

Abstract
Glutamate in the prefrontal cortex (PFC) plays a significant role in several mental illnesses, including schizophrenia, addiction and anxiety. Previous studies on PFC glutamate-mediated function have used techniques that raise questions on the neuronal vs. astrocytic origin of glutamate. The present studies used enzyme-based microelectrode arrays (MEAs) to monitor second-by-second resting glutamate levels in the PFC of awake rats. Locally-applied drugs were employed in an attempt to discriminate between the neuronal or glial components of the resting glutamate signal. Local application of tetrodotoxin (TTX; sodium channel blocker), produced a significant (~40%) decline in resting glutamate levels. In addition significant reductions in extracellular glutamate were seen with locally-applied ω-conotoxin (MVIIC; ~50%; calcium channel blocker), and the mGluR⅔ agonist, LY379268 (~20%), and a significant increase with the mGluR⅔ antagonist LY341495 (~40%), effects all consistent with a large neuronal contribution to the resting glutamate levels. Local administration of D,L-threo-β-benzyloxyaspartate (TBOA; glutamate transporter inhibitor) produced an ~120% increase in extracellular glutamate levels, supporting that excitatory amino acid transporters, which are largely located on glia, modulate clearance of extracellular glutamate. Interestingly, local application of (S)-4-carboxyphenylglycine (CPG; cystine/glutamate antiporter inhibitor), produced small, non-significant bi-phasic changes in extracellular glutamate versus vehicle control. Finally, pre-administration of TTX completely blocked the glutamate response to tail pinch stress. Taken together, these results support that PFC resting glutamate levels in rats as measured by the MEA technology are at least 40-50% derived from neurons. Furthermore, these data support that the impulse flow-dependent glutamate release from a physiologically-evoked event is entirely neuronally derived.
Keywords: basal, chronic, freely moving, TTX, TBOA
I. Introduction
Glutamate neurotransmission is important for maintaining normal brain function such as development, plasticity, learning and memory, and sensory and motor systems (Carlsson and Carlsson, 1990; Grace, 1991; Greenamyre, 1993; Del Arco et al., 1998, 1999). Dysregulation of the glutamate system is involved in neurodegenerative disorders including Parkinson’s disease, Alzheimer’s disease, Huntington’s disease and amyotrophic lateral sclerosis (Doble, 1999; Danbolt, 2001) as well as neuropsychiatric disorders such as schizophrenia, addiction, anxiety, and depression (Mechri et al., 2001; López-Mereno et al., 2008; Mathew et al., 2008; McNally et al., 2008). Because the prefrontal cortex (PFC) is associated with the pathophysiology of several of these disorders and the majority of afferent and efferent fibers are glutamatergic in this brain region, a better understanding of glutamatergic regulation in the PFC is needed.
Measures of extracellular glutamate signaling in the mammalian CNS have been dominated by microdialysis techniques over the past two decades. Microdialysis utilizes semipermeable membranes to control the diffusion of extracellular neurotransmitters along their concentration gradients (Ungerstedt, 1984; Di Chiara et al., 1996) allowing for measurements of multiple analytes at femtomolar concentrations. Microdialysis probes have a diameter of 150-400 μm with an average length of 1-4 mm (Kennedy et al., 2002). Dialysate samples are usually collected every 5-20 minutes (Westerink and Timmerman, 1999). Although recent advances in the detection methods have allowed for sub-minute sampling rates (Lada et al., 1997; Tucci et al., 1997; Rossell et al., 2003), these approaches have not been practical for routine use by many laboratories.
Few microdialysis studies have found evidence that neurons contribute significantly to extracellular glutamate levels under resting (basal) conditions (for review see Timmerman and Westerink, 1997). The general consensus that has emerged from microdialysis studies is that maintenance of extracellular glutamate levels does not follow classical exocytotic release criteria; that is resting glutamate levels have been typically found to be only minimally tetrodotoxin (TTX)- or calcium-dependent (Timmerman and Westerink, 1997; van der Zeyden et al., 2007; Kalivas, 2009). This has led to the conclusion that the origin of resting glutamate is not neuronal, but rather astrocytic, presumably involving a reversal of the high-affinity glutamate transporters (Westerink and Timmerman, 1999; Montiel et al., 2005) or by the cystine-glutamate antiporter (Baker et al., 2002; Kalivas, 2009). Indeed, changes in glial regulation has been estimated to account for ~60% of resting levels (Kalivas, 2009)
More recently, our laboratory has been able to measure glutamate in vivo with enzyme-based microelectrode arrays (MEAs) coupled to amperometric recording techniques (Burmeister et al., 2000; Burmeister and Gerhardt, 2001; Burmeister et al., 2002; Pomerleau et al., 2003; Nickell et al., 2005; Day et al., 2006; Nickell et al., 2007; Quintero et al., 2007; Rutherford et al., 2007; Hascup et al., 2008). Our laboratory, and others using similar microelectrodes, have demonstrated that resting glutamate levels are TTX-dependent thus supporting a neuronal origin for these levels (Kulagina et al., 1999; Day et al., 2006; Oldenziel et al., 2006; Hascup et al., 2008). There are several key differences between the two techniques that may account for the inability of microdialysis to properly study resting glutamate levels. First, the temporal resolution of microdialysis is on the order of minutes, which is too slow to detect glutamatergic neural transmission that occurs over milliseconds to seconds (Kinney et al., 1997). Our enzyme-based MEAs coupled with amperometric recording techniques are capable of detecting the fast glutamate neurotransmission through 1 Hz recording intervals. Second, the large size of the dialysis probe makes it difficult to sample glutamate close to the the synaptic cleft (Westerink and Timmerman, 1999; Drew et al., 2004). Since glutamate has rapid clearance kinetics, being able to sample closer to the synaptic cleft may be necessary to study neuronal glutamate. Third, implantation of the microdialysis probe results in short and long-term trauma, which is evidenced by histological, physiological, biochemical and neurochemical changes in CNS tissue that can occur in a 2.8 mm circumference surrounding the implant site (Clapp-Lilly et al., 1999; Bungay et al., 2003; Hascup et al., 2009). This damage alters neurotransmitter release and uptake near the probe implant site (Yang et al., 1998; Borland et al., 2005). Our MEAs have been shown to cause less damage to the surrounding CNS tissue as compared to microdialysis (Rutherford et al., 2007; Hascup et al., 2009).
In the present study, we used MEA and constant voltage amperometry to elucidate the origin of resting glutamate levels in the PFC. This was done in awake rats since anesthetics such as urethane (Rutherford et al., 2007) and pentobarbital (Dash et al., 2009) are known to decrease resting glutamate levels. The relative contribution of neuron vs. astrocytes to resting glutamate levels were studied by local applications of the sodium channel blocker TTX, the N, P, and Q type calcium channel blocker ω-conotoxin MVIIC, the mGluR group II agonist LY379268, the mGluR group II antagonist LY341495, the non-selective glutamate transporter blocker D,L-threo-β-benzyloxyaspartate (TBOA), and the cystine-glutamate antiporter blocker (S)-4-carboxyphenylglycine (CPG). We also examined the origin of PFC glutamate released in response to tail-pinch stress. Prior studies from our laboratory and others, indicates that this response partly reflects neuronal release of glutamate. (Bagley and Moghaddam, 1997; Lowry et al., 1998; Rutherford et al., 2007; Wassum et al., 2008; Lupinsky et al., 2010).
2. Materials and Methods
2.1 Animals
Male Long Evans (Harlan) rats 3-6 months of age at the time of surgery were used for all experiments. The animals were individually housed in a 12 hour light/dark cycle with ad libitum access to food and water in the Association for Assessment and Accreditation of Laboratory Animal Care International approved animal resource center at the University of Kentucky or McGill’s University Animal Care Committee at the Douglas Mental Health University Institute. Animals were allowed at least one week to acclimate to the environment prior to any experiments. All appropriate animal care (food, water, bedding, cage cleaning, etc.) was performed by the Animal Resource Center staff. There were no procedures involving undue discomfort to the animals. Following surgery, rats were individually housed under the same conditions. Animal care was approved by the University of Kentucky Institutional Animal Care and Use Committee and The Douglas Animal Care, which was in accordance with the Guide for the Care and Use of Laboratory Animals and AAALACI guidelines and the Canadian Council on Animal Care.
2.2. MEA design
MEAs were manufactured, assembled and selected for in vivo recordings as previously described (Burmeister et al., 2000, 2002). Preparation of the MEA for recording has been extensively described in Burmeister et al., 2002 and Hascup et al., 2006. Briefly, the platinum (Pt) recording sites were dip coated with Nafion® (Sigma-Aldrich Corp., St. Louis, MO) to repel anions as previously described (Burmeister and Gerhardt, 2001). Two of the MEA recording sites were coated with a L-glutamate oxidase (EC 1.4.3.11; GluOx) (Seikagaku America, Inc., East Falmouth, MA) coating solution as previously described (Nickell et al., 2005). In order to induce cross-linking of GluOx and increase its adhesion to the MEA, glutaraldehyde and BSA (Sigma-Aldrich Corp., St. Louis, MO) were added to the GluOx solution. The GluOx layer was required by our approach to measure glutamate, as it causes the enzymatic break-down of glutamate to α-ketoglutarate and the electroactive reporter molecule, H2O2. When a potential of +0.7 V vs. a Ag/AgCl reference electrode was applied, the reporter molecule H2O2 was oxidized yielding two electrons at the MEA recording surface. The resulting current was then amplified and recorded by a FAST (fast analytical sensing technology) 16 recording system (Quanteon, LLC, Nicholasville, KY). The remaining two MEA recording sites (self-referencing or sentinel sites) were coated similar to the glutamate recording sites, with the exception that the coating solution did not contain GluOx. This meant the sentinel sites could not record any converted glutamate but could record any electroactive interferents not repelled by Nafion®. When the current recorded on the sentinel sites was subtracted from the current on the glutamate recording sites, the resulting signal represents the extracellular resting glutamate levels in the brain tissue (Burmeister and Gerhardt, 2001; Burmeister et al, 2003; Rutherford et al., 2007).
2.3. Reference electrodes
Miniature Ag/AgCl reference electrodes were prepared by first stripping the Teflon off the silver wire (200 μm bare, 275 μm coated; A-M Systems, Inc., Carlsberg, WA) ¼ inch on each end. One of the stripped ends was soldered to a gold-plated socket (Ginder Scientific, Ottawa, ON) and the other end was coated with AgCl by placing the tip of the stripped sliver wire into a 1.0 M HCl plating bath saturated with NaCl containing a Pt wire and applying +9 V DC using a power supply to the cathode vs. the anode for ~10 minutes.
2.4. Ceramic MEA preparation
The MEA was modified for use in freely moving animals as previously described (Rutherford et al., 2007; Hascup et al., 2008). Briefly, the pedestal was the portion of the system that was chronically implanted on the rat’s head and contained the MEA. During recording sessions, the pedestal was connected to the miniaturized Rat Hat, containing the 4-channel amplifier that was in turn connected to a low torque commutator. The ceramic-based MEAs were prepared for freely moving recordings first by cleaning the MEA with isopropyl alcohol and ddH2O. MEAs were dried for one hour at 40°C to make sure there was no residual water on the sites. Next, the MEAs were coated with Nafion®, cured at 165°C for 4 minutes followed by the coating solutions (either containing or not containing GluOx) as described above.
In order to attach the miniature connector to the modified MEA for freely-moving recordings (together termed the pedestal), connecting wires (30 AWG varnished copper wire, RadioShack) were used. The copper wires were tucked around the miniature connector and the MEA tip was carefully positioned parallel to the connector. Correct positioning of the MEA tip was essential for accurate MEA placement in the brain. Water proof five minute epoxy (The Original Super Glue Corporation) was used to secure the paddle and wires to the round connector, making sure that the exposed part of the sockets on the end of the connector closest to the MEA tip and wires were covered with epoxy. This prevented moisture from penetrating the pedestal. The completed assembly was allowed to cure at room temperature for 24 hours.
2.5. Microelectrode array / cannula assembly
In order to locally apply small quantities of TTX, ω-conotoxin MVIIC, LY379268, LY341495, TBOA, and CPG at the MEA tip, a 26-gauge stainless steel guide cannula (Plastics One, Roanoke, VA) was attached to the MEA assembly using sticky wax (Kerr Corp., Orange, CA). A stainless steel dummy cannula, 0.5 mm longer than the guide, was inserted into the guide and screwed into place, such that the dummy cannula tip was aimed between the four recording sites at a distance of approximately 100 μm away from the MEA tip. Ejections of drugs were performed through a 33-gauge internal cannula, or ejection cannula, which protruded 1 mm past the guide cannula. A stainless steel dummy cannula remained in the guide whenever the animal was not in the recording chamber.
2.6. Electrode calibration
Microelectrodes were calibrated to determine their sensitivity and selectivity for glutamate against ascorbic acid as previously described (Nickell et al., 2005). Briefly, constant potential amperometry was performed using a FAST16 system designed for recording simultaneously from the four-channel microelectrodes with a gain of 2 nA/V (operated at a constant applied potential of +0.7 V vs. a Ag/AgCl reference electrode). The tip of the modified MEA was placed in a continuously stirred solution of 0.05 M PBS (pH 7.4). A recirculating water bath (Gaymar Co.) was used to maintain a constant buffer temperature of 37°C to allow the enzyme layer to function optimally relative to in vivo conditions. Calibrations were performed using final buffer concentrations of 250 μM ascorbic acid and 20, 40, and 60 μM glutamate through additions of aliquots of stock solutions of 20 mM ascorbic acid and 20 mM glutamate. Selectivity ratios for glutamate over ascorbic acid were calculated in addition to the slope (sensitivity), limit of detection (LOD), and linearity (R2) for glutamate for all MEAs. The MEAs were tested during calibration to determine the precision of the four Pt recording sites using dopamine (2 μM final concentration) and H2O2 (8.8 μM final concentration) as test substances. MEAs were only used if all four Pt recording sites had in vitro responses to dopamine and H2O2 that were within 10% of each other. Finally, all administered drugs (TTX, CPG, etc.) were added as test substances to ensure that they would not interfere with the glutamate recordings.
2.7. Electrode implantation
Following a successful calibration of the MEA, rats were anesthetized with 2% isoflurane, and placed in a stereotaxic apparatus (Kopf Instruments, Tujunga, CA). Animal body temperature was maintained at 37°C with a heating pad (Braintree Scientific, Braintree, MA) the animals’ eyes were lubricated with artificial tears (The Butler Company, Columbus, OH) to help maintain moisture and prevent infection. Prior to an incision, the skin directly on top of the animals head was wiped with Betadine solution to keep the incision area clean and to prevent infection, and then reflected, making as small an incision as possible. First, three small holes were drilled in the skull in the adjacent quadrants of where the MEA was implanted for placement of stainless steel skull screws. A fourth hole was drilled contralateral from the recording site for insertion of the miniature Ag/AgCl reference electrode. Next, three small stainless steel screws (Small Parts, Inc) were threaded into the skull to serve as anchors and care was taken so that the screw tips did not touch brain tissue. A 2 mm × 2 mm craniotomy was performed over the PFC area and a glutamate selective MEA pedestal assembly was implanted into the right PFC (MEA tip coordinates - AP: +3.2 mm; ML: -0.8 mm, DV: -5.0 mm vs. bregma), based on the atlas of Paxinos and Watson (1998) with the incisor bar set so that the skull was level (-2.3 mm). The assembly was secured with approximately four layers of dental acrylic (Lang Dental MFG, Wheeling, IL), making sure to cover as much of the MEA as possible. The dental acrylic had a smooth texture and excess acrylic was removed from the skin surface so as not to promote the rat to scratch its head potentially damaging the implant. Rats were allowed to recover for a minimum of two days prior to initial recordings.
2.8. Recording protocol
Typical recording sessions involved allowing the rat to freely roam around the recording chamber for ten minutes to acclimate to their surroundings before connecting the pedestal to the Rat Hat potentiostat. Once the recordings were started, the rat underwent a minimum 60-90 minute acclimation period, or until a stable baseline was established. Following this period, an ejection cannula was inserted. After the ejection cannula was inserted (connected to a 10 μL Hamilton syringe filled with the compound of interest), another acclimation period of ~ 30 minutes was allowed so that baseline was again established. Following this recording period, the effects of locally applied TTX (3 μL; 100 μM; Sigma, St. Louis, MO), TBOA (1 μL; 100 μM; Tocris Cookson Inc., Ellisville, MO), LY379268 (3 μL; 100 μM; Eli Lilly & Co., Indianapolis, IN), LY341495 (3 μL; 100 μM; Eli Lilly & Co., Indianapolis, IN), CPG (3 μL; 50 μM: Tocris Cookson Inc., Ellisville, MO), or ω-conotoxin MVIIC (1 μL; 3 μM: Tocris Cookson Inc., Ellisville, MO) and their respective controls (0.6 mM citrate in saline or saline alone), were studied. All ejection solutions were pH 7.3-7.4. To account for potential variations in exact placement of the cannula and inter-animal variability, vehicle controls were performed in the same rats as their drug counter parts. For studies of glutamate release during tail pinch stress all acclimation and cannula insertion procedures were followed as mentioned above. In animals where tail pinch stress was studied, a five minute tail pinch (clothespin) was performed 60 seconds following local application of TTX or citrate vehicle.
2.9. Data Analysis
The FAST16 recording system saved amperometric data at 1 Hz and drug ejection synchronization marks for all four recording channels. Unless otherwise noted, calibration data, in conjunction with a modified spreadsheet program, was used to determine parameters for the signal differences recorded between data marks. Measures derived from the data file included: 1) resting glutamate, the concentration of glutamate without stimulation and 2) maximum change, the greatest concentration change (increase or decrease) of the signal. A student’s t-test or a 2-way analysis of variance was used to analyze data. Significance was defined as p < 0.05. n=4-7 for all groups, as indicated in the results section.
3. Results
3.1 Resting levels of glutamate in PFC of awake Long Evans rats
We have previously established that resting glutamate levels can be reliably monitored in rats and mice for at least 3-7 days following MEA implantation (Rutherford et al., 2007; Hascup et al., 2008). However, there are substantial rat (and mouse) strain differences in resting glutamate levels as measured by the MEA with amperometry with values ranging from 7-45 μM. Glutamate concentrations in the Long Evans rat PFC are typically 10-20 fold higher than those reported in most microdialysis studies (see Table I), although one reported levels of ~33 μM (Moghaddam, 1993). Additionally, studies conducted using various non-microdialysis techniques have reported resting glutamate levels between 18 μM and 29 μM in various brain areas and rat strains (Kulagina et al., 1999; Oldenziel et al., 2006; Dash et al., 2009). The MEA technology involves measures on a second-by-second interval with a microelectrode that resides directly within the extracellular space of the brain that was closer to the glutamatergic synapses than a microdialysis probe. Also, less damage was produced by the MEA as compared to microdialysis probes (Bungay, 2003; Hascup et al., 2009). Thus, the MEA signals should be larger, in general, due to less dilution and loss of signal due to diffusion (Rice and Cragg, 2008). In addition, the microdialysis methods typically exhibit in vitro recoveries of 10-20%. Therefore, the MEA signals should be in the range of 5-10 times greater than those reported with microdialysis. Figure 1 illustrates the determination of resting glutamate using our recording and measurement technique. In Table I, we have reported a series of values from the literature of several laboratories for extracellular levels of glutamate in the PFC, striatum, and hippocampus compared to our values measured in these brain areas of Long Evans rats and Fischer 344 rats. Clearly, we are in the range of 10 fold greater values as compared to the average levels reported by microdialysis and these are keeping with the expected greater extracellular levels of glutamate that should be measured from the MEA technology. Thus, we feel that the MEA technology is accurately recording resting glutamate values. Although higher than expected, our extracellular resting glutamate values (for rats used in this study, 34.7 μM ± 11.8 μM (n=41))are within the physiologically range since the affinity of the high affinity glial and neuronal glutamate transporters are in the 1-100 μM range, depending on the assay and sub-type (Danbolt, 2001; Peacey et al., 2009).
Table I.
Comparisons of extracellular levels of resting glutamate measured by microdialysis methods in numerous labs to our MEA measurements in awake rats.
| Microdialysis Glutamate (μM) | MEA Glutamate (μM) | |
|---|---|---|
| Striatum | 0.18-1.3 | 7.2 ± 1.2 |
| (Battaglia et al., 1997; Biggs et al., 1997; Segovia et al., 2001) | (Rutherford et al., 2007) | |
| Hippocampus | 0.275-2.12 | 4.7-10.4 |
| (Ueda and Tsuru, 1995; Giovannini et al., 2001; Clinckers et al., 2005; Segovia et al., 2006; Ballini et al., 2008) | (unpublished data) | |
| Prefrontal Cortex | 0.22-33 | 9-45.5 |
| (Moghaddam 1993; Timmerman et al., 1999; Calcagno et al., 2006; Hernandez et al., 2008; Ballini et al., 2008; Lupinsky et al., 2010) | (Rutherford et al., 2007; current study) | |
Figure 1.
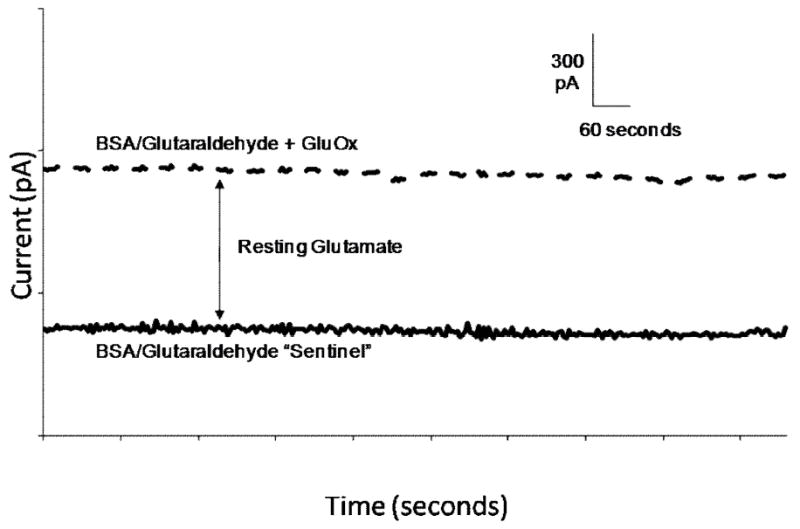
Determination of extracellular resting L-glutamate levels. Representative tracing of GluOx coated recording site (dashed line) and sentinel, or self-referencing site (solid line). The difference between the GluOx and sentinel baselines is an indication of resting glutamate.
3.2 Local Application of TTX
We first wanted to study the effects of a voltage-dependent sodium channel blocker, TTX, which has been previously shown to have little effects on resting glutamate levels in awake rats using microdialysis methods (Timmerman and Westerink, 1997). Local application of the TTX solution (3 μL, 100 μM) into the PFC was seen to produce a transient decrease in extracellular glutamate, which peaked after ~1 minute (Figure 2a). As seen in Figure 2b, local application of TTX significantly (p<0.01) decreased resting glutamate levels in the PFC of Long Evans rats by ~40% (-40.8 ± 10.8%, n=7) compared to citrate control ejections (1.6 ± 4.1%, n=5). These effects support a large neuronal component contributing to resting glutamate levels that was measured by the MEA recordings and not typically observed with microdialysis methods.
Figure 2.
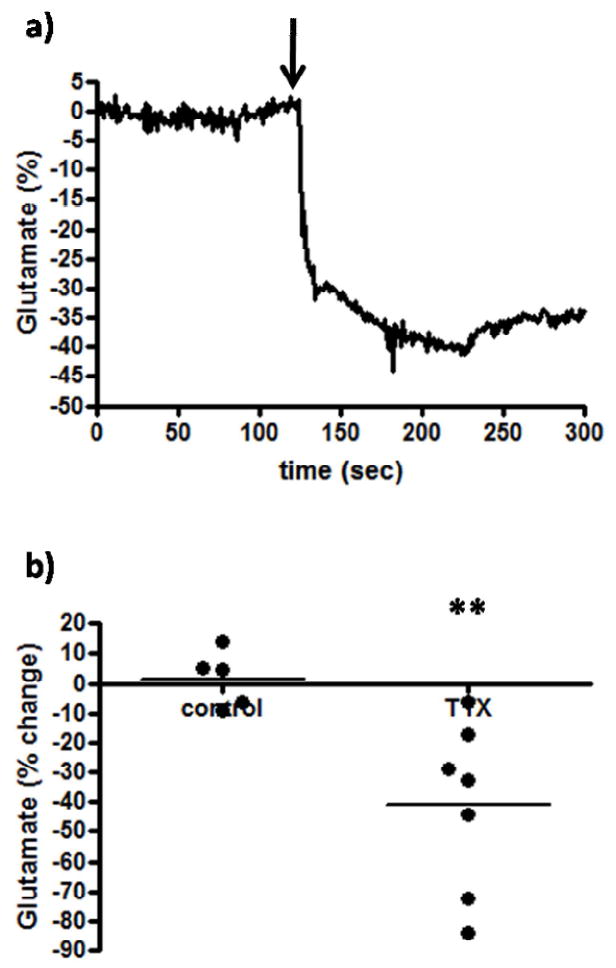
The effects of a sodium channel blocker on extracellular glutamate. After resting glutamate levels were established, TTX (n=7) or citrate control (n=5) significantly decreased (**p<0.01) extracellular glutamate levels. The arrow indicates local administration.
3.3 Local Application of ω-Conotoxin MVIIC
To further evaluate the neuronal contribution to extracellular resting glutamate levels, the N, P, and Q type calcium channel blocker, ω-conotoxin (MVIIC), was locally applied in the awake rats. Locally blocking the calcium channels prevents exocytotic release of neurotransmitters in the surrounding area. Similar to TTX, local application of MVIIC (1 μL, 3 μM) produced a rapid decline in extracellular glutamate levels, which peaked after ~60 seconds (Figure 3a). As seen in Figure 3, MVIIC produced a robust ~50% reduction in extracellular glutamate. We observed that the local application of the calcium channel blocker significantly (p<0.01) decreased (-49.5 ± 12.1 %, n=6) resting glutamate levels in the PFC of awake rats compared to saline control (-0.6 ± 5.8 %, n=5; Figure 3b), therefore supporting the hypothesis that the majority of extracellular glutamate was neuronal in origin. However, while robust, a faster rate of recovery to baseline was observed as compared to the effects of TTX.
Figure 3.
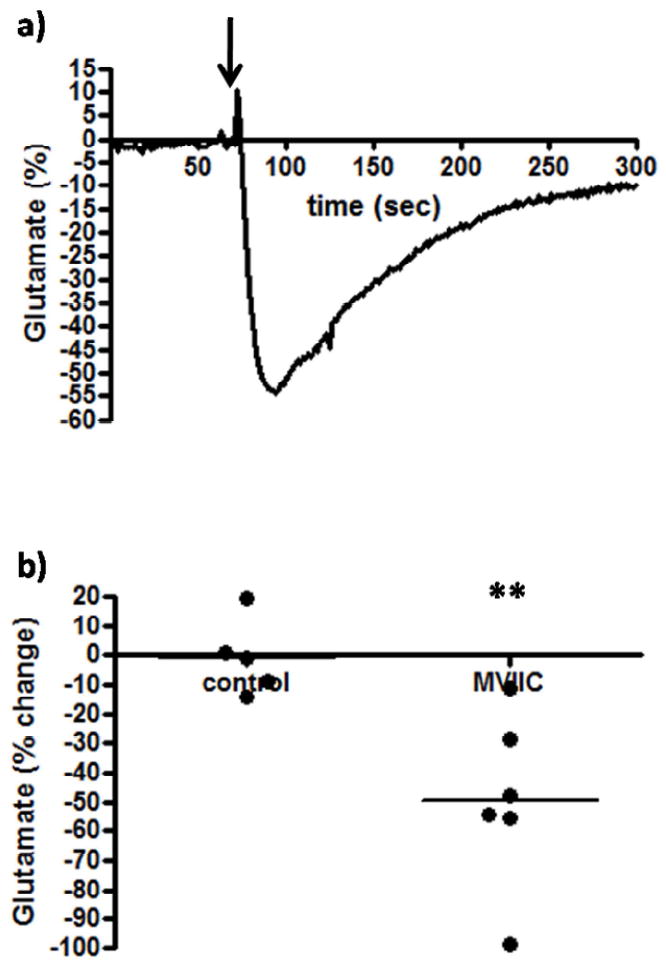
The effects of an N, P, and Q type calcium channel blocker on extracellular glutamate. After resting glutamate levels were established, MVIIC (n=6) or saline control (n=5) was locally applied. The arrow arrow indicates local administration. MVIIC significantly decreased (**p<0.01) extracellular glutamate levels.
3.4 Local Application of the Group II mGluR Agonist LY379268 and Antagonist LY341495
The effects of the group II mGluR agonist, LY379268, on resting glutamate levels were examined in the PFC of Long Evans rats. Group II mGluRs couple to Gi/Go adenylyl cyclase activity and act as inhibitory autoreceptors and were primarily located presynaptically/perisynaptically in the rat PFC (Schoepp 2001). As such, an mGluR group II agonist would be expected to decrease extracellular glutamate levels by inhibiting the release of glutamate into the synaptic cleft and conversely, the mGluR group II antagonist should increase extracellular glutamate levels by facilitating the release of glutamate. Although there was high variability in the controls (likely due to pressure artifact from the cannula placed too close, which would also decrease the effectiveness of the drug), local application of the mGluR group II agonist (LY379268; 3 μL, 100 μM) was seen to produce a small but significant decrease in extracellular glutamate levels in the PFC (Figure 4a). As seen in Figure 4b, LY379268 was seen to significantly (p<0.01) decrease resting glutamate levels by ~20 % as (-20.5 ± 7.8 %, n=7) compared to control (21.9 ± 10.9 %, n=6) ejections. The effects peaked after ~ 2 minutes and stayed constant for ~5 minutes. Local application of the mGluR group II antagonist (LY341495, 3 μL, 100 μM) produced a significant (p<0.05) increase (37.0 ± 12.6 %, n=4) in extracellular glutamate levels in the PFC compared to control (-6.3 ± 3.2 %, n=4) (Figure 5a,b). The effects peaked after ~ 10 seconds and returned to resting levels after ~ 1 minute.
Figure 4.
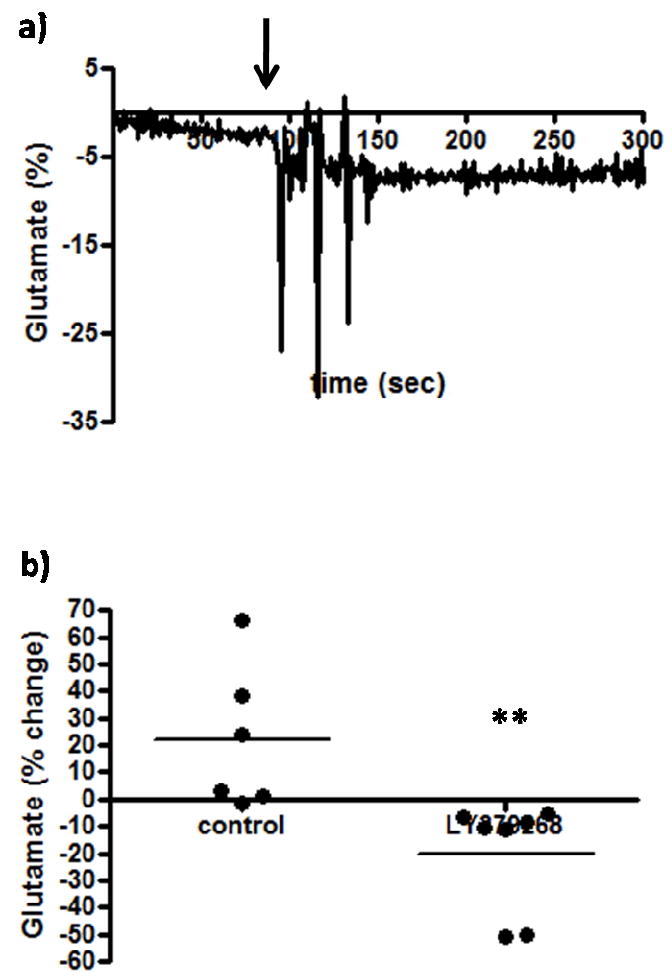
The effects of an mGluR group II agonist on extracellular glutamate. After resting glutamate levels were established, LY379268 (n=7) or physiological saline control (n=6) was locally applied. The arrow indicates local administration. A significant (**p<0.01) decrease was observed with LY379268.
Figure 5.
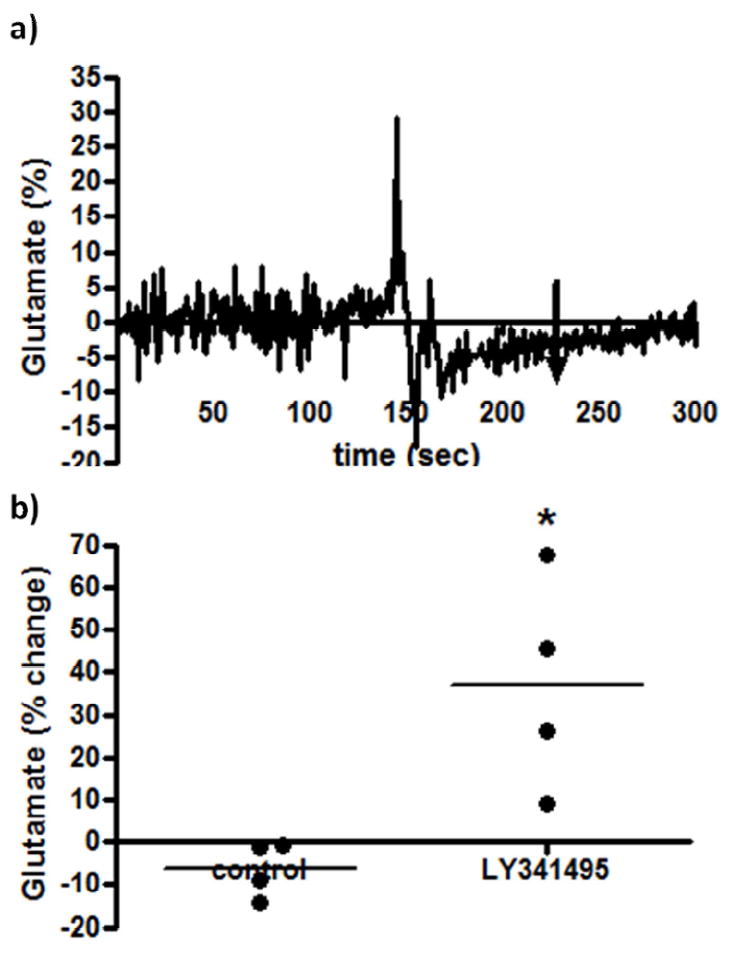
The effects of an mGluR group II antagonist on extracellular glutamate. After resting glutamate levels were established, LY341495 (n=4) or physiological saline control (n=4) was locally applied. The arrow indicates local administration. A significant (*p<0.05) increase was observed with LY341495.
3.5 Local Application of TBOA
In the PFC, glutamate was primarily cleared from the extracellular space through excitatory amino acid transporters (EAATs) located on post-synaptic neurons and glia (Danbolt, 2001). By locally inhibiting these EAATs with TBOA, a non-transportable EAAT inhibitor, we were able to determine if the resting extracellular glutamate measured was influenced by blocking transporter uptake. Local application of TBOA (1 μL, 100 μM) resulted in a significant (p<0.01) ~ 125% increase in glutamate levels (122.4 ± 26.8 %, n=5) compared to saline control (6.9 ± 3.5 %, n=5) (Figure 6a,b), which gradually returned to resting levels approximately 40 minutes following TBOA administration. This finding supports that EAATs play an important role in the regulation of resting extracellular glutamate levels and were required for normal glutamate homeostasis. Important to note is the decrease observed in the representative trace following the increase from TBOA application. This decrease was most likely due to a dilution artifact, as is common with local application and convection-enhanced delivery methods (Occhiogrosso et al., 2003, Burmeister and Gerhardt, 2001, Oldenziel et al., 2006, Hascup et al., 2008).
Figure 6.
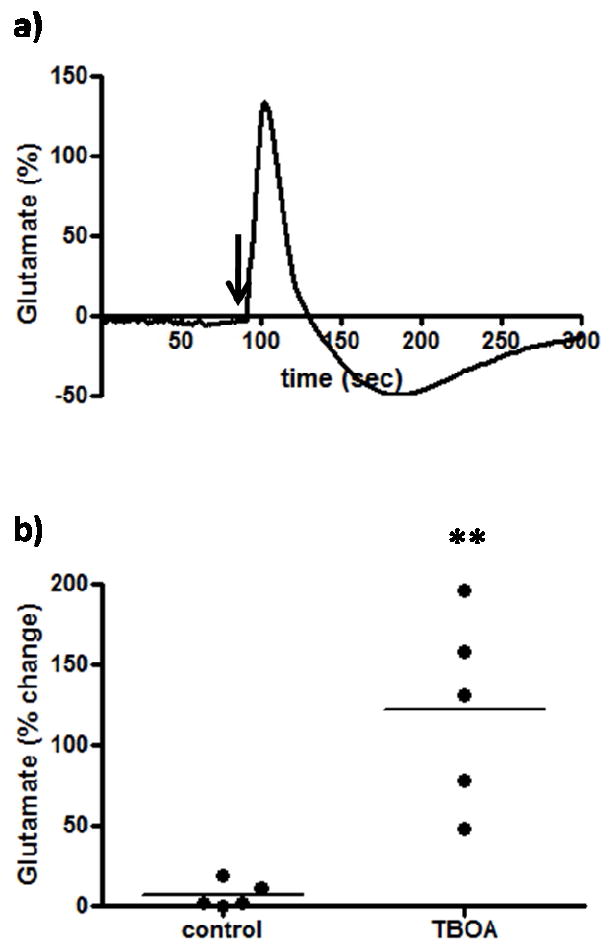
The effects of an uptake inhibitor on extracellular glutamate. After resting glutamate levels were established, TBOA (n=5) or physiological saline control (n=5) was locally applied. The arrow indicates local administration. TBOA significantly (**p<0.01) increased extracellular glutamate.
3.6 Local Application of CPG
The effects of the cystine-glutamate exchanger blocker, CPG, on extracellular glutamate levels were examined to determine the role of the cystine-glutamate exchanger in maintaining resting glutamate levels. Prior studies have shown evidence for possible alterations in the cystine-glutamate exchange that may help account for ~60% of the resting levels of glutamate that are detected by microdialysis (Kalivas, 2009). Upon local application of CPG (3 μL, 50 μM) in the PFC of awake Long Evans rats, we observed a small rapid, but not significant, increase in extracellular glutamate (20.7 ± 8.7 %, n=7) compared to saline controls (6.7 ± 5.8 %, n=7; Figure 7a). This effect of an initial glutamate increase lasted ~2 minutes. The initial increase in glutamate levels was followed by a prolonged decrease in glutamate levels (-6.8 ± 2.7 %, n=7) that was not different from saline controls (6.7 ± 5.8 %, n=7) and lasted approximately 10-15 minutes (Figure 7b). Based on these data, we conclude that the cystine-glutamate exchanger does not have a significant role in the regulation of extracellular resting glutamate that was sampled by the MEA recording technology.
Figure 7.
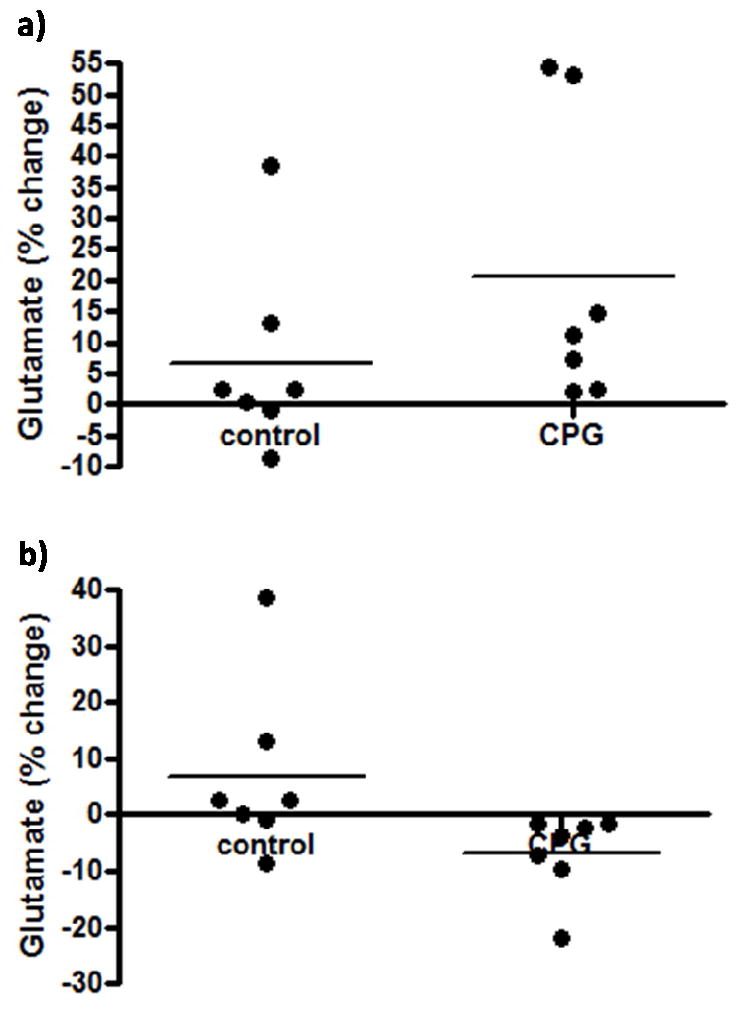
The effects of a cystine-glutamate exchanger blocker on extracellular glutamate. After resting glutamate levels were established, CPG (n=7) or physiological saline control (n=7) was locally applied. CPG non-significantly increased (a) and decreased (b) extracellular glutamate.
3.7 Local Application of TTX Followed by a Tail Pinch Stressor
Finally, we examined the effects of local application of TTX on a physiological event, a five minute tail pinch stress. Prior studies from our laboratory have shown that tail pinch stress, which was a commonly used stressor in awake rats, produces transient increases in extracellular glutamate in the PFC of Long Evans rats (Rutherford et al., 2007). Interestingly, local administration of TTX (3 μL, 100 μM) 60 seconds prior to a tail pinch was seen to completely attenuate tail pinch-induced glutamate release in the PFC of Long Evans rats (Figure 8). As shown in Figure 8, differences in the stress response were immediately apparent. We observed a significant decrease in the glutamate response to stress during minutes 3, 4, and 5 of the stressor and minutes 1-4 following the end of the stressor compared to controls. Interestingly, while local application of TTX was seen to decrease resting levels of glutamate by ~40%, we were able to completely block glutamate release produced by the tail pinch response. We conclude that glutamate release from a tail pinch stressor was due to exocytotic release from neurons, which was completely blocked with pre-treatment of TTX.
Figure 8.
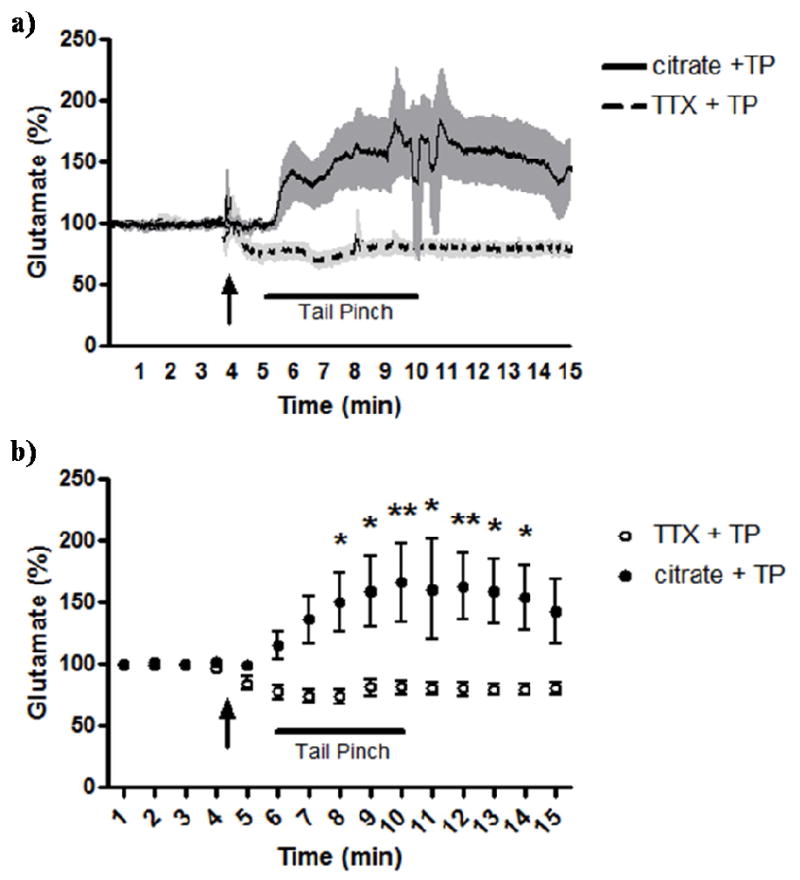
Effects of TTX on tail pinch stress-induced glutamate release. Local application of TTX decreased resting glutamate levels in the PFC and attenuated the glutamate response to stress compared to control. a) Traces showing second-by-second recording of glutamate with TTX administration (dashed line; light shade is SEM) or 0.6 mM citrate vehicle (solid line; shade is SEM) followed by a five minute tail pinch stressor. b) One minute averages of recordings of glutamate with TTX administration (open circle) or citrate vehicle (filled circle) followed by a five minute tail pinch stressor. The arrow indicates local TTX or citrate administration. Bar indicates five minute tail pinch stressor. *p<0.05, **p<0.01. n=6 for both groups.
4. Discussion
In these studies we used a self-referencing recording technique with enzyme-based MEAs that effectively and reliably measured resting glutamate levels to determine how resting glutamate levels were regulated in the PFC of awake Long Evans rats. We locally applied several compounds (individually), while simultaneously measuring glutamate on a second-by-second basis to gain a better understanding of the regulation of extracellular glutamate. We first studied the effects of TTX, which by some is the “gold standard” for studying the potential neuronal contribution of any neurotransmitter signal (Timmerman and Westerink, 1997). Local application of TTX into the rat PFC produced a robust and significant ~40% decrease in resting glutamate levels compared to control, supporting that resting glutamate has a major neuronal component. However, a previous study in our laboratory (Day et al., 2006) reported that local application of TTX completely diminished resting glutamate in the anesthetized rat. The current data supplemented with work in the anesthetized rat by Day et al. (2006) and in the awake mouse by Hascup et al. (2008) supports that while both the anesthetized and awake animal models exhibit resting glutamate levels that are at least in large part sodium channel-dependent as shown by the TTX data, anesthesia may block or inhibit additional pools of glutamate that contribute to resting levels resulting in diminished resting glutamate (Hara and Harris, 2002). This is further supported by the ~58% decrease in glutamate levels we observed in the rat following administration of urethane (Rutherford et al., 2007) and ~42% decrease observed following pentobarbital administration (Dash et al., 2009). This is in sharp contrasts to studies with microdialysis that consistently report that resting glutamate levels in the PFC are TTX-insensitive (Timmerman and Westerink, 1997; Lupinsky et al., 2010). Such discrepancies likely reflect inherent differences between the MEA technology and microdialysis. As discussed earlier, such discrepancies are most likely ascribed to the higher temporal (seconds vs. minutes) and spatial (μm vs. mm) resolution of the MEA compared to microdialysis. Because of this, the MEAs can sample glutamate closer to the synapses residing in the extracellular space with no diffusion barrier. This accounts for our higher levels of glutamate as summarized in Table I. The differences probably also reflect the apparent greater biocompatibility of MEAs over microdialysis probes; whereas substantial scar tissue typically surrounds microdialysis probes, our previous studies indicate that the microenvironment around the MEA is relatively free of such disruption (Hascup et al., 2009).
We next examined the contribution of neuronal glutamate release to extracellular resting glutamate levels by locally applying the N, P, and Q type calcium channel blocker, ω-conotoxin MVIIC, thus preventing exocytotic release. In contrast to recent microdialysis studies using Ca2+-free aCSF (Lupinsky et al., 2010), we observed a significant decrease (~50%) in resting glutamate levels compared to control. Again, the discrepancy between results may be inherent to the techniques themselves as well as the amount of damage associated with the technique. One possible explanation as to why we do not see complete depletion with locally applied MVIIC is because there are internalized neuronal Ca2+ stores (for example from mitochondria and endoplasmic reticulum) that can be recruited when the neuron is unable to import Ca2+ from the extracellular space, allowing for vesicular release via internal Ca2+ stores (Neher and Sakaba, 2008). While it is possible for Ca2+ to enter neurons through means other than Ca2+ channels, such as NMDA and nicotinic acetylcholine receptors (Mulle et al., 1992), recent studies support that it is also possible to mobilize internal stores of Ca2+ without Ca2+ influx (Ryglewski et al., 2007). Previous studies have indicated astrocytic calcium-dependent release of glutamate (Angulo et al., 2004; Shigetomi et al., 2008), however, more recent data from Agulhon and colleagues (2010) showed that neither increasing nor eliminating astrocytic calcium fluxes affects spontaneous or evoked glutamate transmission. Furthermore, given the observed similar effects of TTX and MVIIC (~40% vs. ~50% reduction in extracellular glutamate) we can conclude that resting glutamate is not derived from astrocytes and the associated discrete transient slow inward currents, which are thought to be sodium-independent and calcium-dependent (Parri et al., 2001; Fellin et al., 2004; Shigetomi et al., 2008). Taken together, these findings strongly support that a major contributor to extracellular glutamate levels is neuronal in origin.
Another possible regulatory mechanism contributing to resting glutamate levels involves group II mGluRs, which act as inhibitory autoreceptors. An mGluR group II agonist acting presynaptically would have an inhibitory effect on glutamate release into the synaptic cleft, while an antagonist would facilitate glutamate release. The mGluR group II agonist (LY379268) was shown to significantly (p<0.01) decrease resting glutamate levels by ~20% in the PFC of Long Evans rats compared to control, supporting a presynaptic regulation of tonic extracellular glutamate levels. There was high variability in the LY379268 control ejections, which was likely due to pressure artifact as a result of close placement of the cannula to the MEA tip. However, this would also result in a diminished response of LY379268 (because pressure artifact increases signal whereas LY379268 decreased glutamate). Furthermore, if the greatest outlier from the control ejections (~66%) and its corresponding LY379268 ejection are removed from the study, the significance remains. In further support, the mGluR group II antagonist (LY341495) significantly (p<0.05) increased extracellular glutamate levels by ~40% compared to control. As mentioned earlier, mGluR group II agonists have been shown to decrease extracellular GABA levels (Schaffhauser et al., 1998; Jones et al., 1998; Hanania and Johnson, 1999), which could result in increased glutamate release, however, LY379268 has not show this effect (Greenslade et al., 1999). Because of the ability of GABA to alter glutamate release, it is important to study the effects of GABAergic inhibitors on extracellular glutamate. These studies will be carried out in future studies of the extracellular regulation of glutamate.
Prior studies with microdialysis have shown evidence for regulation of extracellular glutamate by EAATs of which ~90% are localized to glia (Danbolt, 2001). To determine the contribution of glutamate transporter regulation on extracellular resting glutamate levels, TBOA was locally applied in the PFC of Long Evans rats. Glutamate levels significantly (p<0.01) increased by ~125% with local application of TBOA compared to saline control. These data support that EAATs play an important role in extracellular glutamate regulation by helping to maintain normal tonic glutamate levels. Day and colleagues (2006) observed similar findings with an increase of ~175% in glutamate levels with local TBOA application in the anesthetized rat. In addition, microdialysis studies have shown that TBOA will increase extracellular glutamate levels 4-9 fold (Montiel et al., 2005; Tovar-Y-Romo et al., 2009).
Another mechanism proposed to regulate resting glutamate levels is the cystine-glutamate exchanger (Baker et al., 2002; Lorrain et al., 2003; Melendez et al., 2005; Saul’skaya and Mikhailova, 2005; Kalivas, 2009). This antiporter is located mainly on glial cells and acts via uptake of cystine into the glia coupled with the release of glutamate into the extracellular space. CPG has been reported to be a cystine-glutamate exchanger blocker (Baker et al., 2002; Melendez et al., 2005), and as such, a decrease in extracellular glutamate would be expected when CPG was applied. In fact, Baker and colleagues (2002) reported that up to 60% of the glutamate sampled using microdialysis could be derived from the cystine-glutamate exchanger. CPG has also been reported as a competitive group I mGluR antagonist/weak group II mGluR agonist (Brabet et al., 1995; Doherty et al., 1999), which when acting presynaptically on glutamatergic neurons should induce a decrease in extracellular glutamate. Our studies support that local application of CPG in relatively high concentrations that were found to be the most effective by others (Baker et al., 2002) does not produce a significant change in extracellular levels of resting glutamate. In this regard, CPG alone did not produce significant changes in resting glutamate levels observed by Baker et al., 2002. This is further supported by a recent microdialysis study by Lupinsky and colleagues (2010), who also observed non-significant increases in resting glutamate with perfusion of CPG. One possible explanation for the discrepancy between the values previously obtained from microdialysis (~-60%; Baker et al., 2002) and those observed in this study (~-7%) with CPG application could be due to elevated astrocytes and microglia surrounding microdialysis probe implants compared to MEA implants (Hascup et al., 2009). This is also supported by previous studies that showed astrocytic proliferation is important for maintaining glutamate homeostasis following acute brain injuries (Bush et al., 1999; Buffo et al., 2008). Furthermore, it is thought that when astrocytes become reactive (as occurs with brain injury) they disregard their neuroprotective functions, therefore allowing neurons to become more susceptible to neurotoxins and reactive oxygen species (Barger et al., 2007; Fuller et al., 2009). One way that this may occur is by increasing the cystine-glutamate exchanger, thereby decreasing cytosolic astrocytic glutamate leading to decreased production of the primary antioxidant in the brain (glutathione) responsible for maintaining minimal levels of reactive nitrogen and oxygen species. Because glutathione is transferred to neurons to yield glutamate, this could further deplete the neuronal glutamate pool (Barger et al., 2007; Fuller et al., 2009). In short, the brain injury associated with microdialysis probes could by itself produce the effects observed with CPG by leading to increased astrocytes and activated astrocytes, which would in turn upregulate the cystine-glutamate exchanger, increasing extracellular glutamate levels, while decreasing astrocytic glutathione and neuronal pools of glutamate.
Finally, we wanted to determine the source of glutamate from a physiological event-induced change in extracellular glutamate. TTX was locally applied in the PFC and we again observed a decrease in resting glutamate levels. We then carried out a five minute tail pinch stressor 60 seconds after the TTX ejection. While we were not able to reliably show comprehensive depletion of tonic glutamate with local TTX administration, we did observe complete attenuation in the glutamate response to tail pinch stress when the rats were pre-administered TTX but not with pre-administration of citrate control, supporting that physiologically evoked glutamate release is entirely neuronal in origin. This attenuation of the glutamatergic stress response was also shown by Moghaddam (1993) following a 40 minute infusion of TTX and by Lupinsky and colleagues (2010) with continual TTX perfusion, however, both microdialysis studies were unable to alter resting glutamate levels with TTX administration.
Interestingly, while TTX and MVIIC decreased glutamate levels by 90% or greater in some animals, in others these treatments were considerably less effective (as low as 7% and 10% decreases, repectively), implying that the resting glutamate may be completely derived from neurons. This discrepancy is likely due to a limitation of the cannula delivery method. The cannula could shift slightly during implantation, resulting in applications that are slightly closer or further from the MEA that what was intended. This is further supported by the complete block of tail pinch-induced glutamate release. This is likely because the method of local drug delivery in the present studies is not ideal. The cannulation method is contingent on exact placement of the guide cannula and injection cannula. While we try to accurately place the guide cannula, it is possible that it can move slightly during implantation. Additionally, the injection cannula protrudes 1 mm past the guide cannula and may slightly bend or warp during insertion, thereby making the injection closer or further away from the intended injection site. Furthermore, the drugs were ejected in a single bolus rather than perfused over time, which could also lead to more drastic changes in resting glutamate levels. Thus, the drug delivery methods could contribute to the variance of results and the amount of inhibition of resting glutamate that can be achieved. The drug delivery system used in microdialysis studies (perfusion) addresses most of these issues and has some distinct advantages over the cannulation system we employed. However, the amount of tissue damage caused by the microdialysis probe itself, potential dilution of neurotransmitters by prolonged perfusion, as well as TTX-independent resting glutamate levels, supports a need for a better delivery system. There is also some evidence for interhemispheric glutamatergic communication via callosal neurons between left and right PFC (Lupinsky et al., 2010), which may contribute to our inability to completely deplete glutamate levels.
Taken together, the data presented here support tight regulation of extracellular glutamate in the PFC of awake male Long Evans rats. We also determined that the basal glutamate pool sampled using with the MEA technology is at least 40-50% neuronally derived. There are many factors located throughout the synapse that may influence glutamate levels, with the biggest differences observed in this study due to local blockage of calcium channels and sodium channels, and the local manipulation of group II mGluRs. These results support that resting glutamate levels in the PFC of awake Long Evans rats are largely neuronally derived, which has been controversial when determined using other glutamate sampling techniques such as microdialysis. It is likely that the MEA technology is detecting neuronally-derived glutamate due to the decreased damage produced by the electrodes and the ability to sample glutamate levels more rapidly and closer to the synapse.
Acknowledgments
This work was supported by the Canadian Institutes for Health Research, Natural Sciences and Engineering Research Council, NSF EEC-0310723, and NIH CEBRA grant DA017186. Greg A. Gerhardt is sole proprietor of Quanteon, LLC.
Abbreviations used
- MEA
microelectrode array
- GluOx
L-glutamate oxidase
- FAST 16
fast analytical sensing technology
- TTX
tetrodotoxin
- PBS
phosphate buffered saline
- TBOA
D,L-threo-β-benzyloxyaspartate
- CPG
(S)-4-carboxyphenylglycine
References
- Agulhon C, Fiacco TA, McCarthy KD. Hippocampal short- and long-term plasticity are not modulated by astrocyte Ca2+ signaling. Science. 2010;237(5970):1250–1254. doi: 10.1126/science.1184821. [DOI] [PubMed] [Google Scholar]
- Albrecht D, Davidowa H. Action of Urethane on dorsal lateral geniculate neurons. Brain Res Bull. 1989;22:923–927. doi: 10.1016/0361-9230(89)90001-4. [DOI] [PubMed] [Google Scholar]
- Angulo MC, Kozlov AS, Charpak S, Audinat E. Glutamate released from glial cells synchronizes neuronal activity in the hippocampus. J Neurosci. 2004;24(31):6920–6927. doi: 10.1523/JNEUROSCI.0473-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagley J, Moghaddam B. Temporal dynamics of glutamate efflux in the prefrontal cortex and in the hippocampus following repeated stress: effects of pretreatment with saline or diazepam. Neurosci. 1997;77(1):65–73. doi: 10.1016/s0306-4522(96)00435-6. [DOI] [PubMed] [Google Scholar]
- Baker DA, Xi ZX, Shen H, Swanson CJ, Kalivas PW. The orgin and neuronal function of in vivo nonsynaptic glutamate. J Neurosci. 2002;22(20):9134–9141. doi: 10.1523/JNEUROSCI.22-20-09134.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballini C, Corte LD, Pazzagli M, Colivicchi MA, Pepeu G, Tipton KF, Giovannini MG. Extracellular levels of brain aspartate, glutamate and GABA during an inhibitory avoidance response in the rat. J Neurochem. 2008;106(3):1035–1043. doi: 10.1111/j.1471-4159.2008.05452.x. [DOI] [PubMed] [Google Scholar]
- Barger SW, Goodwin ME, Porter MM, Beggs ML. Glutamate release from activated microglia requires the oxidative burst and lipid peroxidation. J Neurochem. 2007;101(5):1205–1213. doi: 10.1111/j.1471-4159.2007.04487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battaglia G, Monn JA, Schoepp DD. In vivo inhibition of veratridine-evoked release of striatal excitatory amino acids by the group II metabotropic glutamate receptor agonist LY354740 in rats. Neurosci Lett. 1997;229(3):161–164. doi: 10.1016/s0304-3940(97)00442-4. [DOI] [PubMed] [Google Scholar]
- Belay A, Collins A, Ruzgas T, Kissinger PT, Gorton L, Csöregi E. Redox hydrogel based bienzyme electrode for L-glutamate monitoring. J Pharmaceutical and Biomedical Analysis. 1998;19(1-2):93–105. doi: 10.1016/s0731-7085(98)00199-x. [DOI] [PubMed] [Google Scholar]
- Biggs CS, Fowler LJ, Whitton PS, Starr MS. Extracellular levels of glutamate and aspartate in the entopeduncular nucleus of the rat determined by microdialysis: regulation by striatal dopamine D2 receptors via the indirect striatal output pathway? Brain Res. 1997;753(1):163–175. doi: 10.1016/s0006-8993(97)00033-4. [DOI] [PubMed] [Google Scholar]
- Boatell ML, Bendahan G, Mahy N. Time-related cortical amino acid changes after basal forebrain lesion: a microdialysis study. J Neurochem. 1995;64:285–291. doi: 10.1046/j.1471-4159.1995.64010285.x. [DOI] [PubMed] [Google Scholar]
- Brabet I, Mary S, Bockaert J, Pin J-P. Phenylglycine derivatives discriminate between mGluR1- and mGluR5-mediated responses. Neuropharm. 1995;34(8):895–903. doi: 10.1016/0028-3908(95)00079-l. [DOI] [PubMed] [Google Scholar]
- Buffo A, Rite I, Tripathi P, Lepier A, Colak D, et al. Origen and progeny of reactive gliosis: a source of multipotent cells in the injured brain. Proc Natl Acad Sci USA. 2008;105:3581–3586. doi: 10.1073/pnas.0709002105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burmeister JJ, Moxon K, Gerhardt GA. Ceramic-based multisite microelectrodes for electrochemical recordings. Anal Chem. 2000;72:187–192. doi: 10.1021/ac9907991. [DOI] [PubMed] [Google Scholar]
- Burmeister JJ, Gerhardt GA. Self-referencing ceramic-based multisite microelectrodes for the detection and elimination of interferences from the measurement of L-glutamate and other analytes. Anal Chem. 2001;73:1037–1042. doi: 10.1021/ac0010429. [DOI] [PubMed] [Google Scholar]
- Burmeister JJ, Pomerleau F, Palmer M, Day BK, Huettl P, Gerhardt GA. Improved ceramic-based multisite microelectrode for rapid measurements of L-glutamate in the CNS. J Neurosci Methods. 2002;119:163–171. doi: 10.1016/s0165-0270(02)00172-3. [DOI] [PubMed] [Google Scholar]
- Burmeister JJ, Gerhardt GA. Ceramic-based multisite microelectrode arrays for in vivo electrochemical recordings of glutamate and other neurochemicals. Trends in Anal Chem. 2003;22(8):498–502. [Google Scholar]
- Bush TG, Puvanachandra N, Horner CH, Polito A, Ostenfeld T, et al. Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron. 1999;23:297–308. doi: 10.1016/s0896-6273(00)80781-3. [DOI] [PubMed] [Google Scholar]
- Calcagno E, Carli M, Invernizzi RW. The 5-HT(1A) receptor agonist 8-OH-DPAT prevents prefrontocortical glutamate and serotonin release in response to blockade of cortical NMDA receptors. J Neurochem. 2006;96(3):853–860. doi: 10.1111/j.1471-4159.2005.03600.x. [DOI] [PubMed] [Google Scholar]
- Carlsson M, Carlsson A. Interaction between glutamatergic and monoaminergic systems within the basal ganglia – implications for schizophrenia and Parkinson’s disease. Trends Neurosci. 1990;13:272–276. doi: 10.1016/0166-2236(90)90108-m. [DOI] [PubMed] [Google Scholar]
- Clapp-Lilly KL, Roberts RC, Duffy LK, Irons KP, Hu Y, Drew KL. An ultrastructural analysis of tissue surrounding a microdialysis probe. J Neurosci Methods. 1999;90:129–142. doi: 10.1016/s0165-0270(99)00064-3. [DOI] [PubMed] [Google Scholar]
- Clinckers R, Gheuens S, Smolders I, Meurs A, Ebinger G, Michotte Y. In vivo modulatory action of extracellular glutamate on the anticonvulsant effects of hippocampal dopamine and serotonin. Epilepsia. 2005;46(6):828–836. doi: 10.1111/j.1528-1167.2005.57004.x. [DOI] [PubMed] [Google Scholar]
- Dash MB, Douglas CL, Vyazovskiy VV, Cirelli C, Tononi G. Long-term homeostasis of extracellular glutamate in the rat cerebral cortex across sleep and waking states. J Neurosci. 2009;29(3):620–629. doi: 10.1523/JNEUROSCI.5486-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day BK, Pomerleau F, Burmeister JJ, Huettl PF, Gerhardt GA. Microelectrode array studies of basal and potassium-evoked release of L-glutamate in the anesthetized rat brain. J Neurochem. 2006;96(6):1626–1635. doi: 10.1111/j.1471-4159.2006.03673.x. [DOI] [PubMed] [Google Scholar]
- Del Arco A, Castaneda TR, Mora F. Amphetamine releases GABA in striatum of the freely moving rat: involvement of calcium and high affinity transporter mechanisms. Nueropharmacology. 1998;37:199–205. doi: 10.1016/s0028-3908(98)00013-6. [DOI] [PubMed] [Google Scholar]
- Del Arco A, González-Mora JL, Armas VR, Mora F. Amphetamine increases extracellular concentrations of glutamate in striatum of the awake rat: involvement of high affinity transporter mechanisms. Neuropharmacology. 1999;38:943–954. doi: 10.1016/s0028-3908(99)00043-x. [DOI] [PubMed] [Google Scholar]
- Doherty AJ, Collingridge GL, Jane DE. Antagonist activity of α–substituted 4-carboxyphenylglycine analogues at group I metabotropic glutamate receptors expressed in CHO cells. Brit J Pharm. 1999;126:205–210. doi: 10.1038/sj.bjp.0702297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew KL, Pehek EA, Rasley BT, Ma YL, Green TK. Sampling glutamate and GABA with microdialysis: suggestions on how to get the dialysis membrane closer to the synapse. J Neurosci Meth. 2004;140:127–131. doi: 10.1016/j.jneumeth.2004.04.039. [DOI] [PubMed] [Google Scholar]
- Dyer RS, Rigdon GC. Urethane affects the rat visual system at subanesthetic doses. Physiol Behav. 1987;41:327–330. doi: 10.1016/0031-9384(87)90396-9. [DOI] [PubMed] [Google Scholar]
- Fellin T, Pascual O, Gobbo S, Pozzan T, Haydon PG, Carmignoto G. Neuronal Synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron. 2004;43:729–743. doi: 10.1016/j.neuron.2004.08.011. [DOI] [PubMed] [Google Scholar]
- Fuller S, Steele M, Münch G. Activated astroglia during chronic inflammation in Alzheimer’s disease-Do they neglect their neurosupportive roles? Mutat Res: Fundam Mol Mech Mutagen (2010) 2009 doi: 10.1016/j.mrfmmm.2009.08.016. [DOI] [PubMed] [Google Scholar]
- Giovannini MG, Rakovska A, Benton RS, Pazzagli M, Bianchi L, Pepeu G. Effects of novelty and habituation on acetylcholine, GABA, and glutamate release from the frontal cortex and hippocampus of freely moving rats. Neurosci. 2001;106(1):43–53. doi: 10.1016/s0306-4522(01)00266-4. [DOI] [PubMed] [Google Scholar]
- Girman SV, Suave Y, Lund RD. Receptive field properties of single neurons in rat primary visual cortex. J Neurophysiol. 1999;82:301–311. doi: 10.1152/jn.1999.82.1.301. [DOI] [PubMed] [Google Scholar]
- Grace AA. Phasic versus tonic dopamine release and the modulation of dopamine system responsivity: a hypothesis for the etiology of schizophrenia. Neuroscience. 1991;41:1–24. doi: 10.1016/0306-4522(91)90196-u. [DOI] [PubMed] [Google Scholar]
- Greenamyre JT. Glutamate – dopamine interactions in the basal ganglia: relationship to Parkinson’s disease. J Neural Transm. 1993;91:255–269. doi: 10.1007/BF01245235. [DOI] [PubMed] [Google Scholar]
- Greenslade RG, Woodhouse S, O’Neill MJ, Bond A, Ward MA, Mitchell SN. Is there a role for GABA in the neuroprotective effects of LY379268, a group II mGluR agonist, in global cerebral ischemia?. Proceedings of the 8th International Conference on In Vivo Method; June 19-23, 1999; New York, NY, USA. 1999. [Google Scholar]
- Hanania T, Johnson KM. Resulation of NMDA-stimulated [14C]GABA and [3H]acetylcholine release by striatal glutamate and dopamine receptors. Brain Res. 1999;844:106–117. doi: 10.1016/s0006-8993(99)01869-7. [DOI] [PubMed] [Google Scholar]
- Hara K, Harris RA. The anesthetic mechanism of urethane: the effects on neurotransmitter-gated ion channels. Anesth Analg. 2002;94:313–318. doi: 10.1097/00000539-200202000-00015. [DOI] [PubMed] [Google Scholar]
- Hascup ER, af Bjerkén S, Hascup KN, Pomerleau F, Huettl P, Strömberg I, Gerhardt GA. Histological studies of the effects of chronic implantation of ceramic-based microelectrode arrays and microdialysis probes in rat prefrontal cortex. Brain Res. 2009;1291:12–20. doi: 10.1016/j.brainres.2009.06.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hascup KN, Rutherford EC, Quintero JE, Day BK, Nickell JR, Pomerleau F, Huettl P, Burmeister JJ, Gerhardt GA. Second-by-Second Measures of L-glutamate and Other Neurotransmitters Using Enzyme-Based Microelectrode Arrays. Chapter 19 of Electrochemical Methods for Neuroscience. 2006 [PubMed] [Google Scholar]
- Hascup KN, Hascup ER, Pomerleau F, Huettl P, Gerhardt GA. Second-by-Second Measures of L-Glutamate in the Prefrontal Cortex and Striatum of Freely Moving Mice. J Pharmacol Exp Ther. 2008;324(2):725–731. doi: 10.1124/jpet.107.131698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández LF, Segovia G, Mora F. Chronic treatment with a dopamine uptake blocker changes dopamine and acetylcholine but not glutamate and GABA concentrations in prefrontal cortex, striatum and nucleus accumbens of the awake rat. Neurochem Int. 2008;52(3):457–69. doi: 10.1016/j.neuint.2007.08.005. [DOI] [PubMed] [Google Scholar]
- Jones NM, Monn JA, Beart PM. type I and II metabotropic glutamate receptors regulate the outflow of [3H]D-aspartate and [14C]γ-aminobutyric acid in rat solitary nucleus. Eur J Pharmacol. 1998;353:43–51. doi: 10.1016/s0014-2999(98)00394-x. [DOI] [PubMed] [Google Scholar]
- Kalivas PW. The glutamate homeostasis hypothesis of addiction. Nat Rev Neurosci. 2009;10(8):561–572. doi: 10.1038/nrn2515. [DOI] [PubMed] [Google Scholar]
- Kulagina NV, Shankar L, Michael AC. Monitoring glutamate and ascorbate in the extracellular space of brain tissue with electrochemical microsensors. Anal Chem. 1999;71:5093–5100. doi: 10.1021/ac990636c. [DOI] [PubMed] [Google Scholar]
- Liachenko S, Tang P, Somogyi GT, Xu Y. Comparison of anaesthetic and non-anaesthetic effects on depolarization-evoked glutamate and GABA release from mouse cerebrocortical slices. Brit J of Pharm. 1998;123:1274. doi: 10.1038/sj.bjp.0701728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liachenko S, Tang P, Somogyi GT, Xu Y. concentration-dependent isoflurane effects on depolarization-evoked glutamate and GABA outflows from mouse brain slices. Brit J of Pharm. 1999;127:131. doi: 10.1038/sj.bjp.0702543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Mereno JA, González-Cuevas G, Mereno G, Navarro M. The pharmacology of the endocannabinoid system: functional and structural interactions with other neurotransmitter systems and their repercussions in behavioral addiction. Addict Biol. 2008;13(2):160–187. doi: 10.1111/j.1369-1600.2008.00105.x. [DOI] [PubMed] [Google Scholar]
- Lorrain DS, Baccei CS, Bristow LJ, Anderson JJ, Varney MA. Effects of ketamine and N-methyl-D-aspartate on glutamate and dopamine release in the rat prefrontal cortex: Modulation by a group II selective metabotropic glutamate receptor agonist LY379268. Neurosci. 2003;117:697–706. doi: 10.1016/s0306-4522(02)00652-8. [DOI] [PubMed] [Google Scholar]
- Lowry JP, Ryan MR, O’Neill RD. Behaviourally induced changes in extracellular levels of brain glutamate monitored at 1s resolution with an implanted biosensor. Anal Commun. 1998;35:87–89. [Google Scholar]
- Lupinsky D, Moquin L, Gratton A. Interhemispheric regulation of the medial prefrontal cortical glutamate stress response in rats. J Neurosci. 2010;30(22):7624–7633. doi: 10.1523/JNEUROSCI.1187-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew SJ, Price RB, Charney DS. Recent advances in the neurobiology of anxiety disorders: implications for novel therapeutics. Am J Med Genet C Semin Med Genet. 2008;148(2):89–98. doi: 10.1002/ajmg.c.30172. [DOI] [PubMed] [Google Scholar]
- McNally L, Bhagwager Z, Hannestad J. Inflammation, glutamate, and glia in depression: a literature review. CNS Spectr. 2008;13(6):501–510. doi: 10.1017/s1092852900016734. [DOI] [PubMed] [Google Scholar]
- Mechri A, Saoud M, Khiari G, d’Amato T, Dalery J, Gaha L. Glutamatergic hypothesis of schizophrenia: clinical research studies with ketamine. Encephale. 2001;27(1):53–59. [PubMed] [Google Scholar]
- Melendez RI, Vuthiganon J, Kalivas PW. Regulation of extracellular glutamate in the prefrontal cortex: focus on the cystine glutamatee exchanger and group I metabotropic glutamate receptors. J Pharm and Exp Therapeutics. 2005;314(1):139–147. doi: 10.1124/jpet.104.081521. [DOI] [PubMed] [Google Scholar]
- Moghaddam B. Stress preferentially increases extraneuronal levels of excitatory amino acids in the prefrontal cortex: comparison to hippocampus and basal ganglia. J Neurochem. 1993;60:1650–1657. doi: 10.1111/j.1471-4159.1993.tb13387.x. [DOI] [PubMed] [Google Scholar]
- Montiel T, Camacho A, Estrada-Sanchez AM, Massieu L. Differential effects of the substrate inhibitor L-trans-pyrrolidine-2,4-dicarboxylate (PDC) and the non-substrate inhibitor DL-threo-β-benzyloxyaspartate (DL-TBOA) of glutamate transporters on neuronal damage and extracellular amino acid levels in rat brain in vivo. Neurosci. 2005;133:667–678. doi: 10.1016/j.neuroscience.2004.11.020. [DOI] [PubMed] [Google Scholar]
- Mulle C, Choquet D, Korn H, Changeux JP. Calcium influx through nicotinic receptor in rat central neurons: its relevance to cellular regulation. Neuron. 1992;8(1):135–143. doi: 10.1016/0896-6273(92)90115-t. [DOI] [PubMed] [Google Scholar]
- Neher E, Sakaba T. Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron. 2008;59:861–872. doi: 10.1016/j.neuron.2008.08.019. [DOI] [PubMed] [Google Scholar]
- Nickell J, Pomerleau F, Allen J, Gerhardt GA. Age-related changes in the dynamics of potassium-evoked L-glutamate release in the striatum of Fischer 344 rats. J Neural Transm. 2005;112:87–96. doi: 10.1007/s00702-004-0151-x. [DOI] [PubMed] [Google Scholar]
- Occhiogrosso G, Edgar MA, Sandberg DI, Souweidane MM. Prolonged convection-enhanced delivery into the rat brainstem. Neurosurgery. 2003;52:388–394. doi: 10.1227/01.neu.0000043696.83722.8d. [DOI] [PubMed] [Google Scholar]
- Oldenziel WH, Dijkstra G, Cremers TIFH, Westerink BHC. In vivo monitoring of extracellular glutamate in the brain with a microsensor. Brain Res. 2006;1118:34–42. doi: 10.1016/j.brainres.2006.08.015. [DOI] [PubMed] [Google Scholar]
- Parri HR, Gould TM, Crunelli V. Spontaneous astrocytic Ca2+ oscillations in situ drive NMDAR-mediated neuronal excitation. Nat Neurosci. 2001;4:803–812. doi: 10.1038/90507. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain: stereotaxic coordinates. Academic Press; New York: 1998. [Google Scholar]
- Peacey E, Miller CC, Dunlop J, Rattray M. The four major N- and C-terminal splice variants of the excitatory amino acid transporter GLT-1 form cell surface homomeric and heteromeric assemblies. Mol Pharmacol. 75(5):1062–1073. doi: 10.1124/mol.108.052829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerleau F, Day BK, Huettl P, Burmeister JJ, Gerhardt GA. Real time in vivo measures of L-glutamate in the rat central nervous system using ceramic-based multisite microelectrode arrays. Ann N Y Acad Sci. 2003;1003:454–457. doi: 10.1196/annals.1300.051. [DOI] [PubMed] [Google Scholar]
- Rocha L, Briones M, Ackermann RF, Anton B, Maidment NT, Evans CJ, Engel J., Jr Pentylenetetrazol-induced kindling: early involvement of excitatory and inhibitory systems. Epilepsy Res. 1996;26:105–113. doi: 10.1016/s0920-1211(96)00046-0. [DOI] [PubMed] [Google Scholar]
- Rutherford EC, Pomerleau F, Huettl P, Strömberg I, Gerhardt GA. Chronic second-by-second measures of L-glutamate in the CNS of freely moving rats. J Neurochem. 2007;102(3):712–722. doi: 10.1111/j.1471-4159.2007.04596.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryglewski S, Pflueger HJ, Duch C. Expanding the neuron’s calcium signaling repertoire: intracellular calcium release via voltage-induced PLC and IP3R activation. PloS Biol. 2007;5(4):e66. doi: 10.1371/journal.pbio.0050066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saul’skaya NB, Mikhailove MO. Vesicular and non-vesicular glutamate release in the nucleus accumbens in conditions of a forced change of behavioral strategy. Neurosci And Behav Phys. 2005;35(7):677–683. doi: 10.1007/s11055-005-0110-5. [DOI] [PubMed] [Google Scholar]
- Sceniak MP, MacIver MB. Cellular actions of urethane on rat visual cortical neurons in vitro. J Neurophysiol. 2006;95:3865–3874. doi: 10.1152/jn.01196.2005. [DOI] [PubMed] [Google Scholar]
- Schaffhauser H, Knoflach K, Pink JR, Bleuel Z, Cartmell J, Geopfert F, Kemp JA, Richards JG, Adam G, Mutel V. Mutiple pathways for regulation of the KCl-induced [3H]GABA release by metabotropic glutamate receptors, in primary rat cortical cultures. Brain Res. 1998;782:91–104. doi: 10.1016/s0006-8993(97)01271-7. [DOI] [PubMed] [Google Scholar]
- Schoepp DD. Unveiling the functions of presynaptic metabotropic glutamate receptors in the central nervous system. J Pharm and Exp Therapeutics. 2001;299(1):12–20. [PubMed] [Google Scholar]
- Segovia G, Del Arco A, Prieto L, Mora F. Glutamate-glutamine cycle and aging in striatum of the awake rat: effects of a glutamate transporter blocker. Neurochem Res. 2001;26(1):37–41. doi: 10.1023/a:1007624531077. [DOI] [PubMed] [Google Scholar]
- Segovia G, Yagüe AG, García-Verdugo JM, Mora F. Environmental enrichment promotes neurogenesis and changes the extracellular concentrations of glutamate and GABA in the hippocampus of aged rats. Brain Res Bull. 2006;70(1):8–14. doi: 10.1016/j.brainresbull.2005.11.005. [DOI] [PubMed] [Google Scholar]
- Shigetomi E, Bowser DN, Sofroniew MV, Khakh BS. Two forms of astrocyte calcium excitability have distinct effects on NMDA receptor-mediated slow inward currents in pyramidal neurons. J Neurosci. 2008;28:6659–6663. doi: 10.1523/JNEUROSCI.1717-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmerman W, Westerink BHC. Brain microdialysis of GABA and glutamate: What does it signify? Synapse. 1997;27:242–261. doi: 10.1002/(SICI)1098-2396(199711)27:3<242::AID-SYN9>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Timmerman W, Cisci G, Nap A, de Vries JB, Westerink BH. Effects of handling on extracellular levels of glutamate and other amino acids in various areas of the brain measured by microdialysis. Brain Res. 1999;833(2):150–160. doi: 10.1016/s0006-8993(99)01538-3. [DOI] [PubMed] [Google Scholar]
- Tovar-Y-Romo LB, Santa-Cruz LD, Zepeda A, Tapia R. Chronic elevation of extracellular glutamate due to transport blockade is innocuous for spinal motoneurons in vivo. Neurochem Int. 2009;54(3-4):186–191. doi: 10.1016/j.neuint.2008.09.015. [DOI] [PubMed] [Google Scholar]
- Ueda Y, Tsuru N. Simultaneous monitoring of the seizure-related changes in extracellular glutamate and gamma-aminobutyric acid concentration in bilateral hippocampi following development of amygdaloid kindling. Epilepsy Res. 1995;20(3):213–219. doi: 10.1016/0920-1211(94)00081-7. [DOI] [PubMed] [Google Scholar]
- Wassum KM, Tolosa VM, Wang J, Walker E, Monbouquette HG, Maidment NT. Silicon Wafer-Based Platinum Microelectrode Array Biosensor for Near Real-Time Measurement of Glutamate in Vivo. Sensors Basel Sensors. 2008;8(8):5023–5036. doi: 10.3390/s8085023. [DOI] [PMC free article] [PubMed] [Google Scholar]