

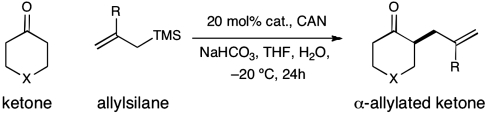
Abstract
The first enantioselective organocatalytic α-allylation of cyclic ketones has been accomplished via singly occupied molecular orbital catalysis. Geometrically constrained radical cations, forged from the one-electron oxidation of transiently generated enamines, readily undergo allylic alkylation with a variety of commercially available allyl silanes. A reasonable latitude in both the ketone and allyl silane components is readily accommodated in this new transformation. Moreover, three new oxidatively stable imidazolidinone catalysts have been developed that allow cyclic ketones to successfully participate in this transformation. The new catalyst platform has also been exploited in the first catalytic enantioselective α-enolation and α-carbooxidation of ketones.
Keywords: asymmetric synthesis, organocatalysis
The enantioselective catalytic α-alkylation of simple ketones remains a fundamental goal in chemical synthesis (1–4). Seminal work from Doyle and Jacobsen (5), Trost and co-workers (6–8), Stoltz and co-workers (9, 10), Braun and co-workers (11, 12), and Hartwig and co-workers (13, 14) has introduced valuable previously undescribed technologies for (i) the enantioselective alkylation of preformed or in situ generated metal enolates (5, 6, 11–17) and (ii) the asymmetric and decarboxylative conversion of allyl keto carbonates to α-allylated ketones (7–10). With these key advances in place, a goal now for asymmetric catalysis has become the direct α-allylation of simple ketone substrates (18, 19), an elusive yet potentially powerful bond construction (Scheme 1).
Scheme 1.

Recently, we questioned whether the catalytic principles of singly occupied molecular orbital (SOMO) activation (20) might be translated to ketonic systems, thereby providing an unreported mechanism for direct and enantioselective α-carbonyl alkylation. Despite the superficial similarities between aldehydes and ketones, these carbonyl families exhibit largely different steric and electronic properties with respect to catalyst interactions. As a consequence, the translation of enantioselective activation modes between these carbonyl subclasses is often difficult (if not unattainable in many cases) (21, 22). Herein, we describe the invention of a previously undisclosed family of organocatalysts that enable cyclic ketones to successfully function within the SOMO-activation platform while being chemically robust to oxidative reagents. Moreover, we document the introduction of a previously undescribed catalytic α-ketone alkylation reaction that is immediately amenable to asymmetric induction.
Enantioselective SOMO Catalysis
Over the last decade, the field of enantioselective synthesis has witnessed tremendous progress in the successful implementation of small organic molecules as asymmetric catalysts. In particular, two modes of carbonyl activation by chiral secondary amines have led to the discovery of a large number of previously undescribed reactions (Scheme 2): (i) Iminium catalysis (23), wherein lowest unoccupied molecular orbital (LUMO) lowering activation is accomplished via the transient condensation of an α,β-unsaturated aldehyde and an amine catalyst, has enabled the enantioselective conjugate addition or cycloaddition of enals with a wide range of external π-nucleophiles or cycloaddition partners and (ii) enamine catalysis (24), a reaction mode wherein a catalytic amount of a chiral enamine exhibits increased propensity to react with a broad selection of electrophiles by raising the highest occupied molecular orbital (HOMO).
Scheme 2.

An evolution of activation modes in organocatalysis.
In view of the established utility of these activation modes for the development of a variety of previously undisclosed, valuable transformations, we recently wondered if the existing two-electron interconversion between iminium and enamine species could be redirected to utilize a 3π electron species and thereby establish a previously undescribed reaction platform that relies on SOMO intermediates (20). In pursuit of this goal, three design elements proved to be extremely important: First, selective oxidation of an equilibrium concentration of enamine in lieu of the aldehyde substrate and amine catalyst was imperative. Known ionization potentials of analogous substrates indicated that such a selective oxidation was indeed feasible (Scheme 3) (20). The second design element required the identification of an amine catalyst that could generically provide high levels of enantiodiscrimination in the subsequent bond-forming processes with π-rich SOMOphiles. Density functional theory (DFT) calculations indicated that the radical cation derived from imidazolidinone catalyst 1 is partitioned away from the bulky tert-butyl group and that the carbon-centered radical populates an (E)-geometry in order to minimize allylic nonbonding interactions with the imidazolidinone framework (Scheme 3) (20). The enantiodetermining event would then be governed by C-5 benzyl shielding of the Re face, leaving the Si face exposed. Finally, the propensity of the SOMO intermediate to engage in stereodefined C─C bond formation with a large variety of π-rich nucleophiles was anticipated.
Scheme 3.

Relevant ionization potentials of reactive species; DFT minimized radical cation; representative transformations.
Importantly, the successful realization of this previously unreported catalytic activation mode has enabled our laboratory to introduce a variety of previously unprecedented enantioselective transformations. Examples include the direct allylic alkylation (20), the α-arylation (20, 25), α-enolation (26), α-vinylation (27), α-carbooxidation of alkenes (28), α-nitro alkylation en route to corresponding amino acids (29), as well as α-chlorination (30) (Scheme 4). Given the high synthetic utility of these enantioenriched aldehyde synthons, we recently recognized the importance (as well as the potential difficulties) in extending this SOMO catalysis concept to ketonic motifs.
Scheme 4.

Selected transformations using SOMO catalysis.
Results and Discussion
The application of our SOMO-activation platform to enantioselective ketone allylation was first evaluated with cyclohexanone, allyltrimethylsilane, ceric ammonium nitrate, and 20 mol% of imidazolidinone 2, an amine catalyst that has previously enabled Diels–Alder reactions with cyclic enones via iminium activation (21, 22). As revealed in Scheme 5, we were happy to find that the critical alkylation step could be achieved in both a direct and enantioselective fashion using imidazolidinone 2; however, competitive catalyst degradation (via methyl furan oxidation) consistently led to poor levels of reaction efficiency (Scheme 5, 42% yield, 93% ee).
Scheme 5.

Initial result of direct ketone allylation.
Heartened by these initial results, we sought to design a second generation catalyst that would be less prone to oxidation while retaining the critical architectural features that confer enantiocontrol. In this context, we hypothesized that either (a) derivatization of the furan methyl group of catalyst 2 to a trifluoromethyl moiety (4), or (b) direct replacement of the furyl ring with heteroaryl systems that are less electron-rich (e.g., benzofuran 5, benzothiophene 6) might render a more robust system. Indeed, imidazolidinones 4, 5 and 6 were all found to catalyze the direct α-allylation of cyclohexanone in good yield while maintaining excellent levels of enantioselectivity (75–85% yield, 88–94% ee). The broad-spectrum utility of this unreported imidazolidinone family 4–6 prompted us to initiate our investigation into substrate scope using all three catalysts.
Experiments that probe the scope of the ketone component have revealed that a variety of four-, five-, and six-membered carbocycles and heterocycles can be readily employed in this asymmetric alkylation reaction (Table 1, entries 1–9, 84–99% ee). Moreover, this protocol is successful with ketones that incorporate alkyl and heteroatom substituents at the β and γ ring positions, an important consideration with respect to natural product synthesis (entries 2, 4, 6, and 8, 85–99% ee). Furthermore, this protocol is generally successful with only two equivalents of ketone, although 20 equivalents are required for the cyclobutane example. Perhaps most notable, we have found that nonsymmetrical cyclic ketones such as 3-pyrrolidinones (entry 6) and 3-furanones (entry 7) afford exclusively the C(4)-alkylation product as determined by GLC (> 200∶1) or HPLC (> 100∶1) analysis. These results are striking given that the 2-enamine isomer should be formed competitively [based on kinetic acidities (31)] and more readily oxidized (based on ionization potentials) (32). With respect to 3-methylcyclohexanone, we assume that the observed sense of regiocontrol can be explained on the basis of steric constraints (entry 8, 99% ee). Curiously, cyclopentanone undergoes exclusive formation of the C2-symmetric 2,5-bis-allylation product (entry 9, 99% ee) (33). Given that 2-allylcyclopentanone (12) is not converted to this C2-bis-allylation adduct when exposed to the same reaction conditions, we presume that the first cyclopentanone alkylation step leads to an iminium/enamine species that undergoes a second oxidation-allylation sequence faster than iminium hydrolysis. It is important to note that the sense of asymmetric induction observed in all cases (Table 1 and Schemes 5–7) is consistent with selective engagement of the SOMOphile with the Si face of the radical cation, in accord with the calculated structure (DFT-3) (34).
Table 1.
 | |||||
| 1 | 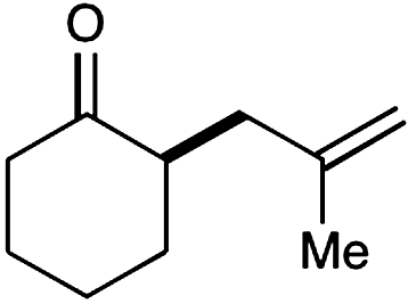 |
2 | 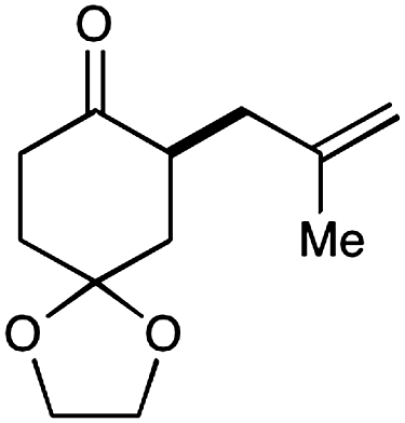 |
3 | 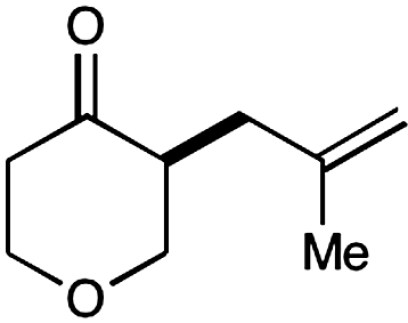 |
| 4, 82%, 91% ee | 4, 85%, 90% ee | 4, 78%, 90% ee | |||
| 4 | 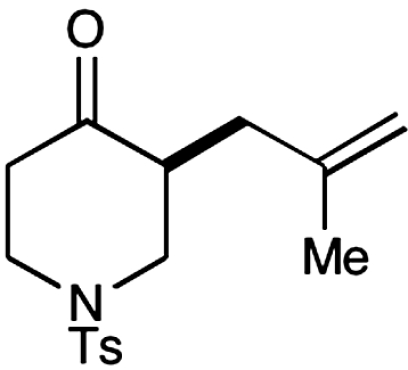 |
5‡ | 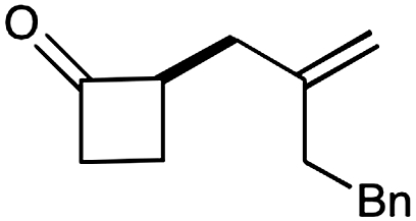 |
6 | 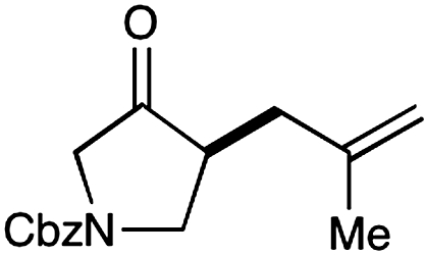 |
| 4, 76%, 90% ee | 5, 66%, 91% ee | 6, 77%, 85% ee | |||
| 7§ | 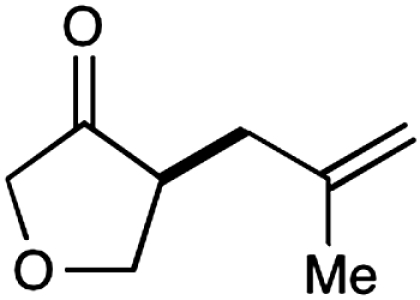 |
8§ | 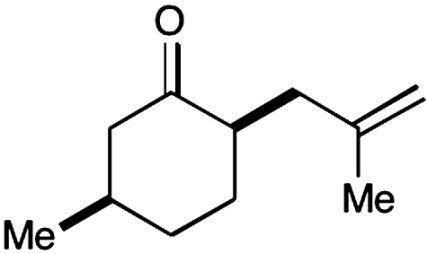 |
9§ | |
| 5, 79%, 84% ee | 5, 70%, 5∶1 dr, 99% ee | 5, 71%, 99% ee | |||
| 10§ | 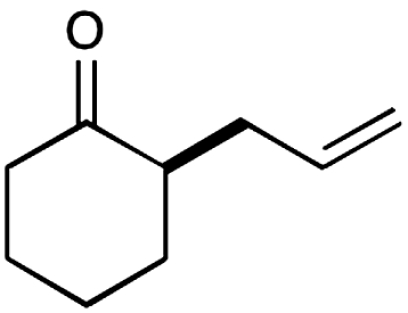 |
11 | 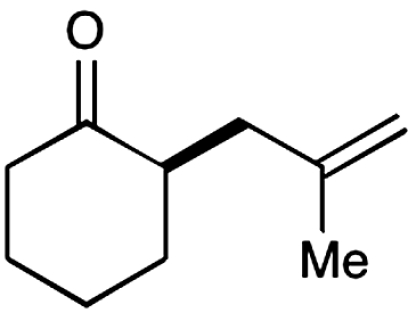 |
12 | 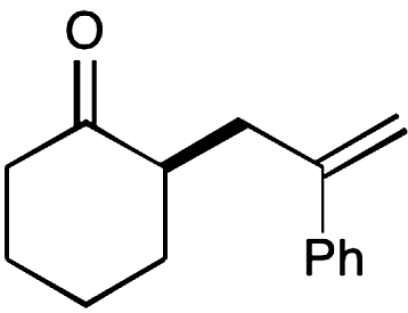 |
| 5, 85%, 91% ee | 4, 82%, 91% ee | 4, 83%, 81% ee | |||
| 13 | 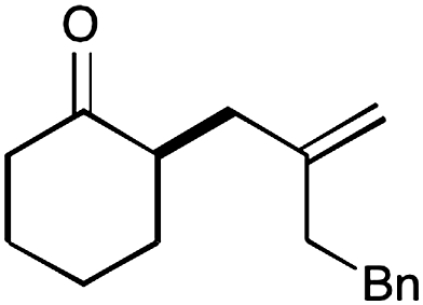 |
14 | 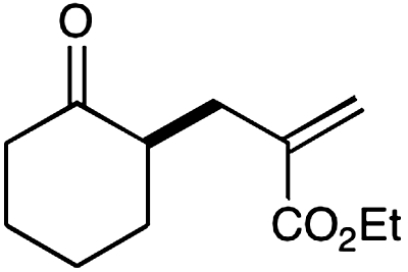 |
15§ | 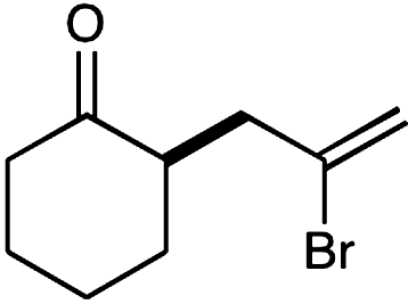 |
| 6, 86%, 96% ee | 6, 80%, 83% ee | 5, 74%, 87% ee | |||
*For each entry: catalyst #, % yield, % ee.
†Enantiomeric excess determined by chiral HPLC, supercritical fluid chromatography, or GLC analysis.
‡Performed with 20 eq of ketone.
§Performed in the absence of NaHCO3.
Scheme 6.

Catalyst design and reaction efficiency.
Scheme 7.

General utility of ketone α-functionalization.
The nature of the allyl silane component in this ketone alkylation reaction has also been evaluated. As revealed in Table 1, this catalytic protocol can accommodate π-rich olefinic silanes (entries 10–13, 82–86% yield, 81–96% ee) as well as electron-deficient silanes (entries 14–15, 74–80% yield, 83–87% ee), including a vinyl bromide (entry 15, 74% yield, 87% ee). Such halogenated olefin adducts should prove to be valuable synthons for use in conjunction with established cross-coupling methodologies (e.g., Buchwald–Hartwig or Stille couplings).
In an effort to further demonstrate synthetic utility, we have sought to apply our ketone-SOMO-activation platform to a series of bond constructions that have only previously been possible with aldehydic substrates. For example, catalyst 6 has successfully enabled the enantioselective α-enolation of cyclohexanone under oxidative conditions (chemical formula 1, Scheme 7, 84% ee). Moreover, the asymmetric α-homobenzylation of cyclohexanone can also be accomplished via a two-step α-carbo-oxidation-hydrogenation sequence using the same imidazolidinone catalyst (chemical formula 2, Scheme 7, 80% ee) (26, 28).
In summary, we have developed a family of oxidatively stable imidazolidinone catalysts 4–6 that are readily employed for the SOMO activation of cyclic ketones. These catalysts allow the direct α-allylation, α-enolation, and α-carbooxidation of carbocyclic ketones with excellent levels of enantiocontrol. Further application of these organocatalytic methodologies will be reported shortly.
Methods
Full experimental details and spectral data are included in SI Text.
General Procedure for the α-Allylation of Ketones.
To a 10-mL oven-dried round-bottom flask equipped with a magnetic stir bar was added cocatalyst trichloroacetic acid or HCl (0.15 mmol, 0.20 eq), imidazolidinone catalyst 4⋅TFA, 5, or 6 (0.15 mmol, 0.20 eq) (as indicated below), NaHCO3 (94.9 mg, 1.13 mmol, 1.50 eq), and ceric ammonium nitrate (1.03 g, 1.88 mmol, 2.50 eq). The reaction vessel was evacuated by high vacuum (0.3 torr) at -78 °C for 1 min. After backfilling with argon, tetrahydrofuran (3.0 mL, 0.25 m) and water (10–20 eq, as indicated below) were added to the reaction vessel, and the reaction mixture was subsequently degassed by repeated evacuation and backfilling with argon (three cycles of 2 min each) at -78 °C. The allyl silane (0.75 mmol, 1.00 eq) was then added via syringe followed by the respective ketone (1.50 mmol, 2.00 eq). The reaction vessel was moved from the -78 °C bath into a -20 °C cooling bath and stirred with a high rate of vortex for 24 h under argon atmosphere. At the end of the indicated reaction time, diethyl ether (5.0 mL) was added to the crude mixture and stirring was continued at -78 °C for 20 min to precipitate the cerium salts. Filtration through a plug of silica gel using diethyl ether as a solvent concentration under reduced pressure furnished the crude product as a yellowish liquid, which was purified by flash chromatography to afford the analytically pure α-allylated ketone.
Method A.
The general procedure was followed using (2R,5R)-5-benzyl-3-methyl-2-(benzofuran-2-yl)imidazolidin-4-one (5) (46 mg, 0.15 mmol, 0.20 eq) and trichloroacetic acid (24 mg, 0.15 mmol, 0.20 eq) in the presence of water (0.27 mL, 15 mmol, 20 eq) without the addition of NaHCO3.
Method B.
The general procedure was followed using (2R,5R)-5-benzyl-3-methyl-2-(benzothiophen-2-yl)imidazolidin-4-one (6) (48 mg, 0.15 mmol, 0.20 eq) and conc. HCl (6.0 μL, 0.20 eq, 0.15 mmol) in the presence of water (0.27 mL, 15 mmol, 20 eq) and NaHCO3 (94.5 mg, 1.12 mmol, 1.50 eq).
Method C.
The general procedure was followed using the trifluoroacetic acid salt of (2R,5R)-5-benzyl-3-methyl-2-(5-trifluoromethylfuran-2-yl)imidazolidin-4-one (4⋅TFA) (63 mg, 0.15 mmol, 0.20 eq) in the presence of water (0.14 mL, 7.5 mmol, 10 eq) and NaHCO3 (94.5 mg, 1.5 eq, 1.12 mmol).
Supplementary Material
Acknowledgments.
The authors thank Hui-Wen Shih for invaluable assistance performing Gaussian DFT calculations. Financial support was provided by the National Institute of General Medical Sciences (R01 01 GM093213-01) and kind gifts from Amgen and Merck.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1002845107/-/DCSupplemental.
References
- 1.Carey FA, Sundberg RJ. Advanced Organic Chemistry Part B: Reactions and Synthesis. 4th Ed. New York: Kluwer Academic/Plenum; 2001. Alkylation of nucleophilic carbon intermediates; pp. 1–47. [Google Scholar]
- 2.Lu Z, Ma S. Metal-catalyzed enantioselective allylation in asymmetric synthesis. Angew Chem Int Edit. 2008;47:258–297. doi: 10.1002/anie.200605113. [DOI] [PubMed] [Google Scholar]
- 3.Trost BM, Crawley ML. Asymmetric transition-metal-catalyzed allylic alkylations: Applications in total synthesis. Chem Rev. 2003;103:2921–2944. doi: 10.1021/cr020027w. [DOI] [PubMed] [Google Scholar]
- 4.Braun M, Meier T. New developments in stereoselective palladium-catalyzed allylic alkylations of preformed enolates. Synlett. 2006:661–676. [Google Scholar]
- 5.Doyle AG, Jacobsen EN. Enantioselective alkylations of tributyltin enolates catalyzed by Cr(salen)Cl: Access to enantiomerically enriched all-carbon quaternary centers. J Am Chem Soc. 2005;127:62–63. doi: 10.1021/ja043601p. [DOI] [PubMed] [Google Scholar]
- 6.Trost BM, Schroeder GM. Palladium-catalyzed asymmetric alkylation of ketone enolates. J Am Chem Soc. 1999;121:6759–6760. doi: 10.1002/chem.200400666. [DOI] [PubMed] [Google Scholar]
- 7.Trost BM, Xu J. Regio- and enantioselective Pd-catalyzed allylic alkylation of ketones through allyl enol carbonates. J Am Chem Soc. 2005;127:2846–2847. doi: 10.1021/ja043472c. [DOI] [PubMed] [Google Scholar]
- 8.Trost BM, Bream RN, Xu J. Asymmetric allylic alkylation of cyclic vinylogous esters and thioesters by Pd-catalyzed decarboxylation of enol carbonate and β-ketoester substrates. Angew Chem Int Edit. 2006;45:3109–3112. doi: 10.1002/anie.200504421. [DOI] [PubMed] [Google Scholar]
- 9.Behenna DC, Stoltz BM. The enantioselective Tsuji allylation. J Am Chem Soc. 2004;126:15044–15045. doi: 10.1021/ja044812x. [DOI] [PubMed] [Google Scholar]
- 10.Mohr JT, Behenna DC, Harned AM, Stoltz BM. Deracemization of quaternary stereocenters by Pd-catalyzed enantioconvergent decarboxylative allylation of racemic β-ketoesters. Angew Chem Int Edit. 2005;44:6924–6927. doi: 10.1002/anie.200502018. [DOI] [PubMed] [Google Scholar]
- 11.Braun M, Laicher F, Meier T. Diastereoselective and enantioselective palladium-catalyzed allylic substitution with nonstabilized ketone enolates. Angew Chem Int Edit. 2000;39:3494–3497. doi: 10.1002/1521-3773(20001002)39:19<3494::aid-anie3494>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 12.Braun M, Meier T. Palladium-catalyzed stereoselective allylic alkylation of lithium enolates. Synlett. 2005:2968–2972. [Google Scholar]
- 13.Graening M, Hartwig JF. Iridium-catalyzed regio- and enantioselective allylation of ketone enolates. J Am Chem Soc. 2005;127:17192–17193. doi: 10.1021/ja0566275. [DOI] [PubMed] [Google Scholar]
- 14.Weix DJ, Hartwig JF. Regioselective and enantioselective iridium-catalyzed allylation of enamines. J Am Chem Soc. 2007;129:7720–7721. doi: 10.1021/ja071455s. [DOI] [PubMed] [Google Scholar]
- 15.Evans PA, Leahy DK. Regioselective and enantiospecific rhodium-catalyzed allylic alkylation reactions using copper(I) enolates: Synthesis of (-)-sugiresinol dimethyl ether. J Am Chem Soc. 2003;125:8974–8975. doi: 10.1021/ja035983p. [DOI] [PubMed] [Google Scholar]
- 16.Yan X-X, et al. Highly enantioselective Pd-catalyzed allylic alkylations of acyclic ketones. Angew Chem Int Edit. 2005;44:6544–6546. doi: 10.1002/anie.200502020. [DOI] [PubMed] [Google Scholar]
- 17.Zheng W-H, Zheng B-H, Zhang Y, Hou X-L. Highly regio-, diastereo-, and enantioselective Pd-catalyzed allylic alkylation of acyclic ketone enolates with monosubstituted allyl substrates. J Am Chem Soc. 2007;129:7718–7719. doi: 10.1021/ja071098l. [DOI] [PubMed] [Google Scholar]
- 18.Dolling U-H, Davis P, Grabowski EJ. Efficient catalytic asymmetric alkylations. 1. Enantioselective synthesis of (+)-indacrinone via chiral phase-transfer catalysis. J Am Chem Soc. 1984;106:446–447. [Google Scholar]
- 19.Andrus MB, Hicken EJ, Stephens JC. Phase-transfer-catalyzed asymmetric glycolate alkylation. Org Lett. 2004;6:2289–2292. doi: 10.1021/ol0491235. [DOI] [PubMed] [Google Scholar]
- 20.Beeson TD, Mastracchio A, Hong J-B, Ashton K, MacMillan DWC. Enantioselective organocatalysis using SOMO activation. Science. 2007;316:582–585. [PubMed] [Google Scholar]
- 21.Northrup AB, MacMillan DWC. The first general enantioselective catalytic Diels-Alder reaction with simple α,β-unsaturated ketones. J Am Chem Soc. 2002;124:2458–2460. doi: 10.1021/ja017641u. [DOI] [PubMed] [Google Scholar]
- 22.Tuttle JB, Ouellet SG, MacMillan DWC. Organocatalytic transfer hydrogenation of cyclic enones. J Am Chem Soc. 2006;128:12662–12663. doi: 10.1021/ja0653066. [DOI] [PubMed] [Google Scholar]
- 23.Lelais G, MacMillan DWC. Modern strategies in organic catalysis: The advent and development of iminium activation. Aldrichim Acta. 2006;39:79–87. [Google Scholar]
- 24.Mukherjee S, Yang JW, Hoffmann S, List B. Asymmetric enamine catalysis. Chem Rev. 2007;107:5471–5569. doi: 10.1021/cr0684016. [DOI] [PubMed] [Google Scholar]
- 25.Conrad JC, Kong J, Laforteza BN, MacMillan DWC. Enantioselective α-arylation of aldehydes via organo-SOMO catalysis. An ortho-selective arylation reaction based on an open-shell pathway. J Am Chem Soc. 2009;131:11640–11641. doi: 10.1021/ja9026902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jang H-Y, Hong J-B, MacMillan DWC. Enantioselective organocatalytic singly occupied molecular orbital activation: The enantioselective α-enolation of aldehydes. J Am Chem Soc. 2007;129:7004–7005. doi: 10.1021/ja0719428. [DOI] [PubMed] [Google Scholar]
- 27.Kim H, MacMillan DWC. Enantioselective organo-SOMO catalysis: The α-vinylation of aldehydes. J Am Chem Soc. 2008;130:398–399. doi: 10.1021/ja077212h. [DOI] [PubMed] [Google Scholar]
- 28.Graham TH, Jones CM, Jui NT, MacMillan DWC. Enantioselective organo-SOMO catalysis: The carbo-oxidation of styrenes. J Am Chem Soc. 2008;130:16494–16495. doi: 10.1021/ja8075633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilson JE, Casarez AD, MacMillan DWC. Enantioselective aldehyde α-nitroalkylation via oxidative organocatalysis. J Am Chem Soc. 2009;131:11332–11334. doi: 10.1021/ja904504j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amatore M, Beeson TD, Brown SP, MacMillan DWC. Enantioselective linchpin catalysis by SOMO catalysis: An approach to the asymmetric α-chlorination of aldehydes and terminal epoxide formation. Angew Chem Int Edit. 2009;48:5121–5124. doi: 10.1002/anie.200901855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garst ME, Bonfiglio JN, Grudoski DA. Specific enolates from α-amino ketones. J Org Chem. 1980;45:2307–2315. [Google Scholar]
- 32. Studies are now under way to determine the mechanistic origins of regioselectivity in these allylic alkylations using SOMO activation.
- 33.Kurth MJ, O’Brien MJ. Desulfurization/α-alkylation of β-keto sulfones. J Org Chem. 1986;50:3846–3848. [Google Scholar]
- 34. DFT calculations were performed at the B3LYP/6-311+G(2d,p)//B3 LYP/6-31G(d) level of theory using Gaussian 03 software: Frisch MJ, et al. (2004) Gaussian 03 Revision C.02, Gaussian Inc., Wallingford, CT.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.