

Abstract
Heterocyclic aromatic amines produce bulky C8 guanine lesions in vivo, which interfere and disrupt DNA and RNA synthesis. These lesions are consequently strong replication blocks. In addition bulky adducts give rise to point and frameshift mutations. The translesion synthesis (TLS) DNA polymerase η is able to bypass slowly C8 bulky adduct lesions such as the widely studied 2-aminofluorene-dG and its acetylated analogue mainly in an error-free manner. Replicative polymerases are in contrast fully blocked by the acetylated lesion. Here, we show that TLS efficiency of Pol η depends critically on the size of the bulky adduct forming the lesion. Based on the crystal structure, we show why the bypass reaction is so difficult and we provide a model for the bypass reaction. In our model, TLS is accomplished without rotation of the lesion into the anti conformation as previously thought.
Keywords: DNA damage, DNA polymerase, translesion DNA synthesis, aromatic amines
Aromatic amines are known to be strong carcinogens. After metabolic activation, aromatic amines react as electrophilic arylnitrenium ions with nucleophilic functionalities of the DNA duplex. Preferred reaction sides are the amino groups of adenine and guanine and particularly the C8-position of guanine (1, 2). The latter reaction gives rise to the so called C8-bulky adduct lesions, which interfere with the replication process leading to mutations (2). Two types of aromatic amine lesions are known. The nonacetylated lesions depicted in Fig. 1A reduce the replication efficiency but are in general faithfully bypassed by high-fidelity polymerases (3). In contrast, the acetylated derivatives (Fig. 1B) block replicative polymerases (3, 4), but can be bypassed with special low-fidelity polymerases (5–9). Moreover, if these acetylated lesions are present in certain repetitive sequences, they are known to induce frameshift mutations (4, 10–12). Recently, in vivo experiments revealed that the low-fidelity Y-family polymerase η is able to replicate DNA containing acetylated aromatic amine lesions such as the acetylated analogue of 2-aminofluorene-dG (AF-dG), 2-acetylaminofluorene-dG (AAF-dG) (13). Replication through the lesion is even for Pol η very difficult and hence slow, but if it occurs it is typically error free.
Fig. 1.

(A) Syn and anti conformation of the nonacetylated C8-dG adducts. (B) The acetylated C8-adducts are restricted to syn-conformation. (C) Synthesized C8-dG adducts: AAB-dG = N-(2′-deoxyguanosin-8-yl)-acetylaminobenzene, AAN = N-(2′-deoxyguanosin-8-yl)-2-acetylaminonaphthalene, AAF-dG: N-(2′-deoxyguanosin-8-yl)-2-acetylaminofluorene, AAA-dG = N-(2′-deoxyguanosin-8-yl)-2-acetylaminoanthracene, AAP-dG: N-(2′-deoxyguanosin-8-yl)-1-acetylaminopyrene.
The distinct mutagenic properties of the acetylated and nonacetylated aromatic amine lesions are caused by their different conformational preferences. While the nonacetylated lesions exist both in syn and anti conformation (14, 15), the corresponding acetylated lesions seem to adopt the syn-conformation with high preference (10, 16, 17). Crystal structures of Ellenberger, Beese, and Patel, showing the nonacetylated AF-dG-lesion inside different polymerases, prove that the lesion is indeed bound in anti conformation, allowing Watson-Crick base pairing with an incoming dCTP (18–20). While bypass of the nonacetylated lesions by high-fidelity polymerases is rather well understood, the mechanism that allows low-fidelity polymerases such as Pol η to replicate through acetylated AAF-dG lesions is still unknown. Computational modeling studies of the Y-family polymerase Dpo4 suggested that the bypass reaction requires rotation of the lesion into the base pairing anti conformation (21). In light of the strong syn preference of the adducted base, this rotation is difficult to envision. Here we report a crystal structure of yeast Pol η in complex with DNA containing the two bulky adducts AAF-dG and 2-acetylaminoanthracene-dG (AAA-dG). The structure shows why Pol η is blocked if it encounters the lesion but they also allow us to construct a model that explains how Pol η finally achieves the bypass. We observe crystallographically that the primer strand can rotate around the bulky moiety, while the AAF-dG is kept in syn conformation. In this situation correct dCTP incorporation opposite the lesion can be envisioned by formation of one hydrogen bond between the dCTP and the lesion. Our crystallographic results were obtained with the wild-type Pol η enzyme which crystallizes with an open polymerase associated domain (PAD) domain. Crystal structures of mutant forms of Pol η in complex with cyclobutane pyrimidine dimer (CPD) lesions show that the PAD domain can move towards the DNA duplex to strengthen the interactions between the duplex and the protein (22). However, the additional protein-DNA interactions do not change the situation in the active site. In fact the rmsd between both protein structures is only 0.6 Å in the vicinity of the lesion, using the main chain atoms of residues 1–220 as a reference frame. This deviation is within the error margin of the structures. Even the side chain of Arg73, which was observed to reorient in the new structure does so only partially. Half of the Arg73 side chains occupy a position indistinguishable from the old structure.
Results
In order to investigate the mechanism of how bulky adduct lesions block Pol η and how finally bypass of acetylated bulky DNA adducts is achieved, we synthesized DNA strands, containing both the AAF-dG and AAA-dG lesions at a predefined site (23, 24). Both lesions were incorporated into 20mer oligonucleotides via automated DNA synthesis (25). The oligonucleotides were finally purified by HPLC to a purity of > 99%. The so obtained DNA strands were used in primer extension studies with yeast Pol η at concentrations ranging from 100–300 nM and in the presence of one or all four deoxynucleoside triphosphates (Fig. 2). The enzyme fully extends undamaged template DNA (Fig. 2A, lane 6). In the presence of AAF-dG and AAA-dG, bypass is indeed possible albeit with strongly reduced efficiency (Fig. 2A, lanes 3, 4, and Fig. S1). In both cases fully extended primer strands are however clearly observed, showing that Pol η is able to read through both lesions. The experiment depicted in Fig. 2A and Fig. S1 shows that bypass is more difficult when the size of the aromatic adduct is increased and that bypass is finally impossible if a large pyrene unit forms the adduct. To determine the base pairing partner of the adducted guanine during bypass, the primer extension experiments were repeated adding 2′,3′-dideoxynucleotides individually to the reaction mixture. As depicted in Fig. 2B, Pol η incorporates, correctly and with high specificity, a dC opposite the AAF-dG and AAA-dG lesions despite the presence of the aromatic unit attached to C8 (Fig. 2B and Fig. S2). Again, in a control experiment with all dNTPs, slow full extension was observed (Fig. 2B, lane 10).
Fig. 2.

Translesion synthesis of different bulky dG DNA adducts catalyzed by Pol η. The primer extension reactions were carried out with fluorescence labeled primers and extension products resolved on a denaturing PAA-gel. (A) Bypass of various bulky adducts in the same sequence context. Reaction conditions are: 300 nM Pol η, 1 μM template DNA, 200 μM dNTPs each, 1 = AAB-dG, 2 = AAN-dG, 3 = AAF-dG, 4 = AAA-dG, 5 = AAP-dG, 6 = undamaged template strand, and m = marker. (B) Incorporation of individual nucleotides with AAF-dG containing oligonucleotides and lesion-free oligonucleotides (200 nM Pol η, 1 μM template DNA, 200 μM of each dideoxynucleotide or all four dNTPs; 1, 6 = ddATP, 2, 7 = ddTTP, 3, 8 = ddGTP, 4, 9 = ddCTP, and 5, 10 = all four dNTPs). (C) Left: Primer extension reaction with 2'-deoxyzebularine-5'-triphosphate (dZTP) and AAF-dG containing oligonucleotides (100 nM Pol η, 1 μM template DNA, 200 μM of each deoxynucleotide of all four dNTPs and dZTP; 1 = dATP, 2 = dTTP, 3 = dGTP, 4 = dCTP, and 5 = dZTP); Right: Primer extension reaction with undamaged template DNA, 6 = dCTP, 7 = dZTP (200 nM Pol η)
To elucidate the mechanism of the observed faithful replication across the AAF/AAA-dG adducts by Pol η, we crystallized primer-template constructs of AAF-dG and AAA-dG with Pol η from Saccharomyces cerevisiae. Nucleotide insertion through the enzyme was prevented by using a primer strand ending with a dideoxy nucleotide which pairs with the nucleobase located directly 3′ of the lesion. Crystals of both complexes diffracted X-rays to about 2.7 Å spacing. The structures were solved by molecular replacement (for details on data collection and structure refinement see SI Text and Table S1). As previously observed for the enzyme, there are two molecules in the asymmetric unit cell of both crystals (26–28), showing the enzyme-DNA complexes in two different states (A and B, Fig. 3 C and D). In the crystal structure, the AAF and AAA moieties of the AAF/AAA-dG lesions are well defined in both complexes in the electron density. In both structures, the aromatic C8-adduct of the fluorene as well as the anthracene are found at structural equivalent positions in respect to the active site of the protein (Fig. 3, Fig. 4, and Figs. S2 and S3). In the two complexes A, the aromatic amine units stack on top of the Watson-Crick base pair formed by the primer ddC and the -1 dG. For both lesions, the bulky aromatic units therefore fully block the active site for the incoming dCTP (complex A) (Fig. 3C). In complex B the situation is surprisingly different (Fig. 3D). First of all, a relatively small movement of the DNA template strand compared to complex A is observed. However, the primer DNA strand is rotated in complex B relative to the aromatic moiety by about 6 Å. Due to this strand rotation the location of the primer end is strongly different in both complexes A and B. This primer rotation brings the dG part of both lesions despite their prevalent syn-conformation closer into the active site. In consequence of the primer motion, the dG-lesion packs above the Watson-Crick base pair formed by the primer end and the template strand. The primer strand displacement results in a partial opening of the active site for the putative dCTP to bind (Fig. 3D and Fig. S4). Nevertheless, one can envision that, based on the observed primer movement, a rather small further movement of the DNA along the trajectory complex A → complex B results in a situation in which the triphosphate can finally bind and nucleotide incorporation can occur (Fig. 3E).
Fig. 3.

Crystal structure of S. cerevisiae DNA Pol η in complex with the AAF-dG. (A) Overall folding topology with the enzyme as cartoon representation and the DNA as stick model (gold), with the AAF-dG adduct in orange. The different domains typical for Y-family DNA polymerases are highlighted by different colors. (B) Difference-omit electron density map (Fobs-DFcalc) of the AAF-dG-lesion with the surrounding protein residues. (C–E) Model for AAF-dG bypass by Pol η. (C) In complex A, the fluorene moiety of the AAF-dG adduct stacks on the previous Watson-Crick base pair, blocking the active site for the incoming nucleotide. (D) In contrast, the DNA in complex B is rotated in respect to the aromatic unit, with the adducted dG moved towards templating position, partially freeing the active site. (E) Hypothetical model of the AAF bypass by Pol η. The dG : dC basepair 3′ of the lesion in the AAF-dG-Pol η complex B was replaced by the AAF-dG adduct and the dCTP triphosphate. The dCTP was placed in the active site according to the structure of Pol η in complex with the cisplatin GG lesion (PDB code 2R8J). Thus the active site for the incoming dCTP opens, which forms one critical hydrogen bond to the adducted nucleobase in syn conformation.
Fig. 4.

Schematic representation of Pol η binding AAF-dG and AAA-dG containing DNA. In the complexes A, the aromatic moieties of AAF-dG (A) and AAA-dG (B) stack on top of the previous G-C base pair, adopting structural equivalent positions. The protein is shown as semitransparent surface representation (gray) with the DNA as stick model (gold) and the lesion highlighted in orange. Active site residues are also depicted as stick model (blue). The catalytic calcium ions in the active site are displayed as red spheres. For complexes B and examples of the electron densities, see Fig. S3 in the SI Text online.
In order to visualize how Pol η copies through the AAF/AAA-dG lesions, we modeled the complex with the bound dCTP (Fig. 3E). Therefore, the primer was shortened by one base and the dC-triphosphate was inserted. In all so far obtained crystal structures of Pol η with a bound triphosphate, the position of the triphosphate is invariant (Fig. S5) (22, 26–28). Indeed, the triphosphate fits perfectly into the active site when the primer is shortened. We then moved the AAF-dG-lesion into the -I position. In this way, we simulate Pol η moving one base pair forward (Fig. 3E). After these manipulations, the mechanism of how the difficult bypass is achieved can be envisioned. In our model, the extension of the template rotation as described above is sufficient to fully open the active site for the incoming dCTP. The dCTP placed in the active is in this model able to form one hydrogen bond with the dG of the lesion. Thus, the crystal structure (Fig. 3 C and D) in combination with the model (Fig. 3E) allows us to decipher the basis for faithful bypass of acetylated bulky adduct lesions. Here, the acetylated bulky adduct lesions do not have to rotate into the anti-conformation to enable Watson-Crick base pairing. Instead, the DNA primer strand rotates, which allows Pol η to decode the bulky dG adduct with dCTP. One critical hydrogen bond is established between the N4 of the triphosphate and the O6 of the dG part of the lesion (Fig. 3E). A similar single H-bond based decoding was recently also observed to allow Pol η to bypass the cisplatin GG lesion (26).
In the bypass hypothesis, Pol η keeps the bulky adduct of the lesion at the same position, but rotates the primer strand around the lesion in order to bring the dG of the lesion into a templating position. The major energetic penalty of this rotational process is the required destacking of the aromatic bulky adduct unit and the last formed Watson-Crick base pair between the primer and the template strand. In complex A, the fluorene and anthracene units stack almost perfectly on top of the base pair (Fig. 4). This stacking interaction is fully sacrificed in complex B. We reasoned that this situation would allow us to obtain biochemical support for our mechanistic proposal by measuring the translesion synthesis process depending on the size of the aromatic unit, which determines the amount of the stacking interaction. The idea behind the experiment is that the required destacking forces should increase with increasing size of the flat aromatic adduct, thus slowing down the bypass process. For the experiment, we prepared three additional bulky adduct lesions with either a small benzene, a larger naphthyl, or an even larger pyrene unit. The primer extension results are depicted in Fig. 2A (lanes 1–5). We indeed observe that the TLS efficiency is strongly reduced if the size of the aromatic adduct is increased. Whereas bypass is surprisingly efficient for the small benzene unit, bypass is impossible when the benzene was replaced by a large pyrene unit.
To further support the proposed TLS mechanism, we performed additional experiments with zebularine triphosphate (dZTP). Because the bypass process was found to rely on the formation of one hydrogen bond between the incoming dCTP and the dG part of the lesion, lesion bypass should be impossible with dZTP where this H-bond cannot form. In agreement with the postulated mechanism, dZTP indeed cannot be incorporated by Pol η opposite the lesion (Fig. 2C, lane 5). In a control experiment with undamaged template DNA, dZTP incorporation opposite dG is observed, albeit with reduced efficiency compared to a dCTP (Fig. 2C, lane 7).
Discussion
Aromatic amines have a high mutagenic and strong carcinogenic potential due to the formation of DNA adducts after their metabolic activation. The compounds react particularly with the C8-position of dG bases and result in C8-dG bulky adducts (2). Two different bulky adduct types (acetylated vs. nonacetylated) with distinct mutagenic properties are known. In this study, lesions derived from aminofluorene and aminoanthracene served as model compounds to investigate the mechanism behind the error-free bypass.
The nonacetylated AF-dG-lesion strongly reduces the efficiency of replicative bypass, but read through by high-fidelity polymerases is accomplished because these lesions can rotate around the glycosidic bond into the anti-conformation (16, 18, 19). Thus, the dG-unit can correctly pair with an incoming dCTP in the active site by forming a Watson-Crick base pair (14, 18, 19). The acetylated bulky adducts in contrast, are not bypassed by high-fidelity polymerases because these lesions are fixed in the syn-conformation (10, 16, 17, 29–32), independent of the size of the aromatic unit attached to the C8 portion of the dG base (29). Faithful bypass of the acetylated lesions in vivo is achieved by low-fidelity TLS polymerases, with Pol η playing a major role (5–7). We obtained two crystal structures of DNA containing the two different acetylated bulky adduct lesions AAF-dG and AAA-dG in complex with Pol η. Both structures show the primer-template constructs in two different positions (complex A, Fig. 3C and B, Fig. 3D). Complex A features the bulky adduct in the active site, blocking the dCTP from entering. In this complex the bulky adduct fully stacks on top of the last primer-template base pair. Complex B in contrast shows the bulky adduct in the process of being bypassed. Here, the enzyme has rotated the DNA primer strand, while keeping the aromatic C8-adduct at a structural equivalent position. This rotational motion partly unblocks the active site but a dCTP can still not enter, which explains why bypass of these lesions is so difficult even for Pol η. A modeling study, in which further movement along this trajectory was performed, shows how the active site might open to enable the nucleotide triphosphate binding in the active site (Fig. 3E). The model shows that a bound dCTP might be able to form one H-bond between the C4-NH2 group of the dCTP and the C6═O group of the AAF/AAA-dG. The starting point of our model is the fact that the triphosphate occupies in all known structures exactly the same position, which allows us to simulate the dCTP bound structure when the primer and the template have move forward (Fig. S5). The formed critical H-bond could provide the basis for Pol η to correctly insert a dC opposite the lesion. Purine triphosphates can not bind due to their larger size. The alternative pyrimidine base dTTP would fit sterically and can indeed be modeled into the active site, but dTTP would be unable to form the critical H-bond. The importance of this hydrogen bond was proven with the artificial triphosphate dZTP lacking the C4-amino group. The dZTP triphosphate is not inserted opposite the lesion, therefore supporting the proposed bypass model (Fig. 2C). Further support comes from a chemical biology study, in which we systematically changed the size of the aromatic adduct, thereby modulating the stacking interactions between the lesion and the last primer-template base pair. Here we observe that smaller sized adducts which establish smaller π-stacking forces are more efficiently bypassed (Fig. S5), even though the rotation barrier around the glycosidic bond is unchanged (29). Bypass of the large pyrene unit is in consequence so strongly blocked because of the large size of the stacking interaction (33, 34).
Two recent crystal structures of Pol η reading through a CPD lesion have shown that if particular crystal contacts are removed by site directed mutagenesis, a further closing of the PAD domain occurs. Closing of the PAD domain, however does not change the situation in the active site, particularly not of the base paring motifs (22, 35), proving that our structures provide a correct insight into the TLS process.
In summary, we conclude that efficient destacking of the aromatic unit on top of the primer-template base pair is a key prerequisite for the efficient bypass reaction by the low-fidelity polymerase Pol η.
Materials and Methods
DNA Preparation.
The AAF-dG phosphoramidite containing an isopropylphenoxyacetyl group at the N2 position for solid phase DNA synthesis was prepared as previously published (23, 24) and incorporated into DNA using ultra mild conditions for DNA synthesis (24). The AAB-dG, AAN-dG, AAA-dG and AAP-dG phosphoramidites were synthesized analogous to Rizzo and coworkers and Gillet, and Schärer and coworkers (23–25) and incorporated into DNA using ultra mild conditions. The oligonucleotides were purified via HPLC, desalted, and characterized by MALDI-TOF mass spectrometry and enzymatic digestion analysis.
Cloning, Protein Expression, and Purification of Pol η.
S. cerevisiae DNA polymerase η was cloned, expressed and purified as previously described (26, 28). In brief, the sequence coding for residues 1–513 of the enzyme was cloned into pDEST007, expressed in E. coli (Rosetta, Novagen) and purified as an amino-terminally tagged Strep-Tag II fusion protein using a Streptactin affinity column (IBA Biotechnology). The protein was further purified using a heparin affinity column. After purification, the enzyme was stored in 20 mM Tris-HCl pH 7.6, 50 mM KCl, 10 mM β-mercaptoethanol, 10% glycerol at 80 °C.
In Vitro Bypass of the Bulky DNA Lesions.
A fluorescent labeled 13mer primer (Metabion) was annealed (95 °C for 4 min followed by cooling down to 4 °C over a period of 45 min) to the unlabeled 20mer template DNA strand, either undamaged or containing the bulky DNA lesion in a 1∶1.5 molar ratio in the reaction buffer [10 mM Tris-HCl, 50 mM NaCl, 10 mM MgCl2, 1 mM dithiothreitol (DTT), pH 7.9]. The final concentration of the primer in the primer extension studies was 1 μM. Primer extensions studies using Pol η were initiated by addition of the nucleotide (final concentration 200 μM, dZTP was purchased at TriLink Biotechnologies) and incubated at 30 °C for 5 min. Reactions were terminated by addition of a solution sodium ethylenediaminetetraacetic (EDTA) (100 mM, 1∶1 volume ratio). The primer extension products were resolved on 22% polyacrylamide-urea gels and visualized using a LAS-3000 imaging system.
Cocrystallization.
For cocrystallization, Pol η (4.5 mg/mL) was incubated with double stranded DNA containing the template with the AAF-dG or the AAA-dG-lesion annealed to primer with a 2′,3′-dideoxybase at the 3′ end (5′-GTG GAT GAGdd) in a 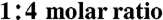 . Crystals were grown by mixing an equal volume of protein-DNA complex with 11%–15% (w/v) PEG 3350, 150–200 mM CaCl2, 100 mM dCTP using the hanging-drop vapor diffusion method. The crystallization plates were incubated at 4 or 18 °C and crystals appeared after 10 to 60 d. Crystals were frozen in artificial mother liquor containing 20% (v/v) butanediol and stored in liquid nitrogen until data collection.
. Crystals were grown by mixing an equal volume of protein-DNA complex with 11%–15% (w/v) PEG 3350, 150–200 mM CaCl2, 100 mM dCTP using the hanging-drop vapor diffusion method. The crystallization plates were incubated at 4 or 18 °C and crystals appeared after 10 to 60 d. Crystals were frozen in artificial mother liquor containing 20% (v/v) butanediol and stored in liquid nitrogen until data collection.
Collection and Processing of X-ray Diffraction Data, Phase Determination, and Structure Refinement.
Diffraction data were measured at the PXI beam line at the Swiss Light Source (SLS), Villigen, Switzerland. The crystals diffracted X-rays to 2.7 Å spacing. The data were processed with the programs XDS and XSCALE (36) and the crystals belong to the same space group, with comparable unit cell constants as the apo-enzyme (27) and cocrystals containing the cisplatin lesion (26, 28). The structures were solved by molecular replacement [PHASER (37)] using the coordinates of the apo-enzyme as search model. In order to reduce model bias, the temperature factors were reset to the Wilson B-factor. Prior to model building in COOT (38) a simulated annealing omit map, removing the area around the lesion, was calculated with PHENIX (39). Restrained refinement was carried out with PHENIX and REFMAC (40), excluding the same set of free reflections as for the previously reported Pol η -DNA complex structures (26, 28). Data processing and refinement statistics are summarized in Table S1. Structural figures were prepared with PyMol (Delano Scientific). The atomic coordinates and structure factors (PDB codes 2XGP and 2XGQ) have been deposited in the PDB at the EBI Macromolecular Structure Database (http://www.ebi.ac.uk/pdbe).
Supplementary Material
Acknowledgments.
We thank the Sonderforschungsbereich 646, 749 and the excellence cluster CIPSM for generous support. S. Schorr thanks the Verband der chemischen Industrie (VCI) for a Kekulé Fellowship. We also thank the beamline scientists at the SLS for their support in setting up the beamline for data collection.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank at the EBI Macromolecular Structure Database (http://www.ebi.ac.uk/pdbe) (PDB ID codes 2XGQ and 2XGP).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1008894107/-/DCSupplemental.
References
- 1.Heflich RH, Neft RE. Genetic toxicity of 2-acetylaminofluorene, 2-aminofluorene and some of their metabolites and model metabolites. Mutat Res. 1994;318:73–114. doi: 10.1016/0165-1110(94)90025-6. [DOI] [PubMed] [Google Scholar]
- 2.Hoffmann GR, Fuchs RP. Mechanisms of frameshift mutations: insight from aromatic amines. Chem Res Toxicol. 1997;10:347–359. doi: 10.1021/tx960128n. [DOI] [PubMed] [Google Scholar]
- 3.Belguise-Valladier P, Fuchs RP. N-2-aminofluorene and N-2-acetylaminofluorene adducts: the local sequence context of an adduct and its chemical structure determine its replication properties. J Mol Biol. 1995;249:903–913. doi: 10.1006/jmbi.1995.0347. [DOI] [PubMed] [Google Scholar]
- 4.Ames BN, Gurney EG, Miller JA, Bartsch H. Carcinogens as frameshift mutagens: metabolites and derivatives of 2-acetylaminofluorene and other aromatic amine carcinogens. Proc Natl Acad Sci USA. 1972;69:3128–3132. doi: 10.1073/pnas.69.11.3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Masutani C, Kusumoto R, Iwai S, Hanaoka F. Mechanisms of accurate translesion synthesis by human DNA polymerase η. Embo J. 2000;19:3100–3109. doi: 10.1093/emboj/19.12.3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Livneh Z. DNA damage control by novel DNA polymerases: translesion replication and mutagenesis. J Biol Chem. 2001;276:25639–25642. doi: 10.1074/jbc.R100019200. [DOI] [PubMed] [Google Scholar]
- 7.Vooradi V, Romano LJ. Effect of N-2-acetylaminofluorene and 2-aminofluorene adducts on DNA binding and synthesis by yeast DNA polymerase η. Biochemistry. 2009;48:4209–4216. doi: 10.1021/bi9000722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matsuda T, Bebenek K, Masutani C, Hanaoka F, Kunkel TA. Low fidelity DNA synthesis by human DNA polymerase η. Nature. 2000;404:1011–1013. doi: 10.1038/35010014. [DOI] [PubMed] [Google Scholar]
- 9.Rothwell PJ, Waksman G. Structure and mechanism of DNA polymerases. Adv Protein Chem. 2005;71:401–440. doi: 10.1016/S0065-3233(04)71011-6. [DOI] [PubMed] [Google Scholar]
- 10.Fuchs RP, Schwartz N, Daune MP. Hot spots of frameshift mutations induced by the ultimate carcinogen N-acetoxy-N-2-acetylaminofluorene. Nature. 1981;294:657–659. doi: 10.1038/294657a0. [DOI] [PubMed] [Google Scholar]
- 11.Fuchs RP, et al. DNA polymerases II and V mediate respectively mutagenic (-2 frameshift) and error-free bypass of a single N-2-acetylaminofluorene adduct. Biochem Soc Trans. 2001;29:191–195. doi: 10.1042/0300-5127:0290191. [DOI] [PubMed] [Google Scholar]
- 12.Shibutani S, Suzuki N, Grollman AP. Mechanism of frameshift (deletion) generated by acetylaminofluorene-derived DNA adducts in vitro. Biochemistry. 2004;43:15929–15935. doi: 10.1021/bi048087e. [DOI] [PubMed] [Google Scholar]
- 13.Bresson A, Fuchs RP. Lesion bypass in yeast cells: Pol η participates in a multi-DNA polymerase process. Embo J. 2002;21:3881–3887. doi: 10.1093/emboj/cdf363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Norman D, et al. NMR and computational characterization of the N-(deoxyguanosin-8-yl)-aminofluorene adduct [(AF)G] opposite adenosine in DNA: (AF)G[syn].A[anti] pair formation and its pH dependence. Biochemistry. 1989;28:7462–7476. doi: 10.1021/bi00444a046. [DOI] [PubMed] [Google Scholar]
- 15.Eckel LM, Krugh TR. 2-Aminofluorene modified DNA duplex exists in two interchangeable conformations. Nat Struct Biol. 1994;1:89–94. doi: 10.1038/nsb0294-89. [DOI] [PubMed] [Google Scholar]
- 16.Fuchs R, Daune M. Physical studies on deoxyribonucleic acid after covalent binding of a carcinogen. Biochemistry. 1972;11:2659–2666. doi: 10.1021/bi00764a017. [DOI] [PubMed] [Google Scholar]
- 17.O’Handley SF, et al. Structural characterization of an N-acetyl-2-aminofluorene (AAF) modified DNA oligomer by NMR, energy minimization, and molecular dynamics. Biochemistry. 1993;32:2481–2497. doi: 10.1021/bi00061a005. [DOI] [PubMed] [Google Scholar]
- 18.Dutta S, et al. Crystal structures of 2-acetylaminofluorene and 2-aminofluorene in complex with T7 DNA polymerase reveal mechanisms of mutagenesis. Proc Natl Acad Sci USA. 2004;101:16186–16191. doi: 10.1073/pnas.0406516101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hsu GW, et al. Observing translesion synthesis of an aromatic amine DNA adduct by a high-fidelity DNA polymerase. J Biol Chem. 2004;279:50280–50285. doi: 10.1074/jbc.M409224200. [DOI] [PubMed] [Google Scholar]
- 20.Rechkoblit O, et al. Mechanism of error-free and semitargeted mutagenic bypass of an aromatic amine lesion by Y-family polymerase Dpo4. Nat Struct Mol Biol. 2010;17:379–388. doi: 10.1038/nsmb.1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang L, Broyde S. A new anti conformation for N-(deoxyguanosin-8-yl)-2-acetylaminofluorene (AAF-dG) allows Watson-Crick pairing in the Sulfolobus solfataricus P2 DNA polymerase IV (Dpo4) Nucleic Acids Res. 2006;34:785–795. doi: 10.1093/nar/gkj479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Silverstein TD, et al. Structural basis for the suppression of skin cancers by DNA polymerase η. Nature. 2010;465:1039–1043. doi: 10.1038/nature09104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Z, Rizzo CJ. Synthesis of the C8-deoxyguanosine adduct of the food mutagen IQ. Org Lett. 2001;3:565–568. doi: 10.1021/ol006968h. [DOI] [PubMed] [Google Scholar]
- 24.Gillet LC, Schärer OD. Preparation of C8-amine and acetylamine adducts of 2′-deoxyguanosine suitably protected for DNA synthesis. Org Lett. 2002;4:4205–4208. doi: 10.1021/ol026474f. [DOI] [PubMed] [Google Scholar]
- 25.Gillet LC, Alzeer J, Schärer OD. Site-specific incorporation of N-(deoxyguanosin-8-yl)-2-acetylaminofluorene (dG-AAF) into oligonucleotides using modified “ultra-mild” DNA synthesis. Nucleic Acids Res. 2005;33:1961–1969. doi: 10.1093/nar/gki335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alt A, et al. Bypass of DNA lesions generated during anticancer treatment with cisplatin by DNA polymerase η. Science. 2007;318:967–970. doi: 10.1126/science.1148242. [DOI] [PubMed] [Google Scholar]
- 27.Trincao J, et al. Structure of the catalytic core of S. cerevisiae DNA polymerase η: implications for translesion DNA synthesis. Mol Cell. 2001;8:417–426. doi: 10.1016/s1097-2765(01)00306-9. [DOI] [PubMed] [Google Scholar]
- 28.Reissner T, Schneider S, Schorr S, Carell T. Crystal structure of a Cisplatin-(1,3-GTG) cross-link within DNA polymerase η. Angew Chem Int Ed. 2010;49:3077–3080. doi: 10.1002/anie.201000414. [DOI] [PubMed] [Google Scholar]
- 29.Florian J, Borden J. Structure and torsional flexibility of the linkage between guanine and fluorene residues in the deoxguanosine-aminofluorene and deoxaguanosine-acetylaminofluorene carcinogenic adducts. Theor Chem Acc. 2005;113:28–34. [Google Scholar]
- 30.Evans FE, Levine RA. NMR study of stacking interactions and conformational adjustments in the dinucleotide-carcinogen adduct 2′-deoxycytidylyl-(3----5)-2′-deoxy-8-(N-fluoren-2-ylacetamido)guanosine. Biochemistry. 1988;27:3046–3055. doi: 10.1021/bi00408a057. [DOI] [PubMed] [Google Scholar]
- 31.Roy D, Hingerty BE, Shapiro R, Broyde S. A slipped replication intermediate model is stabilized by the syn orientation of N-2-aminofluorene- and N-2-(acetyl)aminofluorene-modified guanine at a mutational hotspot. Chem Res Toxicol. 1998;11:1301–1311. doi: 10.1021/tx980106w. [DOI] [PubMed] [Google Scholar]
- 32.Cho BP, Zhou L. Probing the conformational heterogeneity of the acetylaminofluorene-modified 2′-deoxyguanosine and DNA by 19F NMR spectroscopy. Biochemistry. 1999;38:7572–7583. doi: 10.1021/bi990182d. [DOI] [PubMed] [Google Scholar]
- 33.Meyer EA, Castellano RK, Diederich F. Interactions with aromatic rings in chemical and biological recognition. Angew Chem Int Ed. 2003;42:1210–1250. doi: 10.1002/anie.200390319. [DOI] [PubMed] [Google Scholar]
- 34.Choi JY, Guengerich FP. Adduct size limits efficient and error-free bypass across bulky N2-guanine DNA lesions by human DNA polymerase η. J Mol Biol. 2005;352:72–90. doi: 10.1016/j.jmb.2005.06.079. [DOI] [PubMed] [Google Scholar]
- 35.Biertümpfel C, et al. Structure and mechanism of human DNA polymerase η. Nature. 2010;465:1044–1048. doi: 10.1038/nature09196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Crystallogr. 1993;26:795–800. [Google Scholar]
- 37.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr. 2005;61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 38.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of COOT. Acta Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adams PD, et al. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 40.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.