

Abstract
Epoxyeicosatrienoic acids (EETs) are bioactive eicosanoids produced from arachidonic acid by cytochrome P450 epoxygenases. We previously described the expression of CYP-2J epoxygenase in rat trigeminal ganglion neurons and that EETs signaling is involved in cerebrovascular dilation resulting from perivascular nerve stimulation. Herein we evaluate the presence of the EETs signaling pathway in trigeminal ganglion neurons and their role in modulating the release of calcitonin gene-related peptide (CGRP) by trigeminal ganglion neurons. Liquid chromatography tandem mass spectrometry identified the presence of each of the four EETs regio-isomers within primary trigeminal ganglion neurons. Stimulation for one hour with the transient receptor potential vanilloid-1 channel agonist capsaicin (100 nmol/L) or depolarizing K+ (60 mmol/L) increased CGRP release as measured by ELISA. Stimulation-evoked CGRP release was attenuated by 30 min pre-treatment with the EETs antagonist 14,15-epoxyeicosa-5(Z)-enoic acid (14,15-EEZE, 10 μmol/L). K+ stimulation elevated CGRP release 2.9 ± 0.3-fold above control levels, while in the presence of 14,15-EEZE K+-evoked CGRP release was significantly reduced to 1.1 ± 0.2-fold above control release (p<0.01 ANOVA, n=6). 14,15-EEZE likewise attenuated capsaicin-evoked CGRP release from trigeminal ganglion neurons (p<0.05 ANOVA, n=6). Similarly, pre-treatment with the CYP epoxygenase inhibitor attenuated stimulation-evoked CGRP release. These data demonstrate that EETs are endogenous constituents of rat trigeminal ganglion neurons and suggest that they may act as intracellular regulators of neuropeptide release, which may have important clinical implications for treatment of migraine, stroke and vasospasm after subarachnoid hemorrhage.
Keywords: cytochrome P450 epoxygenase, epoxyeicosatrienoic acid, perivascular nerves, CGRP, substance P, TRPV1
Introduction
Neurons of the trigeminal ganglia provide the sensory innervation of the head and face, in addition to contributing to the perivascular nerve supply of the meningeal and cerebral surface vasculature (Edvinsson & Uddman 2005). Stimulation of these perivascular trigeminal afferents results in both peripheral and central release of neuropeptide transmitters, including calcitonin gene-related peptide (CGRP) and substance P (SP), that mediate peripheral vasodilation and inflammation as well as central pain transmission (Edvinsson & Uddman 2005). It is the activation and sensitization of these fibers that underlies the dural inflammation and pain associated with migraine headache (Durham 2006, Edvinsson & Uddman 2005). The identification of the transmitters active within the so-called ‘trigeminovascular system’, and those molecular pathways that modify their activity, has driven the recent progress in acute migraine treatment and remains critical to further progress in primary headache therapy (Durham 2008, Benemei et al. 2007).
We recently demonstrated that the functional regulation of the cerebral vasculature by perivascular vasodilator fibers, including primary trigeminal afferents, involves the epoxyeicosanoid signaling pathway (Iliff et al. 2009). Epoxyeicosatrienoic acids (EETs) are arachidonic acid metabolites of cytochrome P450 (CYP) epoxygenase enzymes resulting from the addition of an epoxide to either one of the four arachidonic acid double bonds, generating the four distinct EETs regiosomers: 5,6-EET, 8,9-EET, 11,12-EET and 14,15-EET (Figure 1A) (Roman 2002, Zeldin 2001). These lipid signaling molecules are most commonly regarded as potent vasodilators in a number of vascular beds, including in the cerebral circulation (Earley et al. 2005, Ellis et al. 1991, Ellis et al. 1990, Iliff et al. 2009) were they are produced both by the vascular endothelium (Medhora et al. 2001) and by perivascular astrocytes (Alkayed et al. 1997, Alkayed et al. 1996), and contribute to the physiological regulation of cerebral blood flow (CBF) (Koehler et al. 2006, Peng et al. 2002, Peng et al. 2004). The cellular levels and biological activity of EETs are regulated through their hydrolysis to less active dihydroxyeicosatrienoic acids (DHETs), a process that is catalyzed by the enzyme soluble epoxide hydrolase (sEH, Figure 1A) (Sadoshima et al. 1986, Spector et al. 2004, Zeldin 2001).
Figure 1. Expression of CYP epoxygenase and soluble epoxide hydrolase in primary trigeminal ganglion neurons.
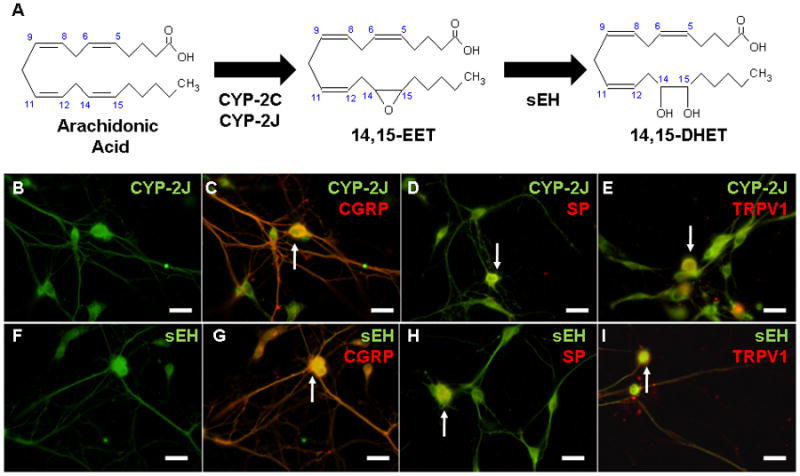
(A) Cytochrome P450 epoxygenase enzymes of the CYP-2C and CYP-2J families convert arachidonic acid to four epoxyeicosatrienoic acid (EET) regio-isomers: 5,6-EET, 8,9-EET, 11,12-EET and 14,15-EET (shown). EETs are degraded by soluble epoxide hydrolase (sEH) to produce dihydroxyeicosatrienoic acids (DHETs). (B–I) Immunocytochemistry of rat primary trigeminal ganglion neurons (TGNs), day 4 in culture. CYP-2J epoxygenase is broadly expressed in primary TGNs (B), including those expressing the neuropeptides calcitonin gene-related peptide (CGRP, C) and substance P (SP, D), in addition to the transient receptor potential vanilloid-1 (TRPV1) cation channel (E). (F) Soluble epoxide hydrolase is expressed in TGNs. sEH-immunoreactivity co-localizes with the neuropeptides CGRP (G) and substance P (H), in addition to the TRPV1 cation channel (I). All images are representative of at least three biological replicates. Scale bars: 60 μm.
Our group reported the expression of two distinct EETs-synthetic CYP epoxygenases, CYP-2J3 and CYP-2J4, in addition to sEH, in the neurons of the rat trigeminal ganglia, including those that contribute to the perivascular innervation of the cerebral surface vasculature (Iliff et al. 2007, Iliff et al. 2009). This finding indicates that these neurons possess the biochemical machinery for the synthesis and regulation of vasoactive EETs. In subsequent experiments utilizing two distinct in vivo models of neurogenic cerebral vasodilation, the increases in CBF resulting from perivascular vasodilator nerve activation were blocked by the EETs antagonist 14,15-epoxyeicosa-5(Z)-enoic acid (14,15-EEZE). These data suggest that EETs signaling contributes to the peripheral neuroeffector actions of these perivascular neurons. The specific role of EETs signaling in the effector actions of primary trigeminal afferents, however, remains unexplored.
Given the established vasodilator effects of EETs in the cerebral circulation (Earley et al. 2005, Ellis et al. 1991, Ellis et al. 1990, Iliff et al. 2009), one possibility is that EETs represent a releasable lipid-based vasoactive paracrine factor, synthesized in, and released from, trigeminal ganglia neurons (TGNs) in parallel with other transmitters such as the vasoactive neuropeptides CGRP or SP. A second possibility is that EETs function as intracellular signaling molecules, regulating the release of other neurotransmitters from TGNs. Both of these proposed roles could account for the expression of EETs-synthetic CYP epoxygenase enzymes in TGNs, in addition to the sensitivity of neurogenic cerebral vasodilation to blockade of the EETs signaling system (Iliff et al. 2007, Iliff et al. 2009). In the present study, we evaluate the potential roles of EETs signaling in the effector function of TGNs, using an in vitro primary cell culture model. Utilizing a sensitive liquid chromatrography tandem mass spectrometry (LC-MS/MS) method, we first assess the potential production and release of EETs species from TGNs. We then examine pharmacologically EETs’ participation in the regulation of neuropeptide release from TGNs, by investigating the contribution of EETs signaling to basal and stimulation-evoked CGRP and SP release from primary TGNs.
Methods
The present experiments were conducted in accordance with the National Institutes of Health Guidelines for the Care and Use of Animals in Research under protocols approved by the Institutional Animal Care and Use Committee of Oregon Health & Science University.
Primary Trigeminal Ganglion Neuron Culture
Postnatal day 2–3 (P2–3) Sprague-Dawley rat pups were deeply anesthetized with isofluorane and rapidly decapitated. Trigeminal ganglia were rapidly dissected bilaterally from each animal and placed in ice-cold Ca2+/Mg2+-free Dulbecco’s phosphate-buffered saline (PBS, Invitrogen). The ganglia were digested first with 0.15% trypsin (Sigma) in Ca2+/Mg2+-free Hanks Balanced Salt Solution (HBSS, Invitrogen) for 30 min at 37 oC, then with 0.2% collagenase (Sigma) in Ca2+/Mg2+-free HBSS for 30 min at 37 oC. Following digestion, ganglia were washed with 0.1% soybean trypsin inhibitor (Sigma) in Ca2+/Mg2+-containing PBS (Invitrogen) followed by a rinse in the plating medium. Ganglia were dissociated by trituration within a 1 ml plastic serological pipette. TGNs were purified on a 35%–60% Percoll (GE Healthcare) gradient as described by Buldyrev et al. (Buldyrev et al. 2006), washed with plating media, then plated onto poly-D-lysine (Sigma) and laminin (Sigma)-coated 6 or 24 well cell culture plates (Falcon). Cultures were grown for four days in Neurobasal-A medium (Invitrogen) supplemented with B-27 (Invitrogen), 0.5 mmol/L glutamine (Invitrogen), 2.5% fetal bovine serum (HyClone), penicillin-streptomycin (Invitrogen) and 50 ng/ml nerve growth factor (Invitrogen).
Immunocytochemistry
For immunocytochemistry, TGNs were plated at a low density (0.5 ganglion/well) on poly-D-lysine and laminin-coated glass coverslips within 24-well cell culture plates. After four days, coverslips were washed three times with warm PBS, fixed with 4% paraformaldehyde in PBS for 10 min, washed three times with PBS, then blocked for 1 hr at room temperature with 5% normal donkey serum (Jackson Immunoresearch), 1% bovine serum albumin (BSA, Sigma) and 0.5% Triton-X (Sigma) in PBS. Coverslips were incubated with primary antibodies in 1% BSA and 0.5% Triton-X in PBS overnight at 4 oC, then washed four times with PBS, and incubated with secondary antibodies in 1% BSA and 0.5% Triton-X in PBS for 1 hr at room temperature. Coverslips were last washed four times with PBS, then mounted on glass slides with Prolong Gold Antifade (Invitrogen).
To evaluate the localization of CYP-2J and sEH enzymes in primary TGNs, primary rabbit polyclonal antibodies raised against recombinant human CYP-2J2 (CYP-2J2rec, 1:500, a gift from Dr. Darryl Zeldin, NIEHS (Wu et al. 1997)) and against residues 340-554 from human sEH (1:100, Santa Cruz) were utilized. To determine CYP-2J or sEH co-localization with the neuropeptides CGRP or SP, double labeling was conducted with a primary goat polyclonal antibody raised against rat CGRP (1:500, Serotec) and a primary guinea pig polyclonal antibody raised against residues 1-11 of rat substance P (1:500, Millipore). Localization of CYP-2J and sEH in capsaicin-responsive trigeminal nociceptors was evaluated by double labeling with an primary guinea pig polyclonal antibody raised against residues 817-838 of the rat transient receptor potential vanilloid-1 (TRPV1) channel (1:1000, Abcam). Secondary detection utilized donkey anti-rabbit ALEXA Fluor-488, donkey anti-goat ALEXA Fluor-594 and donkey anti-guinea pig ALEXA Fluor-594 secondary antibodies (1:800, Invitrogen). Imaging was conducted on a conventional fluorescence microscope (Leica) with image analysis conducted with ImageJ software (NIH). All immunocytochemical localization studies were carried out in at least three independent cell culture runs (n=3 biological replicates) while representative images were selected to demonstrate CYP-2J and sEH immunoreactivity.
Liquid Chromatography-Tandem Mass Spectrometry
We first evaluated the endogenous activity CYP-2J epoxygenase enzymes in primary TGNs. Neurons were plated at a density 8 ganglia per well in 6-well cell culture plates. Un-stimulated neurons from one well from three cell culture runs (n=3 biological replicates) were first washed three times with HEPES-buffered saline (HBS), then scraped and collected in PBS. Cell suspensions were centrifuged at 14,000×g for 10 min at 4 oC to pellet the cells, and the PBS was aspirated. Pellets were re-suspended in 200 μL PBS then frozen at −80°C until the time of processing. Cell pellets were thawed on ice and spiked with 200 pg of d8-14,15-EET and d8-15-HETE internal standard, and 5 μl 1%BHT in methanol was added per ml of sample. 1 ml of KOH (1 mol/L) was added to each sample, after which the samples were hydrolyzed for 30 min at 40°C, cooled for 5 min at room temperature, then neutralized with 1ml HCl (1 mol/L). The pH of the samples was checked and they were further acidified with 88% formic acid to pH=3.0. Samples were extracted 2 times with 2 ml ethyl acetate. The combined organic layers were dried in the SpeedVac, brought up in 1 ml hexane, re-dried, brought up in start solvent (45:55 Acetonitrile:H20), and subjected LC-MS/MS analysis.
In subsequent experiments, we evaluated the effect of either 1 hr treatment with capsaicin (100 nmol/L) or depolarizing K+ (60 mmol/L) upon 5,6-EETs and 14,15-EETs levels in primary TGNs. Primary TGNs were prepared as above, and plated at a density of 10 ganglia per well in 6-well culture plates. Cells were washed three times with HBS, then stimulated for 1 hr at 37 oC with capsaicin or KCl in HBS. Cells were washed three times with HBS then scraped and collected in PBS, then processed as above. The effect of 48 hr treatment with the CYP epoxygenase inhibitor MS-PPOH upon TGN levels of 5,6-EET and 14,15-EET was likewise evaluated. 48 hrs after plating, cells were treated with N-(methylsulfonyl)-2-(2-propynyloxy)-benzenehexanamide (MS-PPOH, 10μmol/L) in culture media for 48 hrs. Cells were washed three times with HBS then scraped and collected in PBS, then processed as above.
The release of 5,6-EETs and 14,15-EETs from primary TGNs following stimulation was additionally evaluated by LC-MS/MS. Primary TGNs were grown in 6-well cell culture dishes (10 ganglia/well) for four days. The cells were washed three times with HBS then stimulated for 1 hr at 37 oC with 100 nmol/L capsaicin or 60 mmol/L KCl in HBS. Following stimulation, the conditioned HBS was collected and subjected to LC-MS/MS analysis of 5,6-EET and 14,15-EET content. For these data, buffer from one well from three distinct cell culture runs was collected (n=3 biological replicates) and frozen at −80°C until the time of processing. Conditioned HBS was thawed on ice, spiked with 200 pg d8-14,15-EET and d8-15-HETE internal standard, and 5 μl 1%BHT in methanol was added per ml of sample. The sample was acidified with 88% formic acid to pH=3.0, determined using pH paper. The media samples were extracted 2 times with 2 ml ethyl acetate and the organic layers were combined and dried under vacuum in a Speed Vac. Sample was brought up in 1 ml of hexane and then re-dried before being brought up in start solvent, then subjected to LC-MS/MS analysis.
DHETs, HETEs and EETs levels were analyzed using the 4000 Q-TRAP triple quadrupole mass spectrometer (Applied Biosystems) with electrospray ionization (ESI) in negative mode. The instrument was interfaced with a Prominence High-Performance Liquid Chromatography (HPLC) unit (Shimadzu) which allows pressures to 10,000 psi. Resolution was obtained with a 2.1×250mm, 5μ BetaBasic C18 HPLC column with guard and gradient elution with water (A) and acetonitrile (B); both with 0.002% acetic acid under the following conditions: 45–60% B over 1 min, linear to 65% B over 15 min, linear to 95% B over 0.10 min, isocratic at 95% B for 4 min, linear to 45% B over 0.1 min, isocratic at 45% B for 10 min. Flow rate was 0.5 ml/min and column temperature was 40°C. ESI source parameters included curtain gas 50, ion spray voltage −4000, temperature 550 °C with ion source gas 1 and gas 2 at 50 and an entrance potential of −10 V. Samples were infused individually and instrument parameters were optimized for multiple reaction monitoring with selective transitions for each EET, DHET and HETE species. The amount of DHETs, EETs or HETEs compounds in the sample was calculated by comparison of the area ratio of the compound with d8-14,15-EET or d8-15-HETE internal standard, then compared to a standard curve generated from blank HBS spiked with known amounts of DHETs, EETs and HETEs. Quantification of cell extract analyte levels (Tables 1 and 2) represents the concentration of analyte detected in reconstituted cell extract (8 or 10 ganglia cell pellet re-suspended in 200 μl PBS). Thus, this value represents a relative, rather than an absolute concentration.
Table 1.
Detection of EETs regio-isomers in primary trigeminal ganglion neurons
| Regio-isomer | 5,6- | 8,9- | 11,12- | 14,15- |
|---|---|---|---|---|
| EET (pg/8 ganglia) | ND | 90.8 ± 17.8 | 177.9 ± 51.6 | 236.2 ± 56.9 |
| DHET (pg/8 ganglia) | 18.2 ± 3.0 | ND | ND | ND |
| % | 3.5 | 17.4 | 34.0 | 45.1 |
ND: not detected. Values are means ± SEM (n = 3 replicates).
Table 2.
Effect of capsaicin and MS-PPOH on cellular and released EETs.
| Cells | Media | |||
|---|---|---|---|---|
| 5,6-EET (pg/10 ganglia) | 14,15-EET (pg/10 ganglia) | 5,6-EET (pg/ml) | 14,15-EET (pg/ml) | |
| Vehicle | 92.1 ± 22.7 | 201.5 ± 29.5 | 186.1 ± 29.4 | 575.4 ± 142.4 |
| Capsaicin | 97.2 ± 18.4 | 214.3 ± 22.2 | 131.6 ± 11.3 | 584.7 ± 66.4 |
| Vehicle | 100.8 ± 9.0 | 242.2 ± 59.1 | ||
| MS-PPOH | 83.7 ± 4.0 | 179.1 ± 10.2 | ||
MS-PPOH: N-(methylsulfonyl)-2-(2-propynyloxy)-benzenehexanamide. Values are means ± SEM (n = 3 replicates).
Neuropeptide Release Assay
The release of the neuropeptides CGRP and SP from primary TGNs was assayed using commercially available enzyme-linked immunosorbent assays (ELISA). TGN cultures were grown for four days on 24-well cell culture plates (2 ganglia/well). To determine basal levels of neuropeptide release, cells were washed three times with warm HBS, then incubated for 1hr at 37oC with fresh HBS. The conditioned buffer was then collected and assayed for neuropeptide content by ELISA. After washing 3 times with HBS, the cells were treated for 30 min at 37oC with pharmacological agents or vehicle in HBS. Cells were then stimulated with either the TRPV1 agonist capsaicin (100 nmol/L) or depolarizing K+ (60 mmol/L) in HBS, with the continued presence of pharmacological agents or vehicle for 1 hr at 37oC. Following stimulation, the conditioned buffer was collected and assayed for neuropeptide content by ELISA. For each cell culture run, two sister culture wells represented each experimental condition. Neuropeptide release was measured independently from each, then averaged to produce a single biological replicate (n=1). On each cell culture plate, the evoked neuropeptide release was normalized to the average basal release measured from an HBS-treated un-stimulated control wells subjected to the same washes and buffer changes as stimulated wells, in order to reduce experimental variability among biological replicates.
Basal and evoked CGRP release was measured using a commercially available rat CGRP ELISA kit (SPIBio) according to the manufacturer’s recommended protocol. Briefly, duplicate 100 μL undiluted conditioned HBS samples from each TGN culture well were assayed. CGRP content was determined photometrically by measuring absorbance at 405 nm on an automated plate reader (Molecular Devices). Absorbance values were corrected by blank subtraction, averaged and converted to CGRP concentration using a 7 point CGRP standard curve. All measured experimental values fell within the linear range of the standard curve.
Stimulation-evoked SP release was likewise measured using a commercially available rat substance P ELISA kit (Cayman). Briefly, duplicate 50 μL conditioned HBS samples from each trigeminal ganglion neuronal culture well were assayed. Samples were diluted 1:100 in ELISA buffer and SP content was determined photometrically by measuring absorbance at 405 nm. Absorbance values were corrected by blank subtraction, averaged and converted to SP concentration using an 8 point SP standard curve. All measured experimental values fell within the linear range of the standard curve.
Chemicals
HEPES-buffered saline consisted of 25 mmol/L HEPES, 140 mmol/L NaCl, 3.5 mmol/L KCl, 2.5 mmol/L CaCl2, 1 mmol/L MgCl2, 3.3 mmol/L D-glucose, and 1% BSA (pH 7.4). Capsaicin (Sigma) was initially dissolved in 100% ethyl alcohol, then diluted in HBS to a 1 μmol/L 10X stock solution (1% ethyl alcohol) and stored at 4 °C until use. Fresh stock was prepared biweekly. 100 nmol/L capsaicin stimulation buffer (0.1% ethyl alcohol) was prepared fresh for each experiment. 14,15-Epoxyeicosa-5(Z)-enoic acid (14,15-EEZE), 14,15-EET, 11,12-EET, 8,9-EET and 5,6-EET (Cayman) were dried using a SpeedVac, dissolved in dimethylsulfoxide (DMSO) to form a 1000X stock solutions. N-(methylsulfonyl)-2-(2-propynyloxy)-benzenehexanamide (MS-PPOH, Cayman Chemical) and 12-[[(tricyclo[3.3.1.13,7]dec-1-ylamino)carbonyl]amino]-dodecanoic acid (AUDA, Cayman Chemical) were dissolved in DMSO to form a 1000X stock solution, and stored at −20 °C until use. Compounds were then diluted 1:1000 in HBS (0.1% DMSO) fresh for each experiment.
Statistics
All values are expressed as the mean ± standard error of the mean (SEM). To account for variability in experimental conditions, TGN density and neuropeptide release between cell culture biological replicates, evoked neuropeptide release was normalized to the average release within two un-stimulated HBS-treated control wells on each cell culture plate (Normalized Neuropeptide Release = Stimulated Release/Control Release). The effect of pharmacological blockade on evoked neuropeptide release was analyzed by one-way analysis of variance (ANOVA) with a post hoc Tukey’s test to evaluate individual group differences. Single treatments were compared to control responses using Student’s t-test. A p value < 0.05 was considered significant.
Results
Expression of CYP-2J and sEH in Primary Trigeminal Ganglion Neurons
In the current study, the expression of CYP-2J and sEH in primary cultured TGNs was first assessed by immunocytochemistry. After four days in culture, primary TGNs exhibited avid process extension and dendritic arborization. Labeling of TGNs with an antibody against human CYP-2J2 which cross-reacts with both rat CYP-2J3 and CYP-2J4 isoforms resulted in strong immunoreactivity throughout virtually all primary neurons. Labeling was most pronounced in the cell soma, although weaker immunoreactivity was evident in neuronal processes (Figure 1B, n=3 replicates). Labeling with an antibody raised against a peptide fragment of human sEH revealed similarly broad expression of this enzyme throughout virtually all primary TGNs (Figure 1F, n=3). Co-labeling with an antibody raised against rat CGRP revealed that nearly all CGRP-positive neurons exhibited CYP-2J- and sEH-immunoreactivity (Figure 1B–C, n=3; Figure 1F–G, n=3). Probing for the expression of the neuropeptide SP revealed a small number of TGNs expressing SP. Of these SP-positive neurons, all expressed CYP-2J (Figure 1D, n=3) and sEH (Figure 1H, n=3). Co-localization was also observed between CYP-2J and the capsaicin-activated cation channel TRPV1 in trigeminal neurons (Figure 1E, n=3; Figure 1I, n=3). These data confirm our previous findings in vivo (Iliff et al. 2009) and demonstrate that primary TGNs generally express both the EETs-synthetic CYP-2J and the EETs-metabolizing sEH enzymes.
Analysis of EETs levels in Primary Trigeminal Ganglion Neurons
The production of EET and DHET metabolites of arachidonic acid in primary TGNs was assayed by LC-MS/MS in three independent cell culture sets plated at a density of 8 ganglia per well. Prior to analysis, cell extracts were subjected to acid hydrolysis to liberate esterified EETs. Hence, all measured values reflect total cellular EETs levels. 8,9-EET (90.8 ± 17.8 pg/well), 11,12-EET (177.9 ± 51.6 pg/well) and 14,15-EET (236.2 ± 56.9 pg/well) were readily detectable in cellular extracts (Table 1, n=3). Although 5,6-EET itself was not detected in these samples, 5,6-DHET was detected (18.2 ± 3.0 pg/well). The lipoxygenase metabolites 11-HETE (45.6 ± 12.0 pg/ml), 12-HETE (40.7 ± 5.1 pg/ml) and 15-HETE (111.2 ± 5.0 pg/ml) were also present within the TGN cellular extract, whereas the CYP hydroxylase metabolite 20-HETE was not detectable. These findings demonstrate that EETs are endogenous constituents of primary TGNs.
In subsequent experiments, TGNs were plated at a density of 10 ganglia per well, and the effect of capsaicin (100 nmol/L) upon cellular levels of 5,6- and 14,15-EETs was evaluated (Table 2, n=3). Treatment for 1 hr with capsaicin did not significantly alter cellular levels of total 5,6- or 14,15-EETs compared to vehicle treatment. Long-term (48 hr) incubation with the CYP epoxygenase inhibitor MS-PPOH (10 μmol/L), however, tended to reduce total cellular 5,6- and 14,15-EETs levels, although this effect did not reach statistical significance (p =0.07 and 0.17, respectively, t-test; Table 2, n=3).
The release of 5,6- and 14,15-EETs by TGNs into the culture media was evaluated following 1 hr stimulation with capsaicin (100 nmol/L). Under un-stimulated conditions, treatment with vehicle for 1 hr revealed the tonic release of both 5,6- or 14,15-EETs which were detected in the conditioned HBS at concentrations of 186.1 ± 29.4 and 575.4 ± 142.4 pg/ml. Stimulation for 1hr with capsaicin did not significantly alter this rate of release (Table 2, n=3).
Role of EETs in Neuropeptide Release from Trigeminal Ganglion Neurons
We next sought to determine whether endogenous EETs regulate the release of other neurotransmitters, such as the neuropeptides CGRP or SP, from primary TGNs. Under un-stimulated conditions after four days in culture, primary TGNs released CGRP at a basal rate of 56.6 ± 12.7 pg/ml/hr. When EETs’ action was inhibited with the EETs antagonist 14,15-EEZE (10 μmol/L), basal CGRP release was significantly reduced to 38.3 ± 8.1 pg/(ml/hr) (Figure 2A, p<0.01, t-test, control vs. 14,15-EEZE, n=14 culture wells per condition), suggesting that EETs up-regulate basal CGRP release from primary TGNs.
Figure 2. Inhibition of CGRP release from primary trigeminal ganglion neurons by 14,15-EEZE.
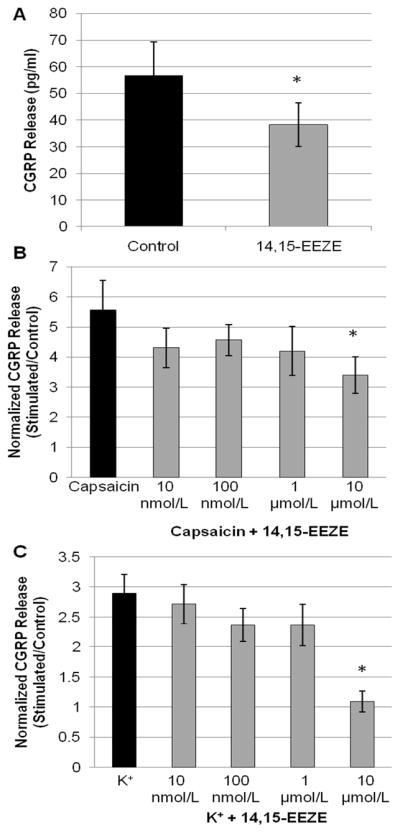
(A) Rat primary TGNs (day 4 in culture) were incubated for 1 hr in blank buffer, then buffer was assayed for calcitonin gene-related peptide (CGRP) release by ELISA. Treatment with the EETs antagonist 14,15-EEZE (10 μmol/L) significantly reduced basal CGRP release (p<0.01, t-test, control vs. 14,15-EEZE; n=14). (B–C) TGNs were stimulated for 1 hr with capsaicin (CAP, 100 nmol/L) or K+ (60 mmol/L) in the continued presence of 14,15-EEZE, then the buffer was assayed for CGRP release. (B) 30 min pre-treatment with the EETs antagonist 14,15-EEZE attenuated capsaicin-evoked CGRP release in a concentration-dependent manner (*p<0.05, ANOVA, treatment vs. capsaicin alone; n=6). (C) 30 min pre-treatment with 14,15-EEZE concentration-dependently attenuated K+-evoked CGRP release (*p<0.01, ANOVA, treatment vs. K+ alone; n=6).
We then determined the effect of EETs on stimulated neuropeptide release from TGNs in response to membrane depolarization or activation of the Ca2+-permeable capsaicin-gated TRPV1 channel (Meng et al. 2009). Following 1 hr stimulation with capsaicin (100 nmol/L) or depolarizing K+ (60 mmol/L), CGRP release increased approximately 5- and 3-fold, respectively, above basal levels measured in sister un-stimulated wells (Figure 2B–C). In order to evaluate the contribution of EETs signaling to CGRP release from TGNs evoked by these two stimuli, cells were pre-treated for 30 min with vehicle or the putative EETs antagonist 14,15-EEZE (10 nmol/L–10 μmol/L), then stimulated with either capsaicin or K+, as above. In the presence of vehicle, capsaicin stimulation increased CGRP release 5.6 ± 1.0-fold over basal levels. Pre-treatment with 14,15-EEZE attenuated capsaicin-evoked CGRP release concentration-dependently, reducing release to 3.4 ± 0.6-fold over basal levels at a concentration of 10 μmol/L (Figure 2B; p<0.05, ANOVA vehicle vs. 14,15-EEZE treatment; n=6 experiments). Tukey’s post hoc analysis identified a significant individual effect of 14,15-EEZE at 10 μmol/L. In the presence of vehicle, depolarizing K+ elevated CGRP release 2.9 ± 0.3-fold over basal levels. Pre-treatment with 14,15-EEZE significantly reduced K+-evoked CGRP release to 1.1 ± 0.2-fold over basal levels (Figure 2C; p<0.01, ANOVA vehicle vs. 14,15-EEZE treatment; n=6 experiments). Post hoc analysis identified specific treatment differences at 10 μmol/L 14,15-EEZE. In total, inhibition of EETs’ actions with the putative EETs antagonist 14,15-EEZE significantly attenuated evoked CGRP release from primary TGNs. This suggests that endogenous EETs signaling is a positive regulator of evoked neuropeptide release from these cells.
In order to further evaluate the contribution of endogenous EETs signaling to evoked neuropeptide release from primary TGNs, the effect of pharmacological inhibition of CYP epoxygenase (EETs ‘synthase’) with MS-PPOH was next examined. To inhibit de novo EETs synthesis, primary neurons were pre-treated for 30 min with MS-PPOH. Additionally, an extended 48 hr pre-treatment with this CYP epoxygenase inhibitor was utilized to deplete the latent pool of esterified EETs present in the phospholipid membrane (Table 2). Analysis of variance revealed that treatment with MS-PPOH significantly altered stimulated CGRP release in response to both 100 nmol/L capsaicin (p<0.05, n=6) and 60 mmol/L K+ (p<0.01, n=5). Specifically, pre-treatment with MS-PPOH for 30 min did not significantly alter the stimulated release of CGRP in response to either capsaicin or K+ compared to vehicle (Figure 3A, n=6). However, 48 hr pre-treatment with MS-PPOH significantly reduced capsaicin-evoked CGRP release from 4.9 ± 0.7-fold increase over basal release with vehicle to a 3.8 ± 0.6-fold increase over basal release with MS-PPOH. Similarly, 48 hr pre-treatment with MS-PPOH reduced K+-evoked neuropeptide release from trigeminal ganglion neurons, from a 3.7 ± 0.6-fold increase over basal release with vehicle treatment to a 2.4 ± 0.1-fold increase over basal release with drug (Figure 3A). These data corroborate the effects of the EETs antagonist 14,15-EEZE upon evoked neuropeptide release from TGNs and thus support the contribution of endogenous EETs signaling to this process. Further, these findings suggest that the bioactive EETs in primary TGNs are not derived from de novo synthesis, but rather are released from a pre-formed pool.
Figure 3. Inhibition of evoked CGRP release by MS-PPOH.
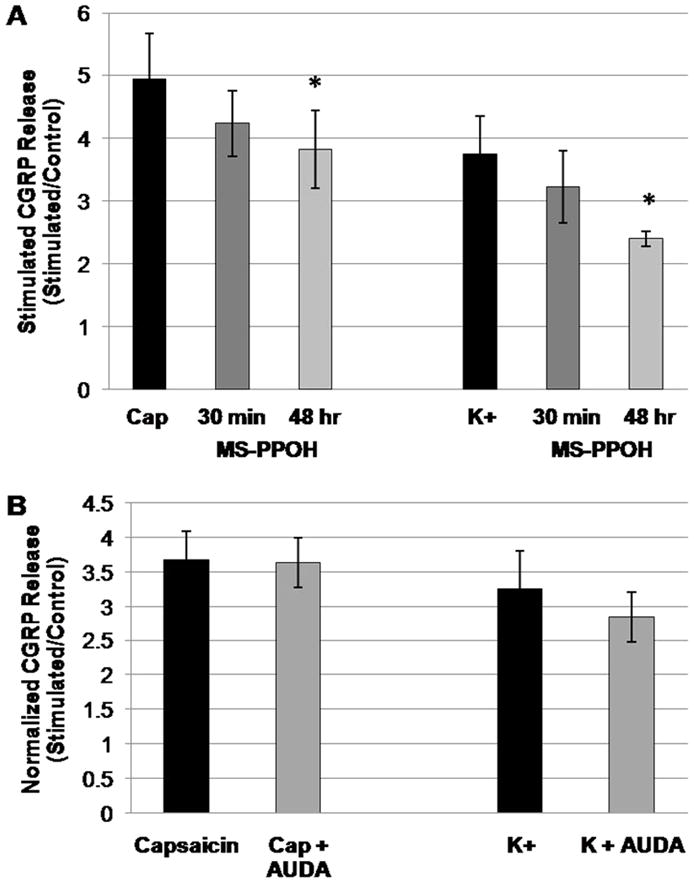
Rat primary TGNs (day 4 in culture) were stimulated for 1 hr with capsaicin (CAP, 100 nmol/L) or K+ (60 mmol/L), then the media were assayed for CGRP release by ELISA. (A) 30 min pre-treatment with the CYP epoxygenase inhibitor MS-PPOH (20 μmol/L) did not significantly alter capsaicin- or K+-evoked CGRP release. Long-term (48 hr) treatment with MS-PPOH significantly attenuated both capsaicin- and K+-stimulated CGRP release (*p<0.05, ANOVA, treatment vs. capsaicin or K+ alone; n=6 and 5, respectively). (B) 30 min pre-treatment with the sEH inhibitor AUDA (1 μmol/L) did not significantly alter the release of CGRP from TGNs following stimulation with either capsaicin or K+ (t-test, n=11 and 6, respectively). All post-treatment stimuli were carried out in the continued presence of MS-PPOH or AUDA, respectively.
We next evaluated the effect of the sEH inhibitor AUDA upon evoked CGRP release from TGNs. After four days in culture, primary TGNs were pre-treated for 30 min with either vehicle or AUDA (1 μmol/L), and then stimulated for 1 hr with either capsaicin (100 nmol/L) or depolarizing K+ (60 mmol/L). The conditioned buffers were removed and assayed for CGRP content by ELISA. Interestingly, 30 min pre-treatment with AUDA did not alter either capsaicin-or K+-evoked CGRP release from primary TGNs (Figure 3B; n=11and 6; p=0.48 and 0.32; respectively).
In order to evaluate whether EETs’ role in the regulation of neuropeptide release is specific to CGRP or rather extends more generally to other trigeminal neuropeptides, we assessed the sensitivity of stimulation-evoked release of SP from TGNs to inhibition of the EETs pathway. When pre-treated with the EETs antagonist 14,15-EEZE (10 μmol/L) for 30 min, capsaicin-evoked SP release was significantly (p<0.05, t-test, n=4 independent cultures) reduced from a 3.0 ± 0.9-fold increase with vehicle treatment to a 1.0 ± 0.3-fold increase with 14,15-EEZE treatment (Figure 4). This finding supports the contribution of EETs signaling to evoked neuropeptide release from trigeminal ganglion neurons and suggests that this action is general, and not limited to CGRP release.
Figure 4. The EETs antagonist 14,15-EEZE blocks evoked substance P release from trigeminal ganglion neurons.

Rat primary TGNs (day 4 in culture) were stimulated for 1 hr with capsaicin (CAP, 100 nmol/L), then the media were assayed for substance P release by ELISA. 30 min pre-treatment with the EETs antagonist 14,15-EEZE (10 μmol/L) blocked capsaicin-stimulated substance P release (*P < 0.05, t-test, treatment vs. capsaicin alone; n=4). Capsaicin stimulation was carried out in the continued presence of 14,15-EEZE.
Direct Action of EETs upon Neuropeptide Release from Trigeminal Ganglion Neurons
We next sought to determine whether the administration of exogenous EETs directly stimulated CGRP release from primary TGNs. After four days in culture, cells were treated for 1 hr with either one of the four EETs regio-isomers: 5,6-EET, 8,9-EET, 11,12-EET, 14,15-EET (1 μmol/L) or vehicle. Analysis of variance revealed that EETs treatment significantly altered CGRP release (p<0.05, n=6). Post hoc Tukey’s analysis for between-group comparison demonstrated that both the 14,15-EET and 5,6-EET regio-isomers significantly increased CGRP release compared to vehicle control. In the presence of vehicle alone, CGRP release was 0.9 ± 0.1-times that of the untreated control wells. Treatment with 14,15-EET and 5,6-EET increased CGRP release 2.0 ± 0.5- and 1.8 ± 0.2-fold over basal release, respectively (Figure 5A, n=6). The 8,9-EET and 11,12-EET regio-isomers tended to stimulate TGN CGRP release (2.0 ± 0.6 and 1.8 ± 0.5-fold increase in CGRP release compared to un-stimulated control, respectively; Figure 5A, n=6) as well, however these effects did not achieve statistical significance in post hoc analysis. These findings demonstrate that exogenous EETs directly stimulate neuropeptide release from primary TGNs.
Figure 5. Direct effects of EETs upon basal and evoked release of CGRP from primary trigeminal ganglion neurons.

(A) Rat primary TGNs (day 4 in culture) were treated for 1 hr with each of the four EETs regio-isomers (1 μmol/L), then the media were assayed for CGRP release by ELISA. EETs treatment stimulated CGRP release from TGNs (p<0.05, ANOVA; n=6), while the specific effects of 5,6-EET and 14,15-EET were determined to be significant by post hoc analysis. (B) 30 min pre-treatment with the 5,6-EET regio-isomer significantly potentiated capsaicin (CAP, 100 nmol/L)-evoked CGRP release (*p<0.01, t-test; n=5). There was as similar trend with the effect of pre-treatment with the 14,15-EET regio-isomer upon capsaicin-stimulated CGRP release, however this did not achieve statistical significance (p=0.20, t-test; n=5). (C) 30 min pre-treatment of primary TGNs with either 5,6-EET or 14,15-EET tended to increase K+ (60 mmol/L)-evoked CGRP release, although this effect likewise did not achieve statistical significance (p=0.09 and 0.19; t-test; n= 5 and 4, respectively).
When TGNs were pre-treated for 30 min with 5,6-EETs or 14,15-EETs (1 μmol/L), then stimulated for 1 hr with either capsaicin (100 nmol/L) or depolarizing potassium (60 mmol/L), evoked CGRP release tended to increase (Figure 5B–C). This effect was modest, however, reaching statistical significance only with the co-administration of 5,6-EET with capsaicin (P<0.01; n=5).
Discussion
In the present study, we demonstrated first that epoxyeicosatrienoic acids are endogenous constituents of primary rat trigeminal ganglion neurons. Moreover, we provided pharmacological evidence that endogenous EETs signaling contributes to the regulation of neuropeptide release from these neurons (represented in Figure 6 in schematic form). We first confirmed the expression of the EETs-synthetic CYP-2J epoxygenase enzymes within primary TGNs, then established that EETs are produced within and released from these neurons. Furthermore, we demonstrated that inhibition of endogenous EETs signaling in TGNs attenuates basal and evoked neuropeptide release, and that exogenous EETs stimulate neuropeptide release.
Figure 6. Intracellular EETs are endogenous regulators of evoked neuropeptide release from primary trigeminal ganglion neurons.
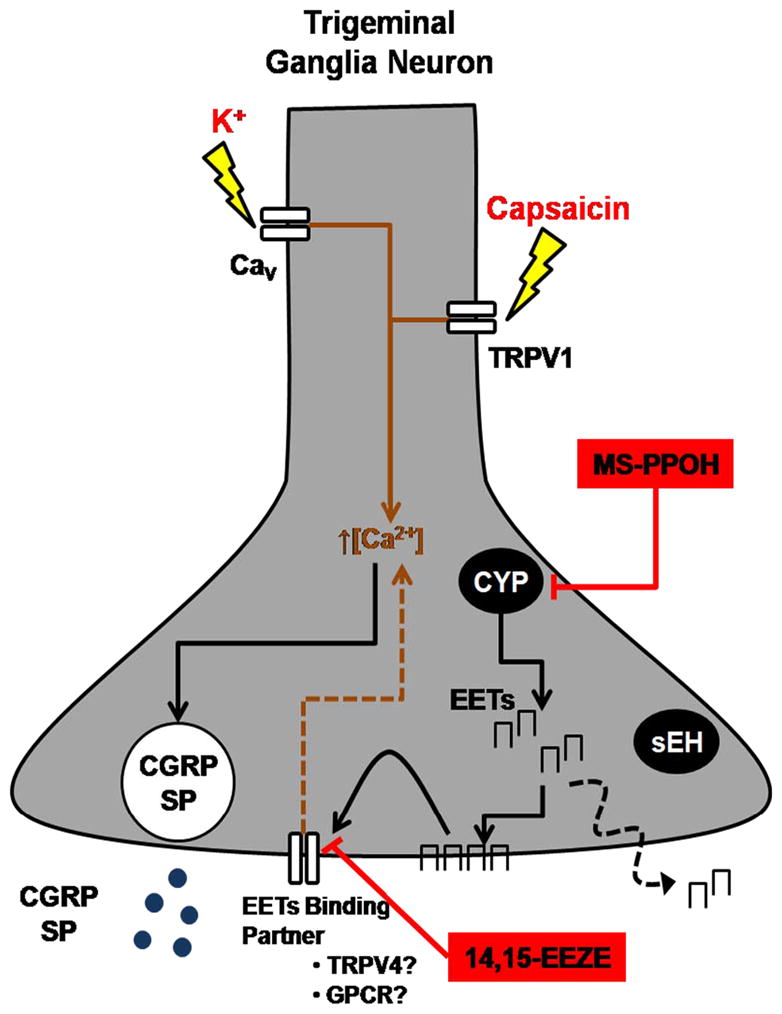
Primary TGNs respond to stimulation by depolarizing K+ or the transient receptor potential vanilloid-1 (TRPV1) agonist capsaicin with the calcium-dependent release of sensory neuropeptides, including calcitonin gene-related peptide (CGRP) and substance P (SP). Primary TGNs express EETs-synthetic cytochrome P450 (CYP) epoxygenase enzymes. EETs are constitutively present in TGNs and are tonically released from these cells. The EETs antagonist 14,15-EEZE inhibits both K +- and capsaicin-stimulated CGRP and SP release. Inhibition of CYP epoxygenase with MS-PPOH also inhibits evoked CGRP release. Administration of EETs alone directly stimulates CGRP release from TGNs. EETs may interact with putative binding partners such as one or more G protein-coupled receptors (GPCRs) or the Ca2+-permeable transient receptor potential vanilloid 4 (TRPV4) cation channel which is expressed on TGNs. Activation of these pathways may underlie the regulation of neuropeptide release from TGNs by endogenous or exogenous EETs. CaV: voltage-gated Ca2+ channel; sEH: soluble epoxide hydrolase.
We recently identified a novel role for neurogenic EETs signaling in the regulation of CBF by extrinsic perivascular vasodilator nerves, describing the expression of a specific family of ‘EETs-synthases’, the CYP-2J epoxygenase enzymes, in the neurons of the rat trigeminal and sphenopalatine ganglia (Iliff et al. 2007, Iliff et al. 2009). In the present study, we sought to determine whether these CYP-2J epoxygenase enzymes are catalytically active in TGNs, producing endogenous EETs. 5,6-EET, 8,9-EET, 11,12-EET and 14,15-EET were each identified in cell extracts from primary TGNs by LC-MS/MS. In the initial experiments, we did not directly detect the 5,6-EET regio-isomer in TGNs. However, the hydrolysis product 5,6-DHET was detectable (Table 1) and likely represents the spontaneous oxidative breakdown product of the unstable 5,6-EET species (Spector & Norris 2007, Zeldin 2001). Subsequent experiments readily detected 5,6-EET, but not 5,6-DHET (Table 2), reflecting refinement in our sample collection and storage procedures between experiments. Among the four EETs regio-isomers, the relative abundance was as follows: 14,15-EET (45%) > 11,12-EET (34%) > 8,9-EET (17%) > 5,6-DHET (4%). Such a profile is consistent with the reported catalytic activity of rat CYP-2J3 (Wu et al. 1997), which is expressed in intact TGNs (Iliff et al. 2009). When characterized within a recombinant expression system, CYP-2J3 was reported to metabolize arachidonic acid to the following EETs regio-isomers: 14,15-EET (41%), 11,12-EET (27%), 8,9-EET (28%) and 5,6-EET (4%) (Wu et al. 1997). Thus, CYP-2J enzymes expressed in TGNs are functionally active, resulting in significant production of endogenous EETs within these cells.
We next sought to define whether EETs produced in TGNs are released or remain within the intracellular compartment by measuring basal and stimulated EETs release into primary TGN media with a sensitive LC-MS/MS method. Under un-stimulated conditions, primary TGNs released both 5,6-EETs and 14,15-EETs in significant quantities. These findings support a potential extracellular action of TGN-derived EETs. However, stimulation of TGNs with 100 nmol/L capsaicin for 1 hr, a stimulus sufficient to evoke both CGRP and substance P release, did not significantly alter the rate of the release of these EETs regio-isomers. This suggests that the release of EETs from TGNs is tonic and not stimulus-dependent.
We then investigated whether endogenous EETs function as regulators of neuropeptide release in TGNs. Although most recent research has focused upon EETs’ contribution to cardiovascular regulation (Roman 2002), the earliest studies describing biological actions of epoxide metabolites of arachidonic acid focused on their role in the regulation of neurohormone release from the hypothalamus and anterior pituitary. Early studies demonstrated that all four EETs regio-isomers, which were constitutively present in both hypothalamic and pituitary tissue, were capable of directly stimulating the release of numerous neurohormones from this neuroendocrine tissue (Canonico et al. 1985, Cashman 1989, Cashman et al. 1987, Negro-Vilar et al. 1985, Snyder et al. 1986, Snyder et al. 1983). Inhibition of EETs synthesis additionally blocked neuroendocrine secretagogue-stimulated hormone release from these tissues (Luini et al. 1985, Capdevila et al. 1983, Cashman & Snowdowne 1992), demonstrating that endogenous hypothalamic and pituitary EETs are positive regulators of neurohormone release.
In light of this established role of EETs signaling in neural cell function, we proposed that endogenous EETs likewise positively regulate neuropeptide release from TGNs. Pre-treatment with the EETs antagonist 14,15-EEZE generally inhibited neuropeptide release from primary TGNs. This included basal CGRP release, capsaicin- and K+-stimulated CGRP release and capsaicin-evoked SP release. Long-term inhibition of EETs synthesis with the CYP epoxygenase inhibitor MS-PPOH attenuated capsaicin- and K+-evoked CGRP release, demonstrating that the effect of 14,15-EEZE upon neuropeptide release from primary TGNs was likely mediated by the EETs signaling pathway. The differential effect of short-term (30 min) versus long-term (48 hr) CYP epoxygenase inhibition upon evoked neuropeptide release may shed light upon the cellular source of bioactive EETs in TGNs. EETs and their DHET hydrolysis products can be re-esterified and stored within the phospholipid bilayer. In both vascular endothelium and brain astrocytes, these membrane-bound esterified EETs form a ready pool from which EETs are released in response to stimulation, a process that is not dependent upon de novo synthesis and is thus resistant to CYP epoxygenase inhibition (Spector et al. 2004, Weintraub et al. 1999). The observation that 30 min treatment with MS-PPOH did not significantly alter the evoked neuropeptide release from TGNs, whereas depletion of cellular EETs pools with long-term CYP epoxygenase inhibition reduced neuropeptide release in a similar manner to 14,15-EEZE, suggests that a similar esterified pool may be the source of bioactive EETs in primary TGNs in vivo. Thus, the effects of the EETs antagonist 14,15-EEZE and the CYP epoxygenase inhibitor MS-PPOH upon neuropeptide release suggest that just as endogenous EETs regulate stimulated release of neurohormones from hypothalamic or pituitary tissue (Capdevila et al. 1983, Cashman & Snowdowne 1992, Luini & Axelrod 1985), endogenous EETs signaling may contribute to evoked neuropeptide release from TGNs.
One surprising finding of the present study was the absence of an effect of sEH inhibition upon evoked neuropeptide release. sEH is a key regulator of EETs metabolism and inhibition of sEH activity elevates bioactive EETs levels and potentiates EETs-mediated effects in several cell systems (Inceoglu et al. 2007, Larsen et al. 2006, Seubert et al. 2006). Thus, our expectation was that sEH inhibition would exert the opposite effect of EETs antagonism with 14,15-EEZE or CYP epoxygenase inhibition with MS-PPOH, elevating evoked CGRP release from primary TGNs. It is noteworthy that although 5,6-, 8,9-, 11,12- and 14,15-EETs regio-isomers were readily detectable in primary TGN extracts, only the 5,6-DHET hydrolysis product was detected. Because 5,6-EET is a poor substrate for sEH, the detected 5,6-DHET analyte likely represents the spontaneous oxidative breakdown product of the unstable 5,6-EET species (Spector & Norris 2007, Zeldin 2001). In subsequent experiments in which the sample processing and storage techniques were refined, no DHET metabolites were detected. The absence of detectable 8,9-, 11,12- and 14,15-DHET species in TGN extracts suggests that although sEH is expressed throughout these neurons both in vivo (Iliff et al. 2007, Iliff et al. 2009) and in vitro (present findings), it may not exert sufficient catalytic activity under basal conditions to affect EETs function following acute inhibition. It is possible that sEH regulates EETs incorporation into the phospholipid membrane pools, as has been described in the vascular endothelium (Fang et al. 2004, Spector et al. 2004). Such activity may be sequestered functionally and/or spatially from a prospective membrane-delimited lipid signaling domain and its inhibition may not acutely alter EETs activity.
In the hypothalamic and pituitary tissue, it was observed that exogenous EETs directly stimulate neurohormone release (Canonico et al. 1985, Capdevila et al. 1983, Cashman 1989, Cashman et al. 1987, Negro-Vilar et al. 1985, Snyder et al. 1986, Snyder et al. 1983). In the present study, we sought to determine whether EETs are sufficient to directly stimulate neuropeptide release from TGNs. Under un-stimulated conditions, inhibition of endogenous EETs signaling with the EETs antagonist 14,15-EEZE significantly reduced basal release of CGRP from TGNs. Similarly, when TGNs were directly stimulated with exogenous EETs (in the absence of capsaicin or depolarizing K+), CGRP release was increased approximately 2-fold above basal levels of release. This suggests that EETs contribute to neuropeptide release independent of TRPV1 channel activation or membrane depolarization.
While these pharmacological data support the role of endogenous EETs signaling in regulating stimulus-evoked neuropeptide release from TGNs, the results from the present LC-MS/MS analysis suggest that EETs’ role in this process may be more complex. Although acute treatment with the EETs antagonist 14,15-EEZE or long-term inhibition of CYP epoxygenase with MS-PPOH attenuated capsaicin- or K+-evoked neuropeptide release, stimulation with capsaicin did not alter EETs levels detected either in cellular extracts or in conditioned media. These data indicate that neurogenic EETs may exert their effects upon stimulus-evoked neuropeptide release via a solely intracellular pathway. However, because EETs are basally released from primary TGNs, an autocrine or paracrine role for EETs in this process cannot be ruled out. Additionally the lack of effect of capsaicin stimulation upon cellular EETs levels supports the above notion that EETs are not produced de novo in response to stimulation, but rather are liberated from a pre-formed pool within the phospholipid membrane. Because the LC-MS/MS analysis was conducted on acid-hydrolyzed samples, the EETs levels reflect whole-cell (including esterified) EETs and thus would not resolve the release of esterified EETs into the cellular free EETs pool.
A conflicting finding in the present study was the observation that while long-term treatment with the CYP epoxygenase inhibitor MS-PPOH, which is thought to deplete cellular EETs pools and was effective in attenuating stimulus-evoked neuropeptide release, did tend to reduce cellular EETs levels, this effect was not statistically significant. If MS-PPOH blocked stimulus-evoked neuropeptide release by depleting cellular EETs stores, why was no significant depletion observed with LC-MS/MS analysis? One possibility is that 48 hour treatment with MS-PPOH produced only a moderate depletion of cellular EETs levels. This might account for both the non-specific reduction in cellular EETs detected by LC-MS/MS as well as the somewhat mild effect of MS-PPOH upon evoked CGRP release compared to that of 14,15-EEZE (Figure 3A).
A second possibility that must be considered is that the observed effects of 14,15-EEZE and MS-PPOH upon stimulus-evoked neuropeptide release are not due to their specific effects upon the EETs signaling pathway. In this scenario, EETs may be present within rat TGNs, however they may not be directly involved in the regulation of neuropeptide release. While the efficacy of two distinct pharmacological agents that act at two distinct points in the EETs signaling pathway argue against this notion, it is nevertheless possible that 14,15-EEZE and MS-PPOH, which share some structural elements may act non-specifically upon one or more molecular targets that impinge upon the neuropeptide release pathway. For example, TRPV1 is activated by certain monoxygenase metabolites of arachidonic acid (Hwang et al. 2000) and thus may be antagonized by certain structurally related compounds. Although there is, currently no experimental evidence for such non-specific interactions between MS-PPOH and other molecular targets including TRPV1, such a possibility cannot be formally ruled out. Hence the conclusive demonstration of EETs’ role in the regulation of neuropeptide release from TGNs awaits the outcome of further molecular studies.
Although the present pharmacological data demonstrate that exogenous EETs directly stimulate neuropeptide release and suggest that endogenous EETs contribute to evoked neuropeptide release from primary TGNs, the molecular basis for EETs’ actions in these processes remains unclear. Experimental evidence suggests that EETs interact with a number of molecular binding partners, including one or more putative G protein-coupled receptors (GPCRs) as well as certain ion channels. EETs activate large-conductance calcium-activate potassium (BK) channels via a GPCR pathway and directly activate ATP-sensitive potassium (KATP) and transient receptor potential vanilloid-4 (TRPV4) cation channels (Earley et al. 2005, Vriens et al. 2005, Watanabe et al. 2003, Roman 2002). In this context, the TRPV4 cation channel is an intriguing candidate to consider. TRPV4 is a Ca2+-permeable cation channel that is activated by a number of different EETs regio-isomers (Earley et al. 2005, Vriens et al. 2005, Watanabe et al. 2003). Functional evidence from rat primary TGNs suggests that TRPV4 is expressed and functional in these neurons (Chen et al. 2008, Chen et al. 2009). In the rat spinal cord dorsal horn, activation of TRPV4 causes the Ca2+-dependent release of both CGRP and SP (Grant et al. 2007). Furthermore, activation of TRPV4 in primary colonic afferents with 5,6-EET triggers the discharge of action potentials and mechanical hyperalgesia (Sipe et al. 2008). Based upon these data, TRPV4 represents a promising molecular candidate linking intracellular EETs signaling to neuropeptide release in TGNs (Figure 6). Further detailed electrophysiological studies will be necessary to conclusively evaluate the potential role of TRPV4 or other molecular pathways in TGN EETs signaling processes.
To interrogate the contribution of the EETs signaling pathway to neuropeptide release from TGNs, we utilized the putative EETs antagonist 14,15-EEZE. One major factor currently hampering the study of the biological activities of EETs is the lack of an identified EETs receptor and the resulting absence of specific pharmacological tools. In the study initially describing the 14,15-EEZE antagonist and characterizing its activity in isolated bovine coronary arteries, the compound competitively antagonized the dilation response to all four EETs regio-isomers (Gauthier et al. 2002). Many subsequent studies have demonstrated the efficacy and specificity of 14,15-EEZE in blocking EETs-mediated processes in a number of cell systems (Gross et al. 2009, Keseru et al. 2008, Liu et al. 2008, Webler et al. 2008, Yang et al. 2008), however, none have defined which EETs binding partners, including putative GPCR(s), TRPV4 or other targets, are specifically antagonized by this analogue. As the structure of 14,15-EEZE and 14,15-EET are identical save for the saturation of the 8,9 and 11,12 olefins (Gauthier et al. 2002), in addition to the fact that this compound is effective in antagonizing a broad spectrum of EET-mediated cellular effects, it is reasonable to assume that 14,15-EEZE antagonizes the binding of all four EETs regio-isomers at all such binding partners.
In summary, the present study identifies first the presence of endogenous EETs in rat TGNs, and second provides evidence suggesting that these compounds may contribute to both basal and evoked neuropeptide release from these neurons (Figure 6). Parallel to EETs’ regulation of neurohormone release in the hypothalamus and anterior pituitary described in early studies (Canonico et al. 1985, Cashman 1989, Cashman et al. 1987, Cashman & Snowdowne 1992, Luini & Axelrod 1985, Negro-Vilar et al. 1985, Snyder et al. 1986, Snyder et al. 1983), exogenous EETs directly stimulate neuropeptide release from primary TGNs while blockade of the EETs signaling pathway attenuates evoked neuropeptide release. Because both neuropeptide release from TGNs at both peripheral and central terminals is a major effector pathway being targeted in the acute treatment of migraine (e.g. the triptans and CGRP receptor antagonists) [3,4], the identification of a signaling pathway that may regulate the release of both CGRP and substance P may provide a novel therapeutic target in the treatment of migraine or other primary headache disorders.
Acknowledgments
The authors thank Dr. Dennis Koop and the Oregon Health & Sciences University Bioanalytical Shared Resource/Pharmacokinetics Core for advice on experimental design and performance of LC-MS/MS analysis; Drs. Paco Herson and Mary Heinricher for thoughtful comments on the manuscript. We thank Dr. Darryl Zeldin of the NIEHS for the generous contribution of anti-CYP-2J2rec antibody. The present study was supported by NINDS F31 NS060498 (JJI) and the Oregon Brain Institute Graduate Student Fellowship for the Neurobiology of Disease funded by Vertex Pharmaceuticals; NINDS RO1 NS044313 (NJA). Drs. Alkayed and Iliff filed a patent application related the relevance of findings reported here for the treatment of migraine.
Abbreviations
- 14,15-EEZE
14,15-epoxyeicosa-5(Z)-enoic acid
- AUDA
12-[[(tricyclo[3.3.1.13,7]dec-1-ylamino)carbonyl]amino]-dodecanoic acid
- BSA
bovine serum albumin
- CAP
capsaicin
- CBF
cerebral blood flow
- CGRP
calcitonin gene-related peptide
- CYP
cytochrome P450
- DHETs
dihydroxyeicosatrienoic acids
- DMSO
dimethylsulfoxide
- DRG
dorsal root ganglia
- EETs
epoxyeicosatrienoic acids
- ESI
electrospray ionization
- HBS
HEPES buffered saline
- HBSS
Hank’s balanced salt solution
- HETEs
hydroxyeicosatetrenoic acids
- LC-MS/MS
liquid chromatography-tandem mass spectrometry
- MS-PPOH
N-(methylsulfonyl)-2-(2-propynyloxy)-benzenehexanamide
- PBS
phosphate buffered saline
- sEH
soluble epoxide hydrolase
- SP
substance P
- TGN
trigeminal ganglion neuron
- TRPV1
transient receptor potential vanilloid-1
- TRPV4
transient receptor potential vanilloid-4
References
- Alkayed NJ, Birks EK, Narayanan J, Petrie KA, Kohler-Cabot AE, Harder DR. Role of P-450 arachidonic acid epoxygenase in the response of cerebral blood flow to glutamate in rats. Stroke. 1997;28:1066–1072. doi: 10.1161/01.str.28.5.1066. [DOI] [PubMed] [Google Scholar]
- Alkayed NJ, Narayanan J, Gebremedhin D, Medhora M, Roman RJ, Harder DR. Molecular characterization of an arachidonic acid epoxygenase in rat brain astrocytes. Stroke. 1996;27:971–979. doi: 10.1161/01.str.27.5.971. [DOI] [PubMed] [Google Scholar]
- Benemei S, Nicoletti P, Capone JA, Geppetti P. Pain pharmacology in migraine: focus on CGRP and CGRP receptors. Neurol Sci. 2007;28(Suppl 2):S89–93. doi: 10.1007/s10072-007-0757-5. [DOI] [PubMed] [Google Scholar]
- Buldyrev I, Tanner NM, Hsieh HY, Dodd EG, Nguyen LT, Balkowiec A. Calcitonin gene-related peptide enhances release of native brain-derived neurotrophic factor from trigeminal ganglion neurons. J Neurochem. 2006;99:1338–1350. doi: 10.1111/j.1471-4159.2006.04161.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canonico PL, Judd AM, Koike K, Valdenegro CA, MacLeod RM. Arachidonate stimulates prolactin release in vitro: a role for the fatty acid and its metabolites as intracellular regulator(s) in mammotrophs. Endocrinology. 1985;116:218–225. doi: 10.1210/endo-116-1-218. [DOI] [PubMed] [Google Scholar]
- Capdevila J, Chacos N, Falck JR, Manna S, Negro-Vilar A, Ojeda SR. Novel hypothalamic arachidonate products stimulatesomatostatin release from the median eminence. Endocrinology. 1983;113:421–423. doi: 10.1210/endo-113-1-421. [DOI] [PubMed] [Google Scholar]
- Cashman JR. 5,6-Epoxyeicosatrienoic acid stimulates growth hormone release in rat anterior pituitary cells. Life Sciences. 1989;44:1387–1393. doi: 10.1016/0024-3205(89)90396-2. [DOI] [PubMed] [Google Scholar]
- Cashman JR, Hanks D, Weiner RI. Epoxy derivatives of arachidonic acid are potent stimulators of prolactin secretion. Neuroendocrinology. 1987;46:246–251. doi: 10.1159/000124827. [DOI] [PubMed] [Google Scholar]
- Cashman JR, Snowdowne KW. Prolactin secretion in anterior pituitary cells: effect of eicosanoids. Eicosanoids. 1992;5:153–161. [PubMed] [Google Scholar]
- Chen L, Liu C, Liu L. The modulation of voltage-gated potassium channels by anisotonicity in trigeminal ganglion neurons. Neuroscience. 2008;154:482–495. doi: 10.1016/j.neuroscience.2008.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Liu C, Liu L, Cao X. Changes in osmolality modulate voltage-gated sodium channels in trigeminal ganglion neurons. Neurosci Res. 2009;64:199–207. doi: 10.1016/j.neures.2009.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durham PL. Calcitonin gene-related peptide (CGRP) and migraine. Headache. 2006;46(Suppl 1):S3–8. doi: 10.1111/j.1526-4610.2006.00483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durham PL. Inhibition of calcitonin gene-related peptide function: a promising strategy for treating migraine. Headache. 2008;48:1269–1275. doi: 10.1111/j.1526-4610.2008.01215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley S, Heppner TJ, Nelson MT, Brayden JE. TRPV4 forms a novel Ca2+ signaling complex with ryanodine receptors and BKCa channels. Circulation Research. 2005;97:1270–1279. doi: 10.1161/01.RES.0000194321.60300.d6. [DOI] [PubMed] [Google Scholar]
- Edvinsson L, Uddman R. Neurobiology in primary headaches. Brain Res Brain Res Rev. 2005;48:438–456. doi: 10.1016/j.brainresrev.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Ellis EF, Amruthesh SC, Police RJ, Yancey LM. Brain synthesis and cerebrovascular action of cytochrome P-450/monooxygenase metabolites of arachidonic acid. Advances in Prostaglandin, Thromboxane, and Leukotriene Research. 1991;21A:201–204. [PubMed] [Google Scholar]
- Ellis EF, Police RJ, Yancey L, McKinney JS, Amruthesh SC. Dilation of cerebral arterioles by cytochrome P-450 metabolites of arachidonic acid. The American Journal of Physiology. 1990;259:H1171–1177. doi: 10.1152/ajpheart.1990.259.4.H1171. [DOI] [PubMed] [Google Scholar]
- Fang X, Weintraub NL, McCaw RB, Hu S, Harmon SD, Rice JB, Hammock BD, Spector AA. Effect of soluble epoxide hydrolase inhibition on epoxyeicosatrienoic acid metabolism in human blood vessels. Am J Physiol Heart Circ Physiol. 2004;287:H2412–2420. doi: 10.1152/ajpheart.00527.2004. [DOI] [PubMed] [Google Scholar]
- Gauthier KM, Deeter C, Krishna UM, Reddy YK, Bondlela M, Falck JR, Campbell WB. 14,15-Epoxyeicosa-5(Z)-enoic acid: a selective epoxyeicosatrienoic acid antagonist that inhibits endothelium-dependent hyperpolarization and relaxation in coronary arteries. Circulation Research. 2002;90:1028–1036. doi: 10.1161/01.res.0000018162.87285.f8. [DOI] [PubMed] [Google Scholar]
- Grant AD, Cottrell GS, Amadesi S, et al. Protease-activated receptor 2 sensitizes the transient receptor potential vanilloid 4 ion channel to cause mechanical hyperalgesia in mice. J Physiol. 2007;578:715–733. doi: 10.1113/jphysiol.2006.121111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross GJ, Gauthier KM, Moore J, Campbell WB, Falck JR, Nithipatikom K. Evidence for role of epoxyeicosatrienoic acids in mediating ischemic preconditioning and postconditioning in dog. American Journal of Physiology. Heart and Circulatory Physiology. 2009;297:H47–52. doi: 10.1152/ajpheart.01084.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SW, Cho H, Kwak J, et al. Direct activation of capsaicin receptors by products of lipoxygenases: endogenous capsaicin-like substances. Proc Natl Acad Sci U S A. 2000;97:6155–6160. doi: 10.1073/pnas.97.11.6155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Close LN, Selden NR, Alkayed NJ. A novel role for P450 eicosanoids in the neurogenic control of cerebral blood flow in the rat. Exp Physiol. 2007;92:653–658. doi: 10.1113/expphysiol.2006/036889. [DOI] [PubMed] [Google Scholar]
- Iliff JJ, Wang R, Zeldin DC, Alkayed NJ. Epoxyeicosanoids as mediators of neurogenic vasodilation in cerebral vessels. Am J Physiol Heart CircPhysiol. 2009;296:H1352–1363. doi: 10.1152/ajpheart.00950.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inceoglu B, Schmelzer KR, Morisseau C, Jinks SL, Hammock BD. Soluble epoxide hydrolase inhibition reveals novel biological functions of epoxyeicosatrienoic acids (EETs) Prostaglandins & Other Lipid Mediators. 2007;82:42–49. doi: 10.1016/j.prostaglandins.2006.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keseru B, Barbosa-Sicard E, Popp R, et al. Epoxyeicosatrienoic acids and the soluble epoxide hydrolase are determinants of pulmonary artery pressure and the acute hypoxic pulmonary vasoconstrictor response. FASEB J. 2008;22:4306–4315. doi: 10.1096/fj.08-112821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehler RC, Gebremedhin D, Harder DR. Role of astrocytes in cerebrovascular regulation. Journal of Applied Physiology (Bethesda, Md. : 1985) 2006;100:307–317. doi: 10.1152/japplphysiol.00938.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen BT, Miura H, Hatoum OA, Campbell WB, Hammock BD, Zeldin DC, Falck JR, Gutterman DD. Epoxyeicosatrienoic and dihydroxyeicosatrienoic acids dilate human coronary arterioles via BK(Ca) channels: implications for soluble epoxide hydrolase inhibition. American Journal of Physiology. Heart andCirculatory Physiology. 2006;290:H491–499. doi: 10.1152/ajpheart.00927.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Li C, Falck JR, Roman RJ, Harder DR, Koehler RC. Interaction of nitric oxide, 20-HETE, and EETs during functional hyperemia in whisker barrel cortex. American Journal of Physiology. Heart and Circulatory Physiology. 2008;295:H619–631. doi: 10.1152/ajpheart.01211.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luini A, Corda D, Axelrod J. Secretagogues elevate cytosolic calcium by stimulating cyclic AMP formation in a corticotropin secreting cell line. Regul Pept Suppl. 1985;4:49–52. doi: 10.1016/0167-0115(85)90218-6. [DOI] [PubMed] [Google Scholar]
- Luini AG, Axelrod J. Inhibitors of the cytochrome P-450 enzymes block the secretagogue-induced release of corticotropin in mouse pituitary tumor cells. Proceedings of the National Academy of Sciences of the United States of America. 1985;82:1012–1014. doi: 10.1073/pnas.82.4.1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medhora M, Narayanan J, Harder D. Dual regulation of the cerebral microvasculature by epoxyeicosatrienoic acids. Trends Cardiovasc Med. 2001;11:38–42. doi: 10.1016/s1050-1738(01)00082-2. [DOI] [PubMed] [Google Scholar]
- Meng J, Ovsepian SV, Wang J, Pickering M, Sasse A, Aoki KR, Lawrence GW, Dolly JO. Activation of TRPV1 mediates calcitonin gene-related peptide release, which excites trigeminal sensory neurons and is attenuated by a retargeted botulinum toxin with anti-nociceptive potential. J Neurosci. 2009;29:4981–4992. doi: 10.1523/JNEUROSCI.5490-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negro-Vilar A, Snyder GD, Falck JR, Manna S, Chacos N, Capdevila J. Involvement of eicosanoids in release of oxytocin and vasopressin from the neural lobe of the rat pituitary. Endocrinology. 1985;116:2663–2668. doi: 10.1210/endo-116-6-2663. [DOI] [PubMed] [Google Scholar]
- Peng X, Carhuapoma JR, Bhardwaj A, Alkayed NJ, Falck JR, Harder DR, Traystman RJ, Koehler RC. Suppression of cortical functional hyperemia to vibrissal stimulation in the rat by epoxygenase inhibitors. Am J Physiol Heart Circ Physiol. 2002;283:H2029–2037. doi: 10.1152/ajpheart.01130.2000. [DOI] [PubMed] [Google Scholar]
- Peng X, Zhang C, Alkayed NJ, Harder DR, Koehler RC. Dependency of cortical functional hyperemia to forepaw stimulation on epoxygenase and nitric oxide synthase activities in rats. J Cereb Blood Flow Metab. 2004;24:509–517. doi: 10.1097/00004647-200405000-00004. [DOI] [PubMed] [Google Scholar]
- Roman RJ. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev. 2002;82:131–185. doi: 10.1152/physrev.00021.2001. [DOI] [PubMed] [Google Scholar]
- Sadoshima S, Fujii K, Yao H, Kusuda K, Ibayashi S, Fujishima M. Regional cerebral blood flow autoregulation in normotensive and spontaneously hypertensive rats--effects of sympathetic denervation. Stroke; a Journal of Cerebral Circulation. 1986;17:981–984. doi: 10.1161/01.str.17.5.981. [DOI] [PubMed] [Google Scholar]
- Seubert JM, Sinal CJ, Graves J, et al. Role of soluble epoxide hydrolase in postischemic recovery of heart contractile function. Circulation Research. 2006;99:442–450. doi: 10.1161/01.RES.0000237390.92932.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipe WEB, Brierley SM, Martin CM, et al. Transient receptor potential vanilloid 4 mediates protease activated receptor 2-induced sensitization of colonic afferent nerves and visceral hyperalgesia. American Journal of Physiology. Gastrointestinal and Liver Physiology. 2008;294:G1288–1298. doi: 10.1152/ajpgi.00002.2008. [DOI] [PubMed] [Google Scholar]
- Snyder G, Lattanzio F, Yadagiri P, Falck JR, Capdevila J. 5,6-Epoxyeicosatrienoic acid mobilizes Ca2+ in anteriorpituitary cells. Biochemical and Biophysical Research Communications. 1986;139:1188–1194. doi: 10.1016/s0006-291x(86)80303-5. [DOI] [PubMed] [Google Scholar]
- Snyder GD, Capdevila J, Chacos N, Manna S, Falck JR. Action of luteinizing hormone-releasing hormone: involvement of novel arachidonic acid metabolites. Proceedings of the National Academy of Sciences of the United States of America. 1983;80:3504–3507. doi: 10.1073/pnas.80.11.3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector AA, Fang X, Snyder GD, Weintraub NL. Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function. Prog Lipid Res. 2004;43:55–90. doi: 10.1016/s0163-7827(03)00049-3. [DOI] [PubMed] [Google Scholar]
- Spector AA, Norris AW. Action of epoxyeicosatrienoic acids on cellular function. Am J Physiol Cell Physiol. 2007;292:C996–1012. doi: 10.1152/ajpcell.00402.2006. [DOI] [PubMed] [Google Scholar]
- Vriens J, Owsianik G, Fisslthaler B, et al. Modulation of the Ca2 permeable cation channel TRPV4 by cytochrome P450 epoxygenases in vascular endothelium. Circulation Research. 2005;97:908–915. doi: 10.1161/01.RES.0000187474.47805.30. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Vriens J, Prenen J, Droogmans G, Voets T, Nilius B. Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature. 2003;424:434–438. doi: 10.1038/nature01807. [DOI] [PubMed] [Google Scholar]
- Webler AC, Michaelis UR, Popp R, Barbosa-Sicard E, Murugan A, Falck JR, Fisslthaler B, Fleming I. Epoxyeicosatrienoic acids are part of the VEGF-activated signaling cascade leading to angiogenesis. American Journal of Physiology. Cell Physiology. 2008;295:C1292–1301. doi: 10.1152/ajpcell.00230.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub NL, Fang X, Kaduce TL, VanRollins M, Chatterjee P, Spector AA. Epoxide hydrolases regulate epoxyeicosatrienoic acid incorporationinto coronary endothelial phospholipids. Am J Physiol. 1999;277:H2098–2108. doi: 10.1152/ajpheart.1999.277.5.H2098. [DOI] [PubMed] [Google Scholar]
- Wu S, Chen W, Murphy E, et al. Molecular cloning, expression, and functional significance of a cytochrome P450 highly expressed in rat heart myocytes. The Journal of Biological Chemistry. 1997;272:12551–12559. doi: 10.1074/jbc.272.19.12551. [DOI] [PubMed] [Google Scholar]
- Yang W, Tuniki VR, Anjaiah S, Falck JR, Hillard CJ, Campbell WB. Characterization of epoxyeicosatrienoic acid binding site in U937 membranes using a novel radiolabeled agonist, 20-125i-14,15-epoxyeicosa-8(Z)-enoic acid. J Pharmacol Exp Ther. 2008;324:1019–1027. doi: 10.1124/jpet.107.129577. [DOI] [PubMed] [Google Scholar]
- Zeldin DC. Epoxygenase pathways of arachidonic acid metabolism. The Journal of Biological Chemistry. 2001;276:36059–36062. doi: 10.1074/jbc.R100030200. [DOI] [PubMed] [Google Scholar]