

Abstract
The actions of glucocorticoids are mediated, in part, by 11β-hydroxysteroid dehydrogenase 1 (11β-HSD1), which amplifies their effects at the pre-receptor level by converting cortisone to cortisol. Glucocorticoids, such as dexamethasone, inhibit vascular smooth muscle cell proliferation; however, the role of 11β-HSD1 in this response remains unknown. Accordingly, we treated human coronary artery smooth muscle cells (HCSMC) with dexamethasone (10−9–10−6 mol/L) and found that after 72 h, dexamethasone increased 11β-HSD1 expression (14.16±1.6 fold, p<0.001) and activity (6.21 ± 1.2 fold, p<0.001) in a dose- and time-dependent manner, which was dependent upon glucocorticoid receptor (GR) activation and C/EBPβ and C/EBPδ signaling. As glucocorticoids are known to negatively regulate GR expression, we examined the effect of decreasing 11β-HSD1 expression on GR expression. In HCSMC transfected with 11β-HSD1 siRNA, GR expression was increased; this effect was associated with protein kinase A activation and CREB phosphorylation. To examine the role of 11β-HSD1 in HCSMC proliferation, we decreased 11β-HSD1 expression and stimulated cells with platelet-derived growth factor (PDGF) (10 ng/ml). Decreased 11β-HSD1 expression was associated with increased cell proliferation in the absence of PDGF compared to scrambled control-transfected cells (236.10 ± 13.11 %, n=4, p<0.001) and this effect was augmented by PDGF. Furthermore, the inhibitory effect of dexamethasone on cellular proliferation was abrogated in 11β-HSD1 siRNA-transfected HCSMC. Downregulation of 11β-HSD1 was associated with decreased p27kip1 expression and increased phosphorylated retinoblastoma protein, consistent with a proliferative response. These findings suggest that 11β-HSD1 plays a role in the effects of glucocorticoids on vascular smooth muscle cell phenotype.
Keywords: 11β-hydroxysteroid dehydrogenase 1, dexamethasone, glucocorticoids, glucocorticoid receptor, proliferation, vascular smooth muscle cells
INTRODUCTION
Glucocorticoids have been shown to modulate vascular reactivity, the response to injury, and atherogenesis. These effects are mediated by glucocorticoid binding to, and activation of, cognate intracellular glucocorticoid receptors (GR) [1]. Glucocorticoids limit vascular remodeling and neointimal proliferation following vascular injury in part, by inhibiting vascular smooth muscle cell proliferation [2–6]. Local application of dexamethasone to rat carotid arteries following balloon injury has been shown to abrogate neointimal proliferation compared to untreated vessels [7]. Similar studies performed in mouse femoral arteries using a drug-eluting polymer cuff revealed that this finding was associated with vascular atrophy owing to a decreased number of vascular smooth muscle cells [8]. Mechanistically, dexamethasone has been shown to affect these responses by inducing cell cycle arrest at the G1 phase in vascular smooth muscle cells [9, 10].
In mammalian blood vessels, local glucocorticoid concentrations are regulated at the pre-receptor level by the two isozymes of 11β-hydroxysteroid dehydrogenases (11β-HSDs), 11β-HSD1 and 11β-HSD2 [11]. The 11β-HSDs are microsomal enzymes of the short-chain alcohol dehydrogenase superfamily that interconvert active glucocorticoids to their inert 11-keto forms [11]. While 11β-HSD2 is expressed in the vascular endothelium and converts active glucocorticoids to their inactive 11-ketosteroids, 11β-HSD1 is a low-affinity NADP(H)-dependent, predominant reductase that regenerates active 11β-hydroxy forms and is expressed mainly in vascular smooth muscle cells [12–14]. Although not previously demonstrated in vascular smooth muscle cells, in adipocytes and transformed cell lines, it has been shown that glucocorticoids up-regulate expression of 11β-HSD1 through an indirect mechanism that involves members of the CCAAT/enhancer-binding protein (C/EBP) family of transcription factors [15–23]. In this manner, glucocorticoids regulate 11β-HSD1 expression and, in turn, 11β-HSD1 serves to amplify the local response to glucocorticoids [12, 24–26].
Experimental evidence has linked 11β-HSD1 to the effects of glucocorticoids on vascular remodeling. Increased 11β-HSD1 activity resulted in elevated glucocorticoid concentrations and inhibition of angiogenesis in murine injury and repair models while inhibition of 11β-HSD1 was found to decrease atherosclerosis in ApoE knockout mice through a mechanism that did not involve altering lipid profiles [27, 28]. To date, the role of 11β-HSD1 in mediating the effects of glucocorticoids in vascular smooth muscle cells and the mechanism(s) by which this occurs remains incompletely characterized. In the present study, therefore, we hypothesized that glucocorticoids increase 11β-HSD1 expression in vascular smooth muscle cells and that 11β-HSD1 expression was necessary to mediate the response to mitogenic stimuli.
MATERIALS AND METHODS
Cell culture
Human coronary artery smooth muscle cells (HCASMC) (Lonza, Walkersville, MD and Promocell, Heidelberg, Germany) were maintained in smooth muscle cell basal medium (SmBM) supplemented with SmGM SingleQuots (Lonza), without antibiotics, at 37°C in 5% CO2. Cells were passaged twice weekly using 0.5% trypsin/EDTA, and experiments were performed on cells from passages 2–5. In selected studies, to decrease 11β-HSD1 expression, HCASMC were transfected with 40 nmol/L Stealth RNAi™ to 11β-HSD1 mRNA (Invitrogen, Carlsbad, CA) or scrambled control (Invitrogen) using Lipofectamine RNAi MAX (Invitrogen) in OptiMEM media (Invitrogen). After 4 h, transfection media was replaced with full growth media, and experiments were performed after 48 h. To decrease glucocorticoid receptor (GR) expression, HCASMC were transfected with 40 nmol/L Stealth RNAi™ to glucocorticoid receptor mRNA (Invitrogen) using the same protocol as for 11β-HSD1 transfection. To decrease C/EBPβ and C/EBP/δ expression, HCASMC were transfected with 40 nmol/L RNAi to C/EBPβ mRNA (Ambion, Foster City, CA) or C/EBPδ mRNA (Ambion), respectively, using the same protocol as for 11β-HSD1 transfection.
RNA extraction and qRT-PCR
Total RNA from HCASMC were extracted with the RNeasy kit (Qiagen, Germantown, MD), following the manufacturer’s instructions. Samples were quantified and checked for purity and quality by A260/A280 measurements. cDNA was synthesized from 1–5 μg of each total RNA sample with random hexamers using the Superscript III First-Strand Synthesis System for RT-PCR (Invitrogen), according to the manufacturer’s instructions. Quantitative real-time PCR (qRT-PCR) was performed on an Applied Biosystems PRISM 7900 HT Sequence Detector (Foster City, CA). TaqMan® Gene Expression Assays for 11β-HSD1, GR, C/EBPβ, C/EBP/δ, and GAPDH were purchased from Applied Biosystems. PCR products were analyzed using the ΔΔCt method with GAPDH as the endogenous control. Cycle parameters were 95°C for 15 min followed by 40 cycles at 95°C for 15 sec, 58°C for 1 min, and 72°C for 1 min.
Western immunoblotting
Cells were lysed directly in RIPA buffer (sodium phosphate pH 7.2 10 mM, sodium chloride 150 mM, Triton × 100 1%, sodium deoxycholate 0.5%, SDS 0.1%) supplemented with protease and phosphatase inhibitors and stored at −80°C prior to analysis. Protein samples (10–35 μg) were separated on 4–15% SDS-polyacrylamide gels and transferred to PVDF membranes. The membranes were incubated with antibodies to 11β-HSD1, 11β-HSD2, mineralocorticoid receptor, anti-C/EBPβ (Abcam, Cambridge, MA); GR, C/EBPδ, phospho-protein kinase A, protein kinase A α-subunit, cAMP Response Element Binding Protein (CREB), hexose-6-phosphate dehydrogenase (Santa Cruz, Santa Cruz, CA); or phospho-GR, phospho-CREB, p27kip1, phospho-Retinoblastoma protein (Rb)(Ser 608), (Cell Signaling, Danvers, MA) overnight at 4°C, and visualized using the ECL detection system (Amersham Biosciences), according to the manufacturer’s instructions. The membranes were then stripped and reprobed with either a polyclonal rabbit anti-β-actin antibody (Sigma-Aldrich) or a polyclonal rabbit anti-β-tubulin antibody (Santa Cruz). A VersaDoc (BioRad) scanning system was used to quantitate band density.
11β-Hydroxysteroid dehydrogenase activity assay
11β-HSD1 activity was measured as previously described [29] with minor modifications. Briefly, HCASMC were incubated with 100 nmol/L cortisone and [1,2(n)3H]-cortisone (Amersham) for 8h, after which steroids were extracted with dichloromethane and resolved by thin-layer chromatography on silica plates. Plates were imaged with iodine vapor, cortisone and hydrocortisone were identified and isolated from the plate, and the corresponding radioactivity was determined by scintillation counting (Beckman Coulter). Results were standardized by total cellular protein levels.
Protein Kinase A activity assay
Protein kinase A activity was measured using PepTag® Non-Radioactive cAMP-Dependent Protein Kinase Assay (Promega) according to the manufacturer’s instructions.
Proliferation Assay
HCSMC were serum starved for 48h in 0.2% fetal calf serum to synchronize the cells. After this time, cells were stimulated with 5% FCS and 10 ng/ml PDGF-BB in the presence or absence of dexamethasone (10−7 mol/L). Cell proliferation was determined by measuring BrdU incorporation into the DNA of dividing cells using the BrdU Cell Proliferation Assay (Calbiochem, San Diego, CA), according to the manufacturer’s instructions. Results were standardized by cell number. Cell viability was determined using the CellTiter 96 AQueous Non-Radioactive Cell Proliferation Assay (Promega, Madison, Wisconsin) according to the manufacturer’s directions.
Cell cytotoxicity and apoptosis assays
The ratio of dead-to-live cells was determined using the MultiTox-Fluor Cytotoxicity Assay (Promega), and caspase 3, 7 activity was quantified using the Caspase-Glo 3/7 Assay (Promega), according to the manufacturer’s instructions.
Murine aorta isolation, immunohistochemistry, and immunofluorescence
All procedures were approved by the Institutional Animal Care and Use Committee at Brigham and Women’s Hospital and Harvard Medical School. Aortas were harvested from C57Bl/6 male mice (Charles River) age 12–16 weeks. The vessels were irrigated with ice-cold phosphate-buffered saline until free of blood, dissected free of adventitia, and cut into 5 mm rings. The rings were placed in smooth muscle cell basal medium (Lonza) and incubated at 37°C in 5% CO2. To decrease 11βHSD-1 expression, 80 nmol/L Stealth RNAi™ to 11β-HSD1 mRNA (Invitrogen, Carlsbad, CA) or scrambled control (Invitrogen) and Lipofectamine RNAi MAX (Invitrogen) was added to the medium. At selected time points, the aortas were embedded in paraffin and 5 μn sections were analyzed by hematoxylin & eosin staining, immunohistochemistry or immunofluorescence as described previously [30, 31] using antibodies to 11β-HSD1 (abcam) and Ki67 (BD Biosciences), respectively.
Data analysis
All experiments were performed a minimum of three times at least in duplicate and data presented are expressed as mean ± standard error of the mean (SEM). The statistical significance of differences was determined by ANOVA followed by Bonferroni’s pairwise comparison using GraphPad Prism 3.0 software (GraphPad Software, Inc, La Jolla, CA). A p value of <0.05 was considered significant.
RESULTS
Dexamethasone increases 11β-HSD1 expression and activity
To determine the effect of dexamethasone on 11β-HSD1 expression in HCSMC, we first assessed steady-state levels of 11β-HSD1 mRNA. Dexamethasone increased 11β-HSD1 mRNA in a dose-dependent manner with the maximum effect observed at 10−7 mol/L (6.5 ± 0.7-fold increase at 18 h vs. control, n=3, p<0.01) (Fig. 1a). As we saw no further increase in 11β-HSD1 mRNA levels with higher concentrations of dexamethasone, we elected to perform experiments on HCSMC treated with dexamethasone 10−7 mol/L. Dexamethasone also increased 11β-HSD1 mRNA in a time-dependent manner with the maximum effect observed at 72 h (116 ± 36-fold increase vs. control, n=3, p<0.01) (Fig. 1b); no further increase in mRNA levels were observed at 96 h. This finding was associated with an increase in 11β-HSD1 protein levels (14.6 ± 1.6-fold increase vs. control, n=3, p<0.001) (Fig. 1c) and 11β-HSD1 reductase activity (Fig. 1d). We found that under these treatment conditions, there was no effect of dexamethasone on 11β-HSD2 or hexose-6-phosphate dehydrogenase protein expression (Supplementary Fig. 1a, 1b).
Fig. 1. Dexamethasone increases 11β-HSD1 expression and activity.

Human coronary artery smooth muscle cells (HCSMC) (a) were treated with increasing concentrations of dexamethasone and 11β-HSD1 mRNA levels were determined by qRT-PCR after 18 h (n=3). HCSMC were treated with dexamethasone (10−7 mol/L) for 0–72 h and 11β-HSD1 (b) mRNA levels were determined by qRT-PCR (n=3), (c) protein expression was examined by Western immunoblotting (n=3), and (d) 11β-HSD1 activity was measured. (n=3). Data are presented as mean ± SEM. A representative blot is shown.*p<0.01 vs. UN, 0. UN, treated; Dex, dexamethasone
The glucocorticoid receptor, C/EBPβ, and C/EBPδ modulate 11β-HSD1 expression
To examine the role of glucocorticoid receptor (GR) activation on the dexamethasone-mediated increase in 11β-HSD1 expression, we first examined GR expression and phosphorylation. While dexamethasone treatment for 24 h decreased total GR protein levels by 45.3 ± 8.6% compared to control cells (n=3, p<0.01) (Fig. 2a), phosphorylation at Ser 211 was increased by 52.4 ± 17.3% compared to control (n=3, p<0.05) (Fig. 2b), indicating activation of the receptor. This increase in phospho-GR was transient, peaking at 24 h. In contrast, we found that under these conditions, dexamethasone had no effect on MR expression in HCSMC (Supplementary Fig. 1c).
Fig. 2. The effect of dexamethasone on glucocorticoid receptor, C/EBPβ and C/EBPδ expression.

Human coronary artery smooth muscle cells (HCSMC) were treated with 10−7 mol/L dexamethasone for 0–72 h and expression of (a) the glucocorticoid receptor (GR) (n=3) and (b) the phospho-GR (n=3) were determined by Western immunoblotting. At the same timepoints, (c) C/EBPβ and (d) C/EBPδ mRNA levels were determined by qRT-PCR (n=3) and corresponding (e) C/EBPβ and (f) C/EBPδ protein levels were assessed by Western blotting (n=3). Data are presented as mean ± SEM. Representative blots are shown. *p<0.05 vs. 0
We next examined the effect of GR activation on expression of members of the CCAAT/enhancer-binding protein (C/EBP) family of transcription factors, as these have been shown to modulate 11β-HSD1 enzyme expression in other cell types [20–23]. Dexamethasone increased C/EBPβ (53.2 ± 11%, vs. control, n=3, p<0.05) (Fig. 2c) mRNA levels and transiently increased C/EBPβ protein levels (80.2 ± 4.7% vs. control, n=3, p<0.01) (Fig. 2d). Dexaethasone also increased C/EBPδ mRNA levels (241.1 ± 19% vs. control, n=5, p<0.001) (Fig. 2e), as well as C/EBPδ protein levels (223.9±6% vs. control, p<0.001) (Fig. 2f).
Next, to demonstrate that GR activation was necessary for the observed increase in 11β-HSD1 expression, we transfected HCASMC with an siRNA to GR mRNA to decrease protein expression by 75.6% (n=3, p<0.001) compared to scrambled control-transfected cells, and examined the effect of GR downregulation on 11β-HSD1 expression. When HCSMC transfected with GR siRNA were treated with dexamethasone, we found a 46.5-fold (n=3, p<0.001) decrease in 11β-HSD1 mRNA (Fig. 3a) and an 8.7-fold decrease (n=3, p<0.001) in 11β-HSD1 protein levels (Fig. 3b) compared to scrambled control-transfected cells treated with dexamethasone. These data indicate that GR activation is necessary for the dexamethasone-dependent increase in 11β-HSD1 expression.
Fig. 3. The glucocorticoid receptor, C/EBPβ, and C/EBPδ regulate the dexamethasone-mediated increase in 11β-HSD1 expression.

Human coronary artery smooth muscle cells (HCSMC) were transfected with an siRNA to decrease glucocorticoid receptor (GR) expression or a scrambled control (SS), treated with dexamethasone (10−7 mol/L), and (a) 11β-HSD1 mRNA levels were determined by qRT-PCR (n=3) and (b) protein expression by Western immunoblotting (n=3). HCSMC were transfected with an siRNA to decrease C/EBPβ or C/EBPδ expression, respectively, or a scrambled control, treated with dexamethasone (10−7 mol/L), and (c) 11β-HSD1 mRNA levels were determined by qRT-PCR (n=3) and (d) protein expression by Western immunoblotting (n=3). mRNA and protein levels were determined. Data are presented as mean ± SEM and representative blots are shown. *p<0.001 vs. SS, **p<0.001 vs. SS + Dex
Similarly, to determine if the transcription factors C/EBPβ or C/EBPδ, which are activated by the GR, were necessary for the observed increase in 11β-HSD1 expression, we first transfected HCSMC with an siRNA to C/EBPβ to decrease C/EBPβ protein levels by 71.5% (n=3, p<0.001) compared to scrambled control-transfected cells. Downregulation of C/EBPβ attenuated the effect of dexamethasone on 11β-HSD1 mRNA (4.6-fold, n=3, p<0.001) and protein levels (7.4-fold, n=3, p<0.001) compared to scrambled control-transfected cells (Fig. 3c, 3d). We next transfected HCSMC with an siRNA to C/EBPδ, which decreased C/EBPδ protein expression by 80.3% (n=3, p<0.01), and found that downregulation of C/EBPδ inhibited the dexamethasone-mediated increase of 11β-HSD1 mRNA (2.1-fold, n=3, p<0.001) and protein levels (2.0-fold, n=3, p=0.01) compared to scrambled control-transfected cells (Fig. 3c, 3d). Together, these data indicate that both C/EBPβ and C/EBPδ are necessary to increase 11β-HSD1 mRNA and protein levels in response to dexamethasone.
11β-HSD1 is involved in the regulation of GR expression
As GR expression is negatively regulated by its ligand, and 11β-HSD1 activity is an important pre-receptor mechanism to generate GR ligand, we also examined the effect of decreasing the expression of 11β-HSD1 on GR expression. When we transfected HCSMC with an siRNA to 11β-HSD1 to decrease protein expression by 64% (p<0.01), we found that compared to scrambled control-transfected cells, downregulation of 11β-HSD1 resulted in an increase in GR protein expression (Fig. 4a). Furthermore, while dexamethasone decreased GR protein expression in scrambled control-transfected cells, this response was abrogated in 11β-HSD1 siRNA-transfected HCSMC suggesting that 11β-HSD1 plays a role in the dexamethasone-mediated downregulation of GR expression (Fig. 4a).
Fig. 4. 11β-HSD1 regulates glucocorticoid receptor expression.

HCSMC were transfected with an siRNA to decrease 11β-HSD1 expression, treated with dexamethasone (10−7 mol/L), and glucocorticoid receptor (GR) (a) protein expression was determined by Western immunoblotting (n=3). (b) Protein kinase A (PKA) and phospho-PKA expression were assessed by Western blotting (n=3), and (c) PKA activity was measured (n=4). (d) Expression of the cyclic AMP response element binding protein (CREB) and phospho-CREB were assayed by Western blotting (n=3). Data are presented as mean ± SEM and representative blots are shown. *p<0.001 vs. SS; **p<0.001 vs. HSD1 siRNA; #p<0.05 vs. SS
The cyclic AMP response element binding protein (CREB) has been shown to regulate transcription of the GR gene in response to glucocorticoid levels [26]. To determine if 11β-HSD1 modulated GR expression by influencing activation of CREB, we first examined expression and activity of protein kinase A, which is known to phosphorylate and activate CREB. In scrambled control-transfected HCSMC, dexamethasone decreased phosphorylation of protein kinase A, which was associated with a decrease in protein kinase A activity (Fig. 4b, 4c). Interestingly, 11β-HSD1 siRNA-transfected HCSMC demonstrated increased protein kinase A phosphorylation and activity compared to scrambled control-transfected cells, and this effect was not influenced significantly by dexamethasone (Fig. 4b, 4c). In line with these findings, dexamethasone treatment decreased CREB phosphorylation in scrambled control-transfected cells, while HCSMC with decreased 11β-HSD1 expression demonstrated increased CREB phosphorylation (Fig. 4d). These findings suggest that 11β-HSD1 expression influences CREB activation as a mechanism by which to regulate GR expression.
Role of 11β-HSD1 in PDGF-stimulated HCSMC proliferation
Glucocorticoids have been shown to regulate vascular smooth muscle cell proliferation; however, the role of 11β-HSD1 in this process remains incompletely characterized. We, therefore, examined the effect of dexamethasone on HCSMC proliferation in cells with normal 11β-HSD1 expression and decreased 11β-HSD1 expression. After 48 h of serum starvation, HCSMC were stimulated with 5% serum and recombinant human PDGF-BB (10 ng/ml) in the presence or absence of dexamethasone (10−7 mol/L). In scrambled control-transfected HCSMC, PDGF-BB increased proliferation as determined by BrdU incorporation (100.38 ± 14.72 vs. 581.32 ± 11.87 arb. units/cell number, n=4, p<0.001), while the addition of dexamethasone to PDGF-BB-stimulated cells decreased cell proliferation (253.42 ± 7.15 arb. units/cell number, n=4, p<0.001). In 11β-HSD1 siRNA-transfected HCSMC, there was an increase in proliferation compared to scrambled control-transfected cells under basal conditions (100.38 ± 14.72 vs. 237.10 ± 12.72 arb. units/cell number, n=4, p<0.001). While PDGF-BB stimulation increased proliferation significantly in 11β-HSD1 siRNA-transfected HCSMC (237.10 ± 12.72 vs. 734.03 ± 29.37 arb. units/cell number, n=4, p<0.001), the addition of dexamethasone had a somewhat attenuated effect in inhibiting proliferation (734.03 ± 29.37 vs. 437.07 ± 36.83 arb. units/cell number, n=4, p<0.001) compared to what was observed in scrambled control-transfected cells (Fig. 5). To demonstrate the potential in vivo relevance of these findings, we harvested aortas from C57Bl/6 wild-type mice (n=12), sectioned the vessels to 5 mm rings, transfected them with siRNA to 11β-HSD1 to decrease protein expression by 52% (p<0.01) or a scrambled control (Fig. 6a), and maintained them in culture for up to 72 h. In selected sections, we stimulated the rings with recombinant rat PDGF-BB (10 ng/ml) in the presence or absence of dexamethasone (10−7 mol/L). Following stimulation with PDGF-BB, co-incubation with dexamethasone decreased cell proliferation, determined by the average number of Ki67 positive fluorescent cells/10 high powered fields (hpf), in scrambled control-transfected aortas (18.21 ± 5.45 vs 7.67± 3.66 cells/hpf, p<0.01). Similar to what was observed with HCSMC, in PDGF-stimulated aorta sections transfected with 11β-HSD1 siRNA that were co-incubated with dexamethasone, there was an attenuated effect in inhibiting proliferation (30.11 ± 3.87 vs. 24.55 ± 4.33 cells/hpf, p=0.07) (Fig. 6b).
Fig. 5. 11β-HSD1 mediates the inhibitory effects of dexamethasone on PDGF-stimulated HCSMC.

Human coronary artery smooth muscle cells (HCSMC) were transfected with an siRNA to decrease 11β-HSD1 expression, synchronized by serum starvation (0.2% serum) for 48 h, and stimulated with 5% fetal calf serum and platelet-derived growth factor-BB (PDGF) (10 ng/ml) in the presence or absence of dexamethasone (10−7 mol/L). Cell proliferation was determined by measuring BrdU incorporation (n=4). Data are presented as mean ± SEM and results are corrected for cell number. *p<0.001 vs. SS; **p<0.001 vs. SS + PDGF; #p<0.001 vs. HSD1 siRNA; ##p<0.001 vs. HSD1 siRNA + PDGF
Fig. 6. 11β-HSD1 mediates the inhibitory effects of dexamethasone on proliferation in PDGF-stimulated murine aortas.
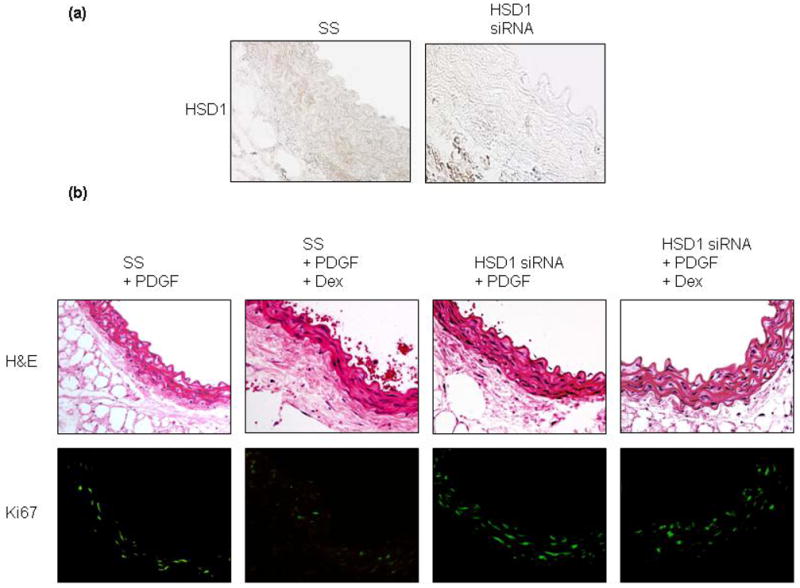
Thoracic aortas from C57Bl/6 male mice (n=12) were isolated, sectioned into 5 mm rings, and transfected with 80 nmol/L Stealth RNAi™ to 11β-HSD1 mRNA or scrambled control (SS) added to smooth muscle basal medium. (a) After 48 h, 11β-HSD1 expression was examined by immunohistochemistry of paraffin embedded sections. (b) The influence of 11β-HSD1 on platelet-derived growth factor-BB (PDGF) (10 ng/ml)-stimulated proliferation, in the presence or absence of dexamethasone (10−7 mol/L), was examined by immunofluorescence labeling of Ki67. Representative hematoxylin & eosin stained sections are provided for comparison (top) and corresponding fluorescent sections are shown (bottom). Images are magnified 200X.
We next sought to determine a mechanism by which to explain the observed differences in proliferation between HCSMC with normal levels of 11β-HSD1 expression and those with decreased 11β-HSD1 expression. We found that there was no difference between the ratio of dead-to-live cells between scrambled control-transfected HCSMC and those with decreased11β-HSD1 expression (0.93 ± 0.08 vs. 0.93 ± 0.15, n=4, p=NS), and we found no difference in apoptosis between the two groups (17.54 ± 6.80 vs. 17.53 ± 4.68 relative light units, n=4, p=NS). These results suggest that the observed differences in cell proliferation could not be attributed to an increase in cell death or apoptosis.
Next, we examined expression of p27kip1 and its downstream target phosphorylated retinoblastoma (Rb) protein as both of these regulate the G1-phase of the cell cycle, are subject to control by glucocorticoids, and have been implicated in mediating vascular smooth muscle cell proliferation. In accordance with previously published reports, we found in scrambled control-transfected cells that PDGF-BB decreased p27kip1 expression and increased phosphorylation of Rb at Ser608, consistent with progression through the cell cycle and proliferation; the addition of dexamethasone to PDGF-stimulated cells increased p27kip1 expression and decreased phosphorylation of Rb, indicating a cell cycle block and inhibition of proliferation. In contrast, in 11β-HSD1 siRNA-transfected HCSMC, p27kip1 expression was decreased and phosphorylation of Rb was increased compared to scrambled-control transfected cells under basal conditions. Here, stimulation with PDGF resulted in a modest, further decrease in p27kip1 expression and the addition of dexamethasone resulted in a minor increase in p27kip1 expression and corresponding decrease in phospho-Rb (Fig. 7a, 7b). Taken together, these data demonstrate that 11β-HSD1 participates in glucocorticoid-mediated inhibition of HCSMC proliferation in response to PDGF, in part, by modulating p27kip1 expression and Rb phosphorylation to facilitate the cell cycle block at G1.
Fig. 7. 11β-HSD1 influences the expression of cell cycle regulators in response to PDGF and dexamethasone.

Human coronary artery smooth muscle cells (HCSMC) were transfected with an siRNA to decrease 11β-HSD1 expression, synchronized by serum starvation (0.2% serum) for 48 h, and stimulated with 5% fetal calf serum and platelet-derived growth factor-BB (PDGF) (10 ng/ml) in the presence or absence of dexamethasone (10−7 mol/L). Expression of (a) p27kip1 (n=3) and (b) phosphorylation of retinoblastoma protein (phospho-Rb) at Ser608 (n=3) were determined by Western immunoblotting. Representative blots are shown
DISCUSSION
In these studies, we found that dexamethasone increases 11β-HSD1 expression and activity in HCSMC in a GR-dependent manner to regulate cell proliferation through p27kip1 and Rb. We also identified a role for 11β-HSD1 in the regulation of GR expression. There are several prior reports that demonstrate that glucocorticoids increase 11β-HSD1 expression in non-vascular cells and one study showing an increase in 11β-HSD1 mRNA levels in rat aortic smooth muscle cells [32, 33]. Our studies now extend these observations to human vascular smooth muscle cells. Furthermore, we demonstrate that 11β-HSD1 expression in response to glucocorticoids is dependent upon GR activation as evidenced by the lack of increase in 11β-HSD1 expression in HCSMC transfected with an siRNA to GR.
The time course of upregulation of 11β-HSD1 expression observed in our studies is consistent with indirect regulation of the 11β-HSD1 promoter by glucocorticoids, paralleling previous reports in human fibroblasts and in A549 cells [15, 23]. The transcriptional activity of glucocorticoids has been linked to the C/EBP family of transcription factors that interact with either the GR or target gene promoters to promote transcription. In addition, C/EBPs, primarily C/EBPβ, mediate the effect of glucocorticoids by inducing transcription of genes that lack GR binding sites [34–36]. We find that C/EBPβ, and to a lesser extent C/EBPδ, are important for the dexamethasone-mediated increase in 11β-HSD1 expression in HCSMC. Our findings are in accord with previously published studies that established the central role of C/EBPs in the regulation of 11β-HSD1 transcription in liver, lung, and adipose cells [20–23].
It is also recognized that glucocorticoids negatively regulate expression of the GR [37, 38]. In our studies, we demonstrate 11β-HSD1 mediates dexamethasone-mediated downregulation of GR expression in HCSMC. Here, we showed that decreased 11β-HSD1 expression, which would decrease levels of active glucocorticoids, was associated with an increase in GR expression compared to control cells and cells exposed to dexamethasone, indicating that 11β-HSD1 plays a role in the regulation of GR expression under ambient conditions. It has been demonstrated that GR expression is controlled at the transcriptional level by the phosphorylation state of CREB [39]. We found that downregulation of GR was associated with decreased CREB phosphorylation in cells with normal levels of 11β-HSD1. Interestingly, we also observed that when we decreased 11β-HSD1 expression, which resulted in an increase in GR expression, there was an associated increase in phospho-CREB levels. We further examined protein kinase A activation as a mechanism of CREB phosphorylation, as a prior study demonstrated that GR transcription is dependent on protein kinase A signaling [40]. Here, we found that increased activation of protein kinase A was associated with the increase in CREB phosphorylation.
As glucocorticoids modulate vascular smooth muscle cell phenotype, we examined the role of 11β-HSD1 in regulating the effect of dexamethasone on vascular smooth muscle cell proliferation. While prolonged exposure to glucocorticoids inhibits proliferation of cultured vascular smooth muscle cells by inducing a cell cycle arrest at the G1 phase [9, 10, 41, 42], the role of 11β-HSD1 in mediating this effect is less well defined. In prior studies performed in an osteosarcoma cell line transfected with 11β-HSD1, expression of 11β-HSD1 was associated with decreased cell proliferation that was attributed to metabolism of endogenous glucocorticoids to increase cortisol levels [43]. This observation supports our findings that HCSMC with decreased 11β-HSD1 expression demonstrate increased proliferation under basal conditions and in response to PDGF as compared to cells with normal 11β-HSD1 expression. The diminished inhibitory effect of dexamethasone in cells with decreased 11β-HSD1 expression suggests that endogenous metabolism of glucocorticoids is involved in modulating cell proliferation. Furthermore, the translational relevance of these findings was confirmed ex vivo in murine aortas with decreased 11β-HSD1 expression.
The precise mechanism by which glucocorticoids inhibit cell proliferation is not yet known; however, it has been shown that glucocorticoids influence the expression of cyclin-dependent kinases and cyclin-dependent kinase inhibitors, including p27kip1 [10]. Accordingly, we investigated the role of 11β-HSD1 in modulating the expression of p27kip1 and the phosphorylation state of Rb. Rb, which lies downstream of p27kip1, has also been linked to the inhibitory effects of dexamethasone on vascular smooth muscle cell proliferation [10, 41, 44]. Consistent with our findings on the effects of 11β-HSD1 expression on proliferation, we found that decreased 11β-HSD1 expression was associated with a downregulation of the cyclin-dependent kinase inhibitor p27kip1, leading to increased phosphorylation of Rb to promote exit from G1 and cell proliferation.
Taken together, our findings indicate that glucocorticoids stimulate a “feed-forward” mechanism to enhance local generation of active glucocorticoids by upregulating the expression and activity of 11β-HSD1 in HCSMC. This increase in 11β-HSD1 expression further serves to regulate the actions of GR activation by modulating GR expression through a protein kinase A–CREB-dependent mechanism. Although this may seem counterintuitive, such a signaling mechanism may serve to redirect glucocorticoids to activate the mineralocorticoid receptor in cells and tissues that don’t express 11β-HSD2. In vascular smooth muscle cells, evidence exists that this mechanism is operative [45]. In HCSMC, we also demonstrate that 11β-HSD1 expression is also necessary to mediate cellular proliferation under basal and growth factor-stimulated conditions. These findings suggest that 11β-HSD1 may serve as a target for therapeutic intervention in vascular disease states characterized by excess vascular smooth muscle cell proliferation, such as restenosis. In fact, selective 11β-HSD1 inhibition has already been shown to limit atherosclerotic neointima formation, and 11β-HSD1−/− mice have a cardioprotective phenotype that is characterized by increased angiogenesis following injury [27, 28]. Our findings suggest further that systemic administration of novel 11β-HSD1 inhibitors that are currently under investigation for obesity and diabetes may have effects on the vasculature [28, 46–49].
Supplementary Material
Acknowledgments
The authors would like to thank Professor Vassilis I. Zannis for thoughtful discussions of the data and Ms. Stephanie Tribuna for expert secretarial assistance.
This work was supported by NIH grants HL61795, HL81587, HL70819, HL89734 (JL) HL81110, and HL700819 (JAL).
References
- 1.Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berk BC, Vallega G, Griendling KK, Gordon JB, Cragoe EJ, Jr, Canessa M, Alexander RW. Effects of glucocorticoids on Na+/H+ exchange and growth in cultured vascular smooth muscle cells. J Cell Physiol. 1988;137:391–401. doi: 10.1002/jcp.1041370302. [DOI] [PubMed] [Google Scholar]
- 3.Longenecker JP, Kilty LA, Johnson LK. Glucocorticoid influence on growth of vascular wall cells in culture. J Cell Physiol. 1982;113:197–202. doi: 10.1002/jcp.1041130203. [DOI] [PubMed] [Google Scholar]
- 4.Longenecker JP, Kilty LA, Johnson LK. Glucocorticoid inhibition of vascular smooth muscle cell proliferation: influence of homologous extracellular matrix and serum mitogens. J Cell Biol. 1984;98:534–540. doi: 10.1083/jcb.98.2.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cavallero C, Di Tondo U, Mingazzini PL, Nicosia R, Pericoli MN, Sarti P, Spagnoli LG, Villaschi S. Cell proliferation in the atherosclerotic plaques of cholesterol-fed rabbits. Part 3. Histological and radioautographic observations on glucocorticoids-treated rabbits. Atherosclerosis. 1976;25:145–152. doi: 10.1016/0021-9150(76)90020-4. [DOI] [PubMed] [Google Scholar]
- 6.Versaci F, Gaspardone A, Tomai F, Ribichini F, Russo P, Proietti I, Ghini AS, Ferrero V, Chiariello L, Gioffre PA, Romeo F, Crea F. Immunosuppressive Therapy for the Prevention of Restenosis after Coronary Artery Stent Implantation (IMPRESS Study) J Am Coll Cardiol. 2006;40:1935–1942. doi: 10.1016/S0735-1097(02)02562-7. [DOI] [PubMed] [Google Scholar]
- 7.Villa AE, Guzman LA, Chen W, Golomb G, Levy RJ, Topol EJ. Local delivery of dexamethasone for prevention of neointimal proliferation in a rat model of balloon angioplasty. J Clin Invest. 1994;93:1243–1249. doi: 10.1172/JCI117078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pires NM, Schepers A, van der Hoeven BL, de Vries MR, Boesten LS, Jukema JW, Quax PH. Histopathologic alterations following local delivery of dexamethasone to inhibit restenosis in murine arteries. Cardiovasc Res. 2005;68:415–424. doi: 10.1016/j.cardiores.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 9.Sanchez I, Goya L, Vallerga AK, Firestone GL. Glucocorticoids reversibly arrest rat hepatoma cell growth by inducing an early G1 block in cell cycle progression. Cell Growth Differ. 1993;4:215–225. [PubMed] [Google Scholar]
- 10.Rogatsky I, Trowbridge JM, Garabedian MJ. Glucocorticoid receptor-mediated cell cycle arrest is achieved through distinct cell-specific transcriptional regulatory mechanisms. Mol Cell Biol. 1997;17:3181–3193. doi: 10.1128/mcb.17.6.3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stewart PM, Krozowski ZS. 11 beta-Hydroxysteroid dehydrogenase. Vitam Horm. 1999;57:249–324. doi: 10.1016/S0083-6729(08)60646-9. [DOI] [PubMed] [Google Scholar]
- 12.Hadoke PW, Iqbal J, Walker BR. Therapeutic manipulation of glucocorticoid metabolism in cardiovascular disease. Br J Pharmacol. 2009;156:689–712. doi: 10.1111/j.1476-5381.2008.00047.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bujalska IJ, Kumar S, Stewart PM. Does central obesity reflect “Cushing’s disease of the omentum”? Lancet. 1997;349:1210–1213. doi: 10.1016/S0140-6736(96)11222-8. [DOI] [PubMed] [Google Scholar]
- 14.Seckl JR, Walker BR. Minireview: 11beta-hydroxysteroid dehydrogenase type 1- a tissue-specific amplifier of glucocorticoid action. Endocrinology. 2001;142:1371–1376. doi: 10.1210/en.142.4.1371. [DOI] [PubMed] [Google Scholar]
- 15.Hammami MM, Siiteri PK. Regulation of 11 beta-hydroxysteroid dehydrogenase activity in human skin fibroblasts: enzymatic modulation of glucocorticoid action. J Clin Endocrinol Metab. 1991;73:326–334. doi: 10.1210/jcem-73-2-326. [DOI] [PubMed] [Google Scholar]
- 16.Jamieson PM, Chapman KE, Edwards CR, Seckl JR. 11 beta-hydroxysteroid dehydrogenase is an exclusive 11 beta- reductase in primary cultures of rat hepatocytes: effect of physicochemical and hormonal manipulations. Endocrinology. 1995;136:4754–4761. doi: 10.1210/en.136.11.4754. [DOI] [PubMed] [Google Scholar]
- 17.Whorwood CB, Donovan SJ, Wood PJ, Phillips DI. Regulation of glucocorticoid receptor alpha and beta isoforms and type I 11beta-hydroxysteroid dehydrogenase expression in human skeletal muscle cells: a key role in the pathogenesis of insulin resistance? J Clin Endocrinol Metab. 2001;86:2296–2308. doi: 10.1210/jc.86.5.2296. [DOI] [PubMed] [Google Scholar]
- 18.Cooper MS, Rabbitt EH, Goddard PE, Bartlett WA, Hewison M, Stewart PM. Osteoblastic 11beta-hydroxysteroid dehydrogenase type 1 activity increases with age and glucocorticoid exposure. J Bone Miner Res. 2002;17:979–986. doi: 10.1359/jbmr.2002.17.6.979. [DOI] [PubMed] [Google Scholar]
- 19.Bujalska IJ, Kumar S, Hewison M, Stewart PM. Differentiation of adipose stromal cells: the roles of glucocorticoids and 11beta-hydroxysteroid dehydrogenase. Endocrinology. 1999;140:3188–3196. doi: 10.1210/en.140.7.3188. [DOI] [PubMed] [Google Scholar]
- 20.Williams LJ, Lyons V, MacLeod I, Rajan V, Darlington GJ, Poli V, Seckl JR, Chapman KE. C/EBP regulates hepatic transcription of 11beta -hydroxysteroid dehydrogenase type 1. A novel mechanism for cross-talk between the C/EBP and glucocorticoid signaling pathways. J Biol Chem. 2000;275:30232–30239. doi: 10.1074/jbc.M001286200. [DOI] [PubMed] [Google Scholar]
- 21.Gout J, Tirard J, Thevenon C, Riou JP, Begeot M, Naville D. CCAAT/enhancer-binding proteins (C/EBPs) regulate the basal and cAMP-induced transcription of the human 11beta-hydroxysteroid dehydrogenase encoding gene in adipose cells. Biochimie. 2006;88:1115–1124. doi: 10.1016/j.biochi.2006.05.020. [DOI] [PubMed] [Google Scholar]
- 22.Bruley C, Lyons V, Worsley AG, Wilde MD, Darlington GD, Morton NM, Seckl JR, Chapman KE. A novel promoter for the 11beta-hydroxysteroid dehydrogenase type 1 gene is active in lung and is C/EBPalpha independent. Endocrinology. 2006;147:2879–2885. doi: 10.1210/en.2005-1621. [DOI] [PubMed] [Google Scholar]
- 23.Sai S, Esteves CL, Kelly V, Michailidou Z, Anderson K, Coll AP, Nakagawa Y, Ohzeki T, Seckl JR, Chapman KE. Glucocorticoid regulation of the promoter of 11beta-hydroxysteroid dehydrogenase type 1 is indirect and requires CCAAT/enhancer-binding protein-beta. Mol Endocrinol. 2008;22:2049–2060. doi: 10.1210/me.2007-0489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hadoke PW, Macdonald L, Logie JJ, Small GR, Dover AR, Walker BR. Intra-vascular glucocorticoid metabolism as a modulator of vascular structure and function. Cell Mol Life Sci. 2006;63:565–578. doi: 10.1007/s00018-005-5427-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Christy C, Hadoke PW, Paterson JM, Mullins JJ, Seckl JR, Walker BR. 11beta-hydroxysteroid dehydrogenase type 2 in mouse aorta: localization and influence on response to glucocorticoids. Hypertension. 2003;42:580–587. doi: 10.1161/01.HYP.0000088855.06598.5B. [DOI] [PubMed] [Google Scholar]
- 26.Walker BR, Yau JL, Brett LP, Seckl JR, Monder C, Williams BC, Edwards CR. 11 beta-hydroxysteroid dehydrogenase in vascular smooth muscle and heart: implications for cardiovascular responses to glucocorticoids. Endocrinology. 1991;129:3305–3312. doi: 10.1210/endo-129-6-3305. [DOI] [PubMed] [Google Scholar]
- 27.Small GR, Hadoke PW, Sharif I, Dover AR, Armour D, Kenyon CJ, Gray GA, Walker BR. Preventing local regeneration of glucocorticoids by 11beta-hydroxysteroid dehydrogenase type 1 enhances angiogenesis. Proc Natl Acad Sci U S A. 2005;102:12165–12170. doi: 10.1073/pnas.0500641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hermanowski-Vosatka A, Balkovec JM, Cheng K, Chen HY, Hernandez M, Koo GC, Le Grand CB, Li Z, Metzger JM, Mundt SS, Noonan H, Nunes CN, Olson SH, Pikounis B, Ren N, Robertson N, Schaeffer JM, Shah K, Springer MS, Strack AM, Strowski M, Wu K, Wu T, Xiao J, Zhang BB, Wright SD, Thieringer R. 11beta-HSD1 inhibition ameliorates metabolic syndrome and prevents progression of atherosclerosis in mice. J Exp Med. 2005;202:517–527. doi: 10.1084/jem.2005011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bujalska IJ, Draper N, Michailidou Z, Tomlinson JW, White PC, Chapman KE, Walker EA, Stewart PM. Hexose-6-phosphate dehydrogenase confers oxo-reductase activity upon 11 beta-hydroxysteroid dehydrogenase type 1. J Mol Endocrinol. 2005;34:675–684. doi: 10.1677/jme.1.01718. [DOI] [PubMed] [Google Scholar]
- 30.Leopold JA, Dam A, Maron BA, Scribner AW, Liao R, Handy DE, Stanton RC, Pitt B, Loscalzo J. Aldosterone impairs vascular reactivity by decreasing glucose-6-phosphate dehydrogenase activity. Nat Med. 2007;13:189–197. doi: 10.1038/nm1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leopold JA, Walker J, Scribner AW, Voetsch B, Zhang YY, Loscalzo AJ, Stanton RC, Loscalzo J. Glucose-6-phosphate dehydrogenase modulates vascular endothelial growth factor-mediated angiogenesis. J Biol Chem. 2003;278:32100–32106. doi: 10.1074/jbc.M301293200. [DOI] [PubMed] [Google Scholar]
- 32.Takeda Y, Miyamori I, Yoneda T, Ito Y, Takeda R. Expression of 11 beta-hydroxysteroid dehydrogenase mRNA in rat vascular smooth muscle cells. Life Sci. 1994;54:281–285. doi: 10.1016/0024-3205(94)00818-3. [DOI] [PubMed] [Google Scholar]
- 33.Tomlinson JW, Walker EA, Bujalska IJ, Draper N, Lavery GG, Cooper MS, Hewison M, Stewart PM. 11beta-hydroxysteroid dehydrogenase type 1: a tissue-specific regulator of glucocorticoid response. Endocr Rev. 2004;25:831–866. doi: 10.1210/er.2003-0031. [DOI] [PubMed] [Google Scholar]
- 34.Yamada K, Duong DT, Scott DK, Wang JC, Granner DK. CCAAT/enhancer-binding protein beta is an accessory factor for the glucocorticoid response from the cAMP response element in the rat phosphoenolpyruvate carboxykinase gene promoter. J Biol Chem. 1999;274:5880–5887. doi: 10.1074/jbc.274.9.5880. [DOI] [PubMed] [Google Scholar]
- 35.Grange T, Roux J, Rigaud G, Pictet R. Cell-type specific activity of two glucocorticoid responsive units of rat tyrosine aminotransferase gene is associated with multiple binding sites for C/EBP and a novel liver-specific nuclear factor. Nucleic Acids Res. 1991;19:131–139. doi: 10.1093/nar/19.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alam T, An MR, Mifflin RC, Hsieh CC, Ge X, Papaconstantinou J. trans-activation of the alpha 1-acid glycoprotein gene acute phase responsive element by multiple isoforms of C/EBP and glucocorticoid receptor. J Biol Chem. 1993;268:15681–15688. [PubMed] [Google Scholar]
- 37.Kalinyak JE, Dorin RI, Hoffman AR, Perlman AJ. Tissue-specific regulation of glucocorticoid receptor mRNA by dexamethasone. J Biol Chem. 1987;262:10441–10444. [PubMed] [Google Scholar]
- 38.Freeman AI, Munn HL, Lyons V, Dammermann A, Seckl JR, Chapman KE. Glucocorticoid down-regulation of rat glucocorticoid receptor does not involve differential promoter regulation. J Endocrinol. 2004;183:365–374. doi: 10.1677/joe.1.05773. [DOI] [PubMed] [Google Scholar]
- 39.Govindan MV. Recruitment of cAMP-response element-binding protein and histone deacetylase has opposite effects on glucocorticoid receptor gene transcription. J Biol Chem. 2010;285:4489–4510. doi: 10.1074/jbc.M109.072728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Doucas V, Shi Y, Miyamoto S, West A, Verma I, Evans RM. Cytoplasmic catalytic subunit of protein kinase A mediates cross-repression by NF-kappa B and the glucocorticoid receptor. Proc Natl Acad Sci U S A. 2000;97:11893–11898. doi: 10.1073/pnas.220413297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reil TD, Kashyap VS, Sarkar R, Freishlag J, Gelabert HA. Dexamethasone inhibits the phosphorylation of retinoblastoma protein in the suppression of human vascular smooth muscle cell proliferation. J Surg Res. 2000;92:108–113. doi: 10.1006/jsre.2000.5942. [DOI] [PubMed] [Google Scholar]
- 42.Reil TD, Sarkar R, Kashyap VS, Sarkar M, Gelabert HA. Dexamethasone suppresses vascular smooth muscle cell proliferation. J Surg Res. 1999;85:109–114. doi: 10.1006/jsre.1999.5665. [DOI] [PubMed] [Google Scholar]
- 43.Rabbitt EH, Lavery GG, Walker EA, Cooper MS, Stewart PM, Hewison M. Prereceptor regulation of glucocorticoid action by 11beta-hydroxysteroid dehydrogenase: a novel determinant of cell proliferation. FASEB J. 2002;16:36–44. doi: 10.1096/fj.01-0582com. [DOI] [PubMed] [Google Scholar]
- 44.Kato JY, Matsuoka M, Polyak K, Massague J, Sherr CJ. Cyclic AMP-induced G1 phase arrest mediated by an inhibitor (p27Kip1) of cyclin-dependent kinase 4 activation. Cell. 1994;79:487–496. doi: 10.1016/0092-8674(94)90257-7. [DOI] [PubMed] [Google Scholar]
- 45.Molnar GA, Lindschau C, Dubrovska G, Mertens PR, Kirsch T, Quinkler M, Gollasch M, Wresche S, Luft FC, Muller DN, Fiebeler A. Glucocorticoid-related signaling effects in vascular smooth muscle cells. Hypertension. 2008;51:1372–1378. doi: 10.1161/HYPERTENSIONAHA.107.105718. [DOI] [PubMed] [Google Scholar]
- 46.Barf T, Vallgarda J, Emond R, Haggstrom C, Kurz G, Nygren A, Larwood V, Mosialou E, Axelsson K, Olsson R, Engblom L, Edling N, Ronquist-Nii Y, Ohman B, Alberts P, Abrahmsen L. Arylsulfonamidothiazoles as a new class of potential antidiabetic drugs. Discovery of potent and selective inhibitors of the 11beta-hydroxysteroid dehydrogenase type 1. J Med Chem. 2002;45:3813–3815. doi: 10.1021/jm025530f. [DOI] [PubMed] [Google Scholar]
- 47.Alberts P, Engblom L, Edling N, Forsgren M, Klingstrom G, Larsson C, Ronquist-Nii Y, Ohman B, Abrahmsen L. Selective inhibition of 11beta-hydroxysteroid dehydrogenase type 1 decreases blood glucose concentrations in hyperglycaemic mice. Diabetologia. 2002;45:1528–1532. doi: 10.1007/s00125-002-0959-6. [DOI] [PubMed] [Google Scholar]
- 48.Alberts P, Nilsson C, Selen G, Engblom LO, Edling NH, Norling S, Klingstrom G, Larsson C, Forsgren M, Ashkzari M, Nilsson CE, Fiedler M, Bergqvist E, Ohman B, Bjorkstrand E, Abrahmsen LB. Selective inhibition of 11 beta-hydroxysteroid dehydrogenase type 1 improves hepatic insulin sensitivity in hyperglycemic mice strains. Endocrinology. 2003;144:4755–4762. doi: 10.1210/en.2003-0344. [DOI] [PubMed] [Google Scholar]
- 49.Alberts P, Ronquist-Nii Y, Larsson C, Klingstrom G, Engblom L, Edling N, Lidell V, Berg I, Edlund PO, Ashkzari M, Sahaf N, Norling S, Berggren V, Bergdahl K, Forsgren M, Abrahmsen L. Effect of high-fat diet on KKAy and ob/ob mouse liver and adipose tissue corticosterone and 11-dehydrocorticosterone concentrations. Horm Metab Res. 2005;37:402–407. doi: 10.1055/s-2005-870228. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.