded with virtually identical results (71% yield, 15.4∶84.6 e.r.) compared to the 0.25-g scale reaction (Fig. 5). The minor isomer was then selectively converted to the bis(phosphate) under kinetic resolution conditions to yield 5 in > 1.9∶98.1 e.r. (52.4% isolated yield from 4). The resulting optically pure monophosphate was then subjected to deprotection with sodium metal and liquid ammonia to yield the disodium salt of d-myo-inositol-6-phosphate in near quantitative yield.
Fig. 5.

Reagents and conditions: (1) Et2NP(OBn)2, peptide 3, 10 Å M.S., CHCl3, 4 °C; (2) 30% H2O2, 0 °C, 71% yield, 15.1∶84.9. Conditions (1) and (2) were repeated for the kinetic resolution of 5, 74.0% yield, > 1.9∶98.1 e.r.; (3) NH3, Na, -78 °C; (4) DOWEX 50WX8-500, C6H11NH2, 95%.
Finally, we assigned the absolute configuration of the bis(cyclohexylamine) salt 11 (d-myo-inositol-6-phosphate), which exhibited the opposite optical rotation (levorotary) relative to known reports in the literature for d-myo-inositol-4-phosphate (dextrorotary). Notably, the magnitude of rotation was smaller than the two disparate values reported in the literature [synthetic 11: [α]D = -0.3 (c = 2.0, H2O, pH 8–9); literature ent-11: [α]D = +2 (c = 2, H2O) (31) and [α]D = +1.1 ( c = 5, H2O) (32)]. We and others have found that optical rotations of synthetic myo-inositol phosphates are highly sensitive to sample preparation.§ With such notoriously low optical rotations with the inositol phosphates, the process of reproducing the magnitude is not trivial. Thus, in an effort to unambiguously establish the optical purity of our sample and in an effort to aid in the characterization of future syntheses of inositol phosphates, we devised a two-step protection strategy such that our sample could be compared to a racemic sample by chiral normal-phase high-pressure liquid chromatography (HPLC) (Fig. 6). We observe only a minute difference in the enantiomeric ratio of compounds 5 and 14 after this synthetic sequence, confirming the optical purity of our synthetic d-myo-inositol-6-phosphate. Furthermore, as a testament to the robustness of this method, the entire synthetic sequence through 14 (Figs. 5 and 6) was repeated, with the benzyl protecting groups with essentially identical results.
Fig. 6.

Reagents and conditions: (1) Ac2O, pyridine, CDCl3; (2) BnBr, 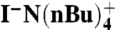 , K2CO3, CD3CN, 41%, 2.1∶97.9 e.r.
, K2CO3, CD3CN, 41%, 2.1∶97.9 e.r.
Conclusions
We have completed a streamlined enantioselective synthesis of d-myo-inositol-6-phosphate based on a catalytic asymmetric method developed herein. For this purpose, we developed an unprecedented peptide catalyst containing the nonproteinogenic amino acid L-Atz. We have demonstrated that such peptides can exhibit considerable levels of enantioselectivity in a kinetic resolution setting, in particular when hydrogen bond donors or acceptors are present. We now intend to apply these catalysts to the synthesis and modification of additional complex natural products (35, 36), employing ubiquitous phosphoramidite reagents.
Materials and Methods
General Methods.
Proton NMR spectra were collected on Bruker 400- or 500-MHz spectrometers at 25 °C. Proton chemical shifts are reported in ppm (δ) relative to internal tetramethylsilane (TMS, δ 0.0 ppm) or with the solvent reference relative to TMS employed as the internal standard (CDCl3, δ 7.26 ppm; CD3OD, δ 3.31 ppm; D2O, δ 4.79 ppm). Data are reported as follows: chemical shift {multiplicity [singlet (s), doublet (d), triplet (t), quartet (q), and multiplet (m)], coupling constants [Hz], integration}. Broad peaks are denoted by (br) before the chemical shift multiplicity. Carbon NMR spectra were collected on Bruker 400 (101 MHz) or 500 (126 MHz) NMR spectrometers with complete proton decoupling. Carbon chemical shifts are reported in ppm (δ) relative to TMS with respective solvent resonances as the internal standard (CDCl3, δ 77.16). A drop of methanol was added as an internal standard (δ 49.5) for carbon NMR experiments performed in D2O. Where indicated, carbon-phosphorous couplings are reported as follows: chemical shift, multiplicity, and coupling constant. Phosphorous NMR spectra were recorded on Bruker 400 (162 MHz) and 500 (202 MHz) NMR spectrometers with complete proton and carbon decoupling. Phosphorous chemical shifts are reported in ppm (δ) relative to 85% H3PO4 as an external standard. Infrared spectra were obtained using a Thermo Electron Corporation Nicolet 6700 FT-IR instrument. Thin-layer chromatography (TLC) was performed using silica gel 60 Å F254 precoated plates (0.25 mm thickness) and TLC Rf values are reported herein. Analytical TLC plates were developed utilizing cerium ammonium molybdate (CAM) or visualized by UV absorbance. Preparatory TLC was performed using 20 × 20 cm Silica Gel 60 Å F254 precoated plates (0.25 mm thickness) and visualized by UV absorbance. Flash chromatography was performed using Silica Gel 60 Å (32–63 μM). Specific rotations were determined with a Perkin Elmer Polarimeter 341 at 20 °C on the sodium D line (path length 10.0 cm). High-resolution mass spectra were obtained from institutional providers with the method of ionization reported herein. Phosphitylations were monitored by 31P and 1H NMR using the following procedure: 100–200 μL of reaction mixture (including molecular sieves) were added to 600 μL of CDCl3. Chemical shifts representing the phosphoramidite (δ 147.8 ppm, 1P) and the corresponding inositol phosphite [monophosphite δ 142.4 ppm (s, 1P), bisphosphite δ 142.5 ppm (s, 2P)] were integrated to determine reaction progress. The following diagnostic 1H NMR signals were used: starting material [δ 4.19 (t, J = 2.6, 1H) and 3.99 (t, J = 9.5, 2H)], monophosphite [δ 4.12 (t, J = 2.3, 1H) and 4.05 (td, J = 9.6, 1.4 1H)] and bisphosphite [δ 3.44 (t, J = 9.4, 1H)]. Representative NMR spectra, which include resonances for myo-inositol phosphite intermediates, can be found in SI Appendix. Analytical reverse-phase HPLC was performed employing a single-wavelength UV detector equipped with a Waters® Sunfire RP C-18 (4.6 × 150 mm) column on a gradient of 0–100% acetonitrile in water with 0.1% TFA over 36 min. Preparative reverse-phase peptide purifications were performed on a BioTage SP4 instrument (C18HS 25 + 3, 0–100% MeOH in water with 0.1% TFA). Measurements of enantiomeric excess were carried out via analytical normal-phase HPLC equipped with a diode array detector and employing Chiralcel® columns. Specific methods are described below. Reactions were performed under inert atmosphere (N2 or argon) employing flame- or oven-dried glassware. All solvents were either distilled or taken from a solvent purification system. Chloroform (Aldrich, > 98.8%, American Chemical Society grade) was dried according to a previously reported procedure (37) as follows: Chloroform stabilized with 0.5–1% ethanol was washed two times with equal volumes of deionized water, dried with anhydrous K2CO3, refluxed over P2O5, collected, and stored for up to 5 days wrapped in aluminum foil. Molecular sieves (10 Å, Universal Oil Products type, powder) were used as provided by Fluka without further preparation. Dibenzyl N,N-diethylphosphoramidite (90% pure) was purchased from Alfa Aesar and used without further purification.
Synthesis of Atz-Containing Peptides.
All peptides were prepared on solid support using the appropriate preloaded Wang resin and Fmoc amino acid chemistry. Fmoc deprotections were performed with 20% piperidine in DMF for 30 min, except for the deprotection following the second amino acid, which was performed with 50% piperidine for 5 min. Amino acid couplings were performed with 5 equivalents of desired Fmoc amino acid, 5 equivalents O-(Benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU), and 10 equivalents Hünig’s base in DMF for 3 h. Boc-L-Atz was incorporated using a double coupling procedure, with sequential couplings of 30 min each (2 equiv Boc-Atz, 2 equiv HBTU, 5 equiv of Hünig’s base). Peptides were cleaved from solid support with 9∶1 MeOH:DMF and 5 equiv of DBU (based upon resin loading). After 3 h, the cleavage cocktail was transferred to a round bottom flask and the remaining DBU was quenched with 5 equiv TFA (based upon resin loading) and concentrated in vacuo and purified as described in the general methods.
Characterization of Peptide 3.
1H NMR (CDCl3, 400 MHz) δ 7.64 (br d, J = 6.6, 1H), 7.30–7.00 (m, 16H), 6.83 (br d, J = 6.0, 1H), 5.50 (br d, J = 7.6, 1H), 4.85–4.63 (m, 3H), 4.53 (br t, 1H), 4.42 (m, 1H), 4.25 (br s, 1H), 3.69 (s, 3H), 3.58 (m, 1H), 3.35 (m, 1H), 3.30–2.95 (m, 6H), 2.80–2.60 (m, 2H), 2.27–1.94 (m, 4H), 1.77 (s, 9H), 1.17 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 173.1, 171.6, 170.8, 170.2, 154.8, 140.5, 136.5, 135.9, 129.2, 129.0, 128.6, 128.6, 128.5, 127.1, 127.0, 126.2, 80.7, 77.3, 74.7, 69.3, 60.6, 55.2, 54.5, 54.0, 53.6, 52.5, 50.5, 37.9, 37.8, 37.5, 33.0, 32.0, 28.3, 28.1, 27.0; IR (film, cm-1) 3283, 3087, 3064, 3028, 2977, 2934, 1739, 1685, 1642, 1541, 1520, 1497, 1446, 1392, 1367, 1332, 1279, 1249, 1188, 1170, 1113, 1090, 1057, 1030. Exact mass calculated for [C47H61N9O9]H+ requires m/z 896.467, found 896.467; [α]D = -14.4 (c = 1.0, CHCl3). HPLC tR = 24.2 min Sunfire RP C-18 column (Waters® 4.6 × 150 mm) on a gradient of 0–100% acetonitrile with 0.1% TFA over 36 min.
6-Dibenzylphosphate-1,3,5-tri-O-PMB-myo-inositol (5).
To a flame-dried flask was added 80 mL freshly distilled chloroform. Molecular sieves (10 Å, 13.32 g) were added to the stirring chloroform, after which heat evolved. Please note, 10-Å molecular sieves should be added to the chloroform solution and not vice versa. CAUTION! HCl gas and heat evolve upon addition of sieves. The reaction was stirred at room temperature for 30 min under a stream of nitrogen before adding 1,3,5-tri-O-PMB-myo-inositol (1 g, 1.85 mmol) and peptide 3 [Boc-Atz-Hyp(But)- hPhe-Phe-Phe-OMe, 82.8 mg, 0.093 mmol]. The solution was cooled to 0 °C in an ice bath and dibenzyl N,N-diethylphosphoramidite (90% pure) was then added (0.836 mL, 2.40 mmol); the reaction and bath were then moved to a 4 °C cold room. The reaction was monitored by 31P and 1H NMR. After 48 h, the ratio of starting material the mono- and the bis- was 0.15∶1∶0.17, whereupon the reaction was quenched with 0.8 mL MeOH. After stirring for 20 min the reaction mixture was vacuum filtered through a Celite pad, cooled to 0 °C, and vigorously stirred with 30% H2O2 (2 mL, 18.5 mmol). After 1 h the reaction was poured into a separatory funnel containing ice. Residual H2O2 was quenched with the addition of saturated aqueous Na2SO3 and checked for peroxides using potassium iodide starch paper. CAUTION! EXOTHERMIC QUENCH! The contents were shaken and the organic phase removed. The aqueous layer was then extracted twice with CH2Cl2, and the combined organics were dried with anhydrous Na2SO4 and concentrated. The resulting residue was purified by flash chromatography (2–4% ethanol in diethyl ether) yielding 5 as a colorless oil (1.048 g, 70.8% yield), which exhibited 69.1% enantiomeric excess (ee, chiral HPLC, see below).
Kinetic Resolution of 5.
To a flame-dried flask was added 50 mL of freshly distilled chloroform. To this flask was added 9.41 g of 10 Å molecular sieves. In another flask, 1.048 g (1.31 mmol) of 5 (69.1% ee) was dissolved in 4 mL freshly distilled chloroform. After stirring the sieves for 30 min under a stream of nitrogen, the solution of 5 was transferred via syringe to the suspension of sieves, and the flask containing 5 was washed with 3 mL additional chloroform. To the mixture of sieves and 5 was added 58.6 mg (0.0654 mmol) peptide catalyst 3. The flask was then cooled in an ice bath, and to it was added 100.4 μL dibenzyl N,N-diethylphosphoramidite (0.29 mmol, 90% pure). The reaction and bath was placed in a cold room (4 °C) for 6 h, where 31P and 1H NMR indicated complete consumption of phosphoramidite. The reaction was filtered through a Celite pad, and the filtered sieves were washed with 50 mL CH2Cl2. The filtrate was cooled to 0 °C, and 0.6 mL of 30% H2O2 was added. The reaction was stirred vigorously for 1 h, poured into a separatory funnel containing ice, and quenched with saturated aqueous Na2SO3. The organic layer was removed, and the aqueous layer was washed twice more with CH2Cl2. The combined organics were dried with anhydrous Na2SO4, filtered, and concentrated in vacuo. The resulting residue was purified by flash chromatography (2–4% ethanol in diethyl ether) to yield a colorless oil (0.775 g, 74.0% yield), which showed 96.2% ee as determined by chiral HPLC. 1H NMR (400 MHz, CDCl3) δ 7.28–7.44 (m, 16H), 6.90–6.84 (m, 6H), 5.02–4.86 (m, 4H), 4.78 (d, J = 10.6, 1H), 4.77 (dt, J = 9.5, 9.4, 1H), 4.70 (d, J = 10.6, 1H), 4.63 (d, J = 11.6, 1H), 4.59 (d, J = 11.6, 1H), 4.54 (d, J = 11.5, 1H), 4.52 (d, J = 11.5, 1H), 4.10 (t, J = 2.5, 1H), 4.06 (td, J = 9.6, 2.01, 1H), 3.81 (s, 3H), 3.78 (s, 3H), 3.75 (s, 3H), 3.40–3.32 (m, 2H), 3.17 (dd, J = 9.6, 2.4, 1H), 2.38 (d, J = 2.0, 1H), 2.33 (s, 1H); 13C NMR (101 MHz, CDCl3) δ 159.5, 159.4, 159.1, 136.3, 136.3, 136.2, 136.2, 130.7, 129.8, 129.7, 129.7, 129.6, 129.6, 128.4, 128.4, 128.2, 128.2, 127.8, 114.0, 113.9, 113.7, 80.9 (d, JC-P = 3.6), 79.2 (d, JC-P = 7.0), 78.2, 77.8 (d, JC-P = 2.5), 73.9, 71.9, 71.8, 69.2 (d, JC-P = 3.5), 69.2 (d, JC-P = 3.7), 66.3, 55.3; 31P NMR (162 MHz, CDCl3) δ -1.50; IR (film, cm - 1) 3396, 3064, 3034, 3002, 2954, 2935, 2906, 2836, 1612, 1586, 1514, 1456, 1364, 1302, 1249, 1175, 1152, 1077, 1035, 1023; TLC Rf = 0.14 (2% ethanol in diethyl ether). Exact mass calculated for [C44H49O12P]H+ requires m/z 801.303, found 801.301; HPLC tR = 29.8 min employing a Chiralcel® OD column eluting with 35% ethanol in hexanes at a flow rate of 0.5 mL/ min.; [α]D = +12.9 (c = 1.0, CHCl3).
Supplementary Material
ACKNOWLEDGMENTS.
We are grateful to the National Institutes of Health (GM-068649) for financial support. We also thank Drs. Bianca Sculimbrene and Yingju Xu for preliminary studies.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/1001111107/DCSupplemental.
*We have presumed that Pmh-based catalysis by P(V) proceeds via a covalent catalysis mechanism.
†For an example of stereochemical and mechanistic dichotomy in superficially similar group transfer situations, see ref. 27.
‡The species that undergoes secondary kinetic resolution in situ when reactions are run to higher levels of conversion is the intermediate phosphite. The degree of secondary kinetic resolution is difficult to quantify with this intermediate due to its sensitivity to silica gel purification.
§Observations in the literature report strong pH-dependent values for optical rotations of phosphoinositides and inositol phosphates. Similarly, the adjustments of the solutions of the samples to identical pH is difficult, due in part to difficulties in achieving identical ionizations states for the samples; see (33, 34).
References
- 1.Irvine RF, Schell MJ. Back in the water: The return of the inositol phosphates. Nat Rev Mol Cell Biol. 2001;2:327–338. doi: 10.1038/35073015. [DOI] [PubMed] [Google Scholar]
- 2.Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006;443:651–657. doi: 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- 3.Onnebo SMN, Saiardi A. Inositol pyrophosphates get the Vip1 treatment. Cell. 2007;129:647–649. doi: 10.1016/j.cell.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 4.Prestwich GD. Phosphoinositide signaling: From affinity probes to pharmaceutical targetsy. Chem Biol. 2004;11:619–637. doi: 10.1016/j.chembiol.2004.03.025. [DOI] [PubMed] [Google Scholar]
- 5.Billington DC. The Inositol Phosphates: Chemical Synthesis and Biological Significance. New York: VCH; 1993. [Google Scholar]
- 6.Sculimbrene BR, Miller SJ. Discovery of a catalytic asymmetric phosphorylation through selection of a minimal kinase mimic: A concise total synthesis of D-myo- inositol-1-phosphate. J Am Chem Soc. 2001;123:10125–10126. doi: 10.1021/ja016779+. [DOI] [PubMed] [Google Scholar]
- 7.Sculimbrene BR, Morgan AJ, Miller SJ. Enantiodivergence in small-molecule catalysis of asymmetric phosphorylation: Concise total syntheses of the enantiomeric D-myo-inositol-1-phosphate and D-myo-inositol-3-phosphate. J Am Chem Soc. 2002;124:11653–11656. doi: 10.1021/ja027402m. [DOI] [PubMed] [Google Scholar]
- 8.Morgan AJ, Komiya S, Xu YJ, Miller SJ. Unified total syntheses of the inosi tol polyphosphates: D-I-3,5,6P (3), D-I-3,4,5P (3), D-I-3,4,6P (3), and D-I-3,4,5,6P (4) via catalytic enantioselective and site-selective phosphorylation. J Org Chem. 2006;71:6923–6931. doi: 10.1021/jo0610816. [DOI] [PubMed] [Google Scholar]
- 9.Sculimbrene BR, Xu YJ, Miller SJ. Asymmetric syntheses of phosphatidylinositol-3-phosphates with saturated and unsaturated side chains through catalytic asymmetric phosphorylation. J Am Chem Soc. 2004;126:13182–13183. doi: 10.1021/ja0466098. [DOI] [PubMed] [Google Scholar]
- 10.Xu YJ, Sculimbrene BR, Miller SJ. Streamlined synthesis of phosphatidylinositol (PI), PI3P, PI3,5P(2), and deoxygenated analogues as potential biological probes. J Org Chem. 2006;71:4919–4928. doi: 10.1021/jo060702s. [DOI] [PubMed] [Google Scholar]
- 11.Kayser-Bricker KJ, Jordan PA, Miller SJ. Catalyst-dependent syntheses of phosphatidylinositol-5-phosphate-DiC8 and its enantiomer. Tetrahedron. 2008;64:7015–7020. doi: 10.1016/j.tet.2008.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morgan AJ, Wang YK, Roberts MF, Miller SJ. Chemistry and biology of deoxy-myo-inositol phosphates: Stereospecificity of substrate interactions within an archaeal and a bacterial IMPase. J Am Chem Soc. 2004;126:15370–15371. doi: 10.1021/ja047360x. [DOI] [PubMed] [Google Scholar]
- 13.Wang YLK, et al. The temperature dependence of the inositol monophosphatase K-m correlates with accumulation of di-myo-inositol 1,1′ -phosphate in Archaeoglobus fulgidus. Biochemistry. 2006;45:3307–3314. doi: 10.1021/bi052467y. [DOI] [PubMed] [Google Scholar]
- 14.Wang YLK, et al. Insights into the structural specificity of the cytotoxicity of 3-deoxyphosphatidylinositols. J Am Chem Soc. 2008;130:7746–7755. doi: 10.1021/ja710348r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Longo CM, Wei Y, Roberts MF, Miller SJ. Asymmetric Synthesis of L,L- and L,D-Di-myo-inositol-1,1′-phosphate and their Behavior as Stabilizers of Enzyme Activity at Extreme Temperatures. Angew Chem, Int Ed. 2009;48:1–5. doi: 10.1002/anie.200900480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caruthers MH. Chemical synthesis of DNA and DNA analogs. Acc Chem Res. 1991;24:278–284. [Google Scholar]
- 17.Sculimbrene BR. Boston: Boston College; 2004. Catalytic asymmetric phosphorylation. PhD thesis. [Google Scholar]
- 18.Mal’tseva TV, Ivanova EM, Korobeinicheva IK. The relationsip between chemical-shift P-31 nuclear magnetic resonance, valent stretching frequency of the bond P = O of a phosphoryl fraction in diphenylphosphoazolydes and diphenylphos- phorylammonium cations and their reactivity. Izv Sib Otd Akad Nauk SSSR, Ser Khim Nauk. 1985:112–116. [Google Scholar]
- 19.Pirrung MC. Histidine kinases and two-component signal transduction systems. Chem Biol. 1999;6:R167–R175. doi: 10.1016/S1074-5521(99)80044-1. [DOI] [PubMed] [Google Scholar]
- 20.Beaucage SL, Caruthers MH. Deoxynucleoside phosphoramidites—A new class of key intermediates for deoxypolynucleotide synthesis. Tetrahedron Lett. 1981;22:1859–1862. [Google Scholar]
- 21.Vlasuk GP, Webb TR, Abelman MM, Pearson DA, Miller TA. 5492895. US Patent. 1999 Sep 21;
- 22.Stec WJ, Zon G. Stereochemical studies of the formation of chrial internucleotide linkages by phosphoramidite coupling in the synthesis of oligodeoxyribonucleotides. Tetrahedron Lett. 1984;25:5279–5282. [Google Scholar]
- 23.Dahl BH, Nielsen J, Dahl O. Mechanistic studies on the phosphoramidite coupling reaction in oligonucleotide synthesis. I. Evidence for nucleophilic catalysis by tetrazole and rate variations with the phosphorous substituents. Nucleic Acids Res. 1987;15:1729–1743. doi: 10.1093/nar/15.4.1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dahl BH, Nielsen J, Dahl O. Substituent influence on the reaction-rate of deoxyribonucleoside phosphoramidites with deoxyribonucleosides catalyzed by tetrazole. Nucleosides Nucleotides. 1987;6:457–460. [Google Scholar]
- 25.Hayakawa Y, Kataoka M. Preparation of short oligonucleotides via the phosphoramidite method using a tetrazole promoter in a catalytic manner. J Am Chem Soc. 1997;119:11758–11762. [Google Scholar]
- 26.Brady PB, Morris EM, Fenton OS, Sculimbrene BR. Efficient catalyst turnover in the phosphitylation of alcohols with phosphoramidites. Tetrahedron Lett. 2009;50:975–978. [Google Scholar]
- 27.Fiori KW, Puchlopek ALA, Miller SJ. Enantioselective sulfonylation reactions mediated by a tetrapeptide catalyst. Nat Chem. 2009;1:630–634. doi: 10.1038/nchem.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schreiber SL, Schreiber TS, Smith DB. Reactions that proceed with a combination of enantiotopic group and diastereotopic face selectivity can deliver products with every high enantiomeric excess: Experimental support of a mathematical model. J Am Chem Soc. 1987;109:1525–1529. [Google Scholar]
- 29.Desiraju GR, Steiner T. 1st Ed. Oxford: Oxford Univ Press; 1999. The weak hydrogen bond. [Google Scholar]
- 30.Iyer RP, Egan W, Regan JB, Beaucage SL. 3H-1,2-Benzodithiole-3-one 1,1- Dioxide as an improved sulfurizing reagent in the solid-phase synthesis of oligodeoxyri- bonucleoside phosphorothioates. J Am Chem Soc. 1990;112:1253–1254. [Google Scholar]
- 31.Sureshan KM, Watanabe Y. An efficient route to optically active inositol derivatives via the resolution of myo-inositol 1,3,5-orthoformate: A short synthesis of D-myo-inositol-4-phosphate. Tetrahedron: Asymmetry. 2004;15:1193–1198. [Google Scholar]
- 32.Vacca JP, et al. The total synthesis of myo-inositol polyphosphates. Tetrahedron. 1989;45:5679–5702. [Google Scholar]; Vacca JP, et al. Corrigendum. Tetrahedron. 1991;47:907. [Google Scholar]
- 33.Ozaki S, Kondo Y, Shiotani N, Ogasawara T, Watanabe Y. Synthesis and some properties of D-myo-inositol 1,4,5-tris(dihydrogen phosphate) J Chem Soc, Perkin Trans. 1992;1:729–737. [Google Scholar]
- 34.Mayr GW, Dietrich W. The only inositol tetrakisphosphate detectable in avian erythrocytes is the isomer lacking phosphate at position-2: A NMR study. FEBS Lett. 1987;213:278–282. doi: 10.1016/0014-5793(87)81505-3. [DOI] [PubMed] [Google Scholar]
- 35.Lewis CA, Miller SJ. Site-selective derivatization and remodelling of erythromycin A by using simple peptide-based chiral catalysts. Angew Chem, Int Ed. 2006;45:5616–5619. doi: 10.1002/anie.200601490. [DOI] [PubMed] [Google Scholar]
- 36.Lewis CA, Longcore KE, Miller SJ, Wender PA. An approach to the site- selective diversification of apoptolidin A with peptide-based catalysts. J Nat Prod. 2009;72:1864–1869. doi: 10.1021/np9004932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Armarego WLF, Perrin DD. Purification of Laboratory Chemicals. 4th Ed. Amsterdam: Elsevier; 1997. p. 143. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.