

Abstract
A dihedral energy correction (CMAP) term has been recently included in the CHARMM force field to obtain a more accurate description of the peptide backbone. Its importance in improving dynamical properties of proteins and preserving their stability in long molecular-dynamics simulations has been established for several globular proteins. Here we investigate its role in maintaining the structure and function of two potassium channels, Shaker Kv1.2 and KcsA, by performing molecular-dynamics simulations with and without the CMAP correction in otherwise identical systems. We show that without CMAP, it is not possible to maintain the experimentally observed orientations of the carbonyl groups in the selectivity filter in Shaker, and the channel loses its selectivity property. In the case of KcsA, the channel retains some selectivity even without CMAP because the carbonyl orientations are relatively better preserved compared to Shaker.
Introduction
Since its release in 1998, the CHARMM22 force field has been extensively used in molecular dynamics (MD) simulations of proteins (1). In a recent study, it was found that the CHARMM22 force field leads preferentially to π-helical conformations of peptides rather than the more commonly observed α-helix (2). The problem stems from the sinusoidal form of the dihedral terms, which is too limiting and hence unable to reproduce the results of ab initio calculations in alanine dipeptide (3). This problem was resolved by MacKerell et al. by introducing ϕ, ψ dihedral crossterms and a two-dimensional dihedral energy-grid correction map, which was called CMAP (4,5). We note that while the form of the correction is suggested by ab initio calculations in the gas phase, it has been necessary to optimize it further to obtain improved agreement for the ϕ and ψ dihedral angles when compared with the crystallographic data (4).
Since then, importance of the CMAP correction in long MD simulations has been confirmed in several studies of globular proteins. In a 25-ns MD study of hen lysozyme, Buck et al. showed that using CHARMM22 led to substantial deviations in the calculated root-mean-square Cα fluctuations from the crystallographic B-factors. Similar deviations were found for the order parameter S2 from the nuclear magnetic resonance data (6). Inclusion of the CMAP correction dramatically improved the MD results, bringing them to quantitative agreement with the data (6). Long MD simulations of bovine pancreatic trypsin inhibitor with and without the CMAP correction resulted in the protein becoming unstable early on in the latter case while remaining stable for the entire 30-ns simulation in the former case (7). Similar results favoring the inclusion of CMAP were obtained in MD simulations of small peptides (8). Such corrections to the dihedral terms have also been considered in the AMBER force field, resulting in improved agreement between the simulation results and experimental data (9).
So far, the effect of the CMAP correction in MD simulations of membrane proteins has not been investigated properly. A large number of MD simulations of the KcsA potassium channel have been performed without CMAP using its high-resolution crystal structure (10), and the results appear to be consistent with experimental observations (reviewed in (11–15)). That is, no glaring discrepancies have been found that would suggest inclusion of the CMAP correction might improve the results. In contrast, a recent MD study of the Shaker potassium channel Kv1.2 without CMAP (16) indicated that the carbonyl groups in the selectivity filter were not always oriented toward the filter axis, as observed in the crystal structure (17). To prevent flipping of the carbonyl groups, the backbone dihedral angles of the filter residues were restrained using a small harmonic force (16). This result is important in two respects. First it shows that, although the TVGYG sequence of residues in the filter is conserved among the potassium channels, this does not guarantee that the filter dynamics will also be similar in all respects. Secondly, the requirement for additional dihedral restraints suggests that the CMAP correction may be needed in MD simulations of some potassium channels.
The above observations call for a comparative study of the Shaker and KcsA potassium channels using the CHARMM22 force field with and without the CMAP correction. Here we perform such a study focusing on the structure and function of the selectivity filter. In all cases, the channel systems are carefully equilibrated using several protocols, and the orientations of the carbonyl groups in the filter are traced to see whether they remain oriented toward the filter axis or flip back. For the equilibrated structures, the selectivity of the S1 binding site in the filter (see Fig. 1) is studied via thermodynamic integration, where the K+ ion at S1 is transformed to a Na+ ion. From the difference of the free energy changes at the S1 site and in bulk water, the selectivity free energy ΔΔG(K+ → Na+) is determined. In all cases, reverse transformations are also performed to ensure that there are no significant hysteresis effects. Our results show that inclusion of the CMAP correction is essential for a proper description of the selectivity filter in Shaker.
Figure 1.
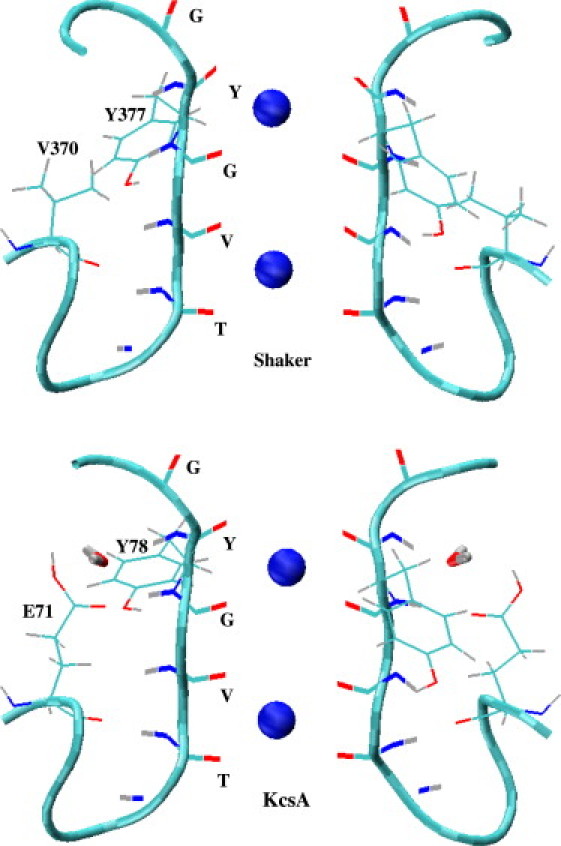
Crystal structures of the selectivity filter in Shaker and KcsA potassium channels. The CO and NH groups of TVGYG residues, the side chains of Tyr-377 and Val-370 in Shaker and Tyr-78 and Glu-71 in KcsA are shown explicitly. Two K+ ions occupy the S1 and S3 binding sites, which are indicated by spheres in the figures (S1 refers to the binding site at the top, coordinated by the carbonyls of Y and G residues). The water molecule near the Tyr-78 residue in KcsA is also shown.
Methods
Model system and MD simulations
The crystal structures of the selectivity filter in the Shaker (17) and KcsA (10) potassium channels are shown in Fig. 1. The TVGYG sequence of the filter residues is identical in both cases, and the filter structures are seen to be very similar. Nevertheless there are two important differences between the two structures, which are found to have implications on the function of the filter. The first is that the Val-370 residue in Shaker is replaced by Glu-71 in KcsA. The side chain of Glu-71 is assumed to be protonated (18,19) and makes a hydrogen bond with the amide group of Tyr-78 (Fig. 1), which helps in stabilizing the upper part of the filter in KcsA. Secondly, as shown in Fig. 1, there is a water molecule in KcsA near the Tyr-78 residue, whereas there are no water molecules near the corresponding Tyr-377 residue in the Shaker structure. As will be seen below, this water molecule plays an important role in stabilizing the filter structure in KcsA. To delineate the role of this water molecule, we have removed it from the KcsA structure and repeated the MD simulations for the resulting system. Considering the possibility that this water molecule may not have been observed in the lower-resolution crystal structure of Shaker, we have included it in a similar position as in KcsA and repeated the Shaker MD simulations for this system as well.
The simulation systems for the Shaker and KcsA potassium channels are constructed using the VMD suite of software (20). The pore domain of the crystal structure of Shaker (PDB ID: 2A79, residues 312–421) is embedded in a lipid bilayer consisting of 96 1-palmitoyl-2-oleoyl-phosphatidylethanolamine molecules and solvated with 8900 water molecules, 8 K+ and 24 Cl– ions. The extra Cl– ions are needed to neutralize the positive charges on the channel protein and thus avoid any simulation artifacts associated with periodic boundaries. Three of the K+ ions are placed in the cavity and the S1 and S3 binding sites as observed in the crystal structure. Initially the protein coordinates are fixed and the system is equilibrated with 1 atm pressure coupling until the correct water and lipid densities are obtained. At this point, the x and y dimensions of the simulation box is fixed (at 76 and 72 Å, respectively), and pressure coupling is applied only in the z direction. At the second stage of equilibration, the side chains of the protein atoms are gradually relaxed in a 2-ns MD simulation while the backbone atoms are still constrained. In the final stage, the backbone atoms are relaxed in several steps using the restraints (in kcal/mol/Å2): 100, 50, 20, 10, 5, 2.5, 1.5, 1.0, 0.6, 0.3, and 0.1. Reduction of k from 0.1 to 0 has not caused any visible changes in the KcsA results (see Figs. 4 and 5), and therefore it is not included in the Shaker results. We have experimented with different simulation times for these restraints to make sure that the final results are independent of how the system is equilibrated (the exceptions are the first three steps, for which 200 ps is used uniformly).
Figure 4.
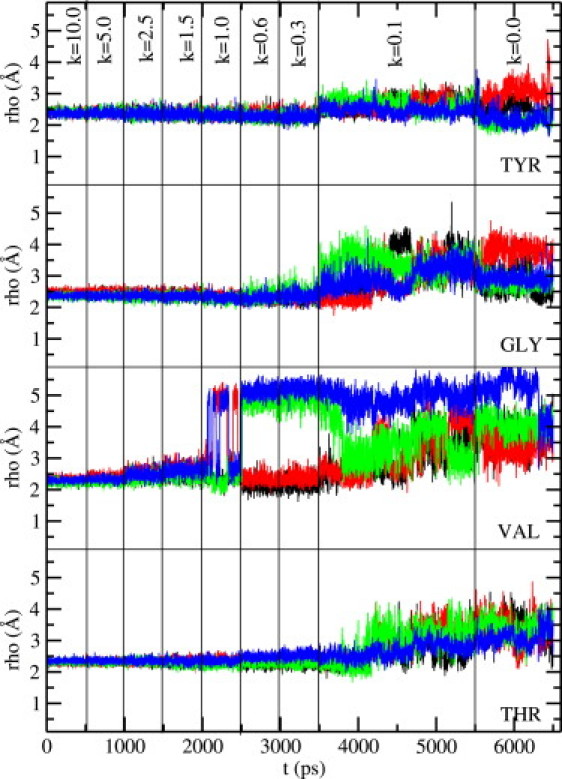
Same as Fig. 2, but for the KcsA potassium channel (without CMAP).
Figure 5.
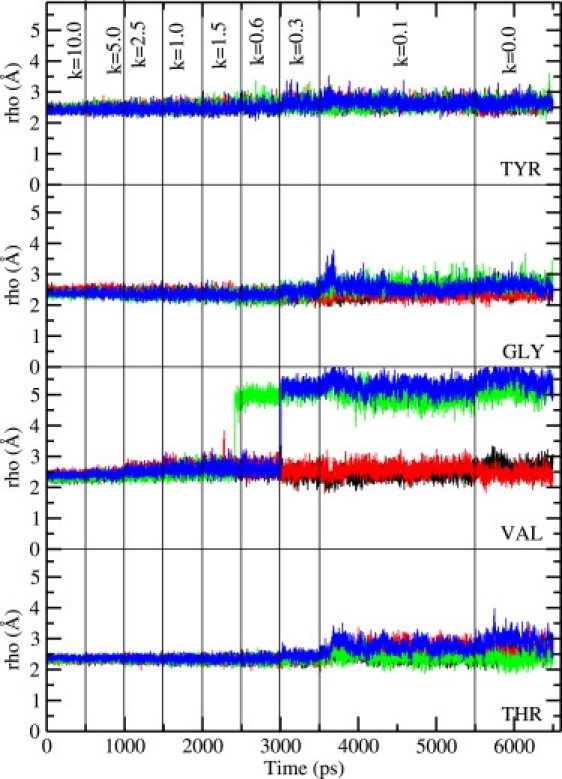
Same as Fig. 3, but for the KcsA potassium channel (with CMAP).
The simulation system for the KcsA channel is constructed in a similar manner. The crystal structure (PDB ID: 1K4C) is embedded in a bilayer of 105 phosphatidylethanolamine molecules and solvated with 9640 water molecules, 6 K+ and 18 Cl– ions. This system is equilibrated using the same protocol as in the case of Shaker. For both potassium channels, identical procedures are employed in constructing two simulation systems, one with the original CHARMM22 (without CMAP) and another with the CMAP correction included.
MD simulations are carried out using the latest version (2.6) of the NAMD code (21), which allows inclusion of the CMAP correction terms in the CHARMM22 force field. CHARMM22 provides a complete set of parameters for all the atoms in the system and uses the TIP3P model for water molecules. An NpT ensemble is used with periodic boundary conditions. Pressure is kept at 1 atm using the Langevin piston method with a damping coefficient of 5 ps−1 (22). Similarly, temperature is maintained at 298 K through Langevin damping with a coefficient of 5 ps−1. Electrostatic interactions are computed using the particle-mesh Ewald algorithm. The list of nonbonded interactions is truncated at 13.5 Å, and a switching cut-off distance of 10 Å is used for the Lennard-Jones interactions. A time-step of 2 fs is employed in all simulations. Trajectory data is written at 1-ps intervals during both equilibration and production runs.
Free energy calculations
The selectivity of the model channels against permeation of Na+ ion is studied using the thermodynamic integration (TI) method (23). For this purpose, the K+ ion at the S1 binding site is alchemically transformed into Na+ ion, and the resulting free energy difference is calculated from TI using
| (1) |
where H(λ) = (1 − λ)H0 + λH1, with H0 and H1 representing the Hamiltonians of the initial and final states, respectively (e.g., if the initial state is a K+ ion, in the final state it becomes a Na+ ion at the same position). A restraining force of 5 kcal/mol/Å2 is applied on the ion during the free energy simulations. We have checked the influence of this restraint on the results in a test case, and found no perceptible change in the free energy difference for different values of the restraining force. The integral in Eq. 1 is performed using a seven-point Gaussian quadrature, which has been shown to yield sufficiently accurate results (24). At each TI window, the system is equilibrated for 150 ps, followed by 350 ps of a production run. To check against hysteresis effects, we have also performed the reverse transformation. The free energy change is determined from the average of the forward and backward transformations as
| (2) |
To determine the selectivity free energy, an identical calculation is performed in bulk water and the result is subtracted from that in the binding site, yielding
| (3) |
A positive result for ΔΔG(K+ → Na+) indicates that the channel is selective for K+ ions, and a negative one indicates it is Na+-selective.
Results
Structure of the selectivity filter
The main question regarding the structure of the selectivity filter is whether the carbonyl groups forming the binding sites retain their orientations toward the channel axis as observed in the crystal structure, or flip back. To address this issue we have plotted the ρ coordinate of the carbonyl oxygens of Tyr, Gly, Val, and Thr residues—which measures their distance from the filter axis—as a function of time during equilibration. As mentioned in Methods, we have experimented with different equilibration times for the restraints employed in relaxing the system. Shaker simulations are found to be quite robust, leading to similar final results regardless of the simulation time used. In Figs. 2 and 3 we show the results obtained from a 3.5-ns MD simulation, where the backbone restraints are reduced from 10 to 0.1 kcal/mol/Å2 in 8 steps as indicated on the figures. As seen in Fig. 2, without the CMAP correction, the carbonyl oxygens are not very stable and start flipping back as soon as the restraints are relaxed, consistent with the observations of Khalili-Araghi et al. (16). Among the valine residues, three of the four carbonyls flip and among tyrosine, two carbonyls flip. Substantial fluctuations are observed among the carbonyls of the threonine and glycine residues, though they mostly retain their axial orientations. Inclusion of the CMAP correction (Fig. 3) is seen to prevent these flipping events and suppress the fluctuations in carbonyl orientations, leading to a filter structure that is in more conformity with the crystal structure. Only one valine carbonyl is seen to flip toward the end of the simulation, which remains flipped when the simulation is continued for another 0.5 ns. Such an event has also been observed in KcsA simulations and is thought to be related to the inactivation of the channel (25).
Figure 2.
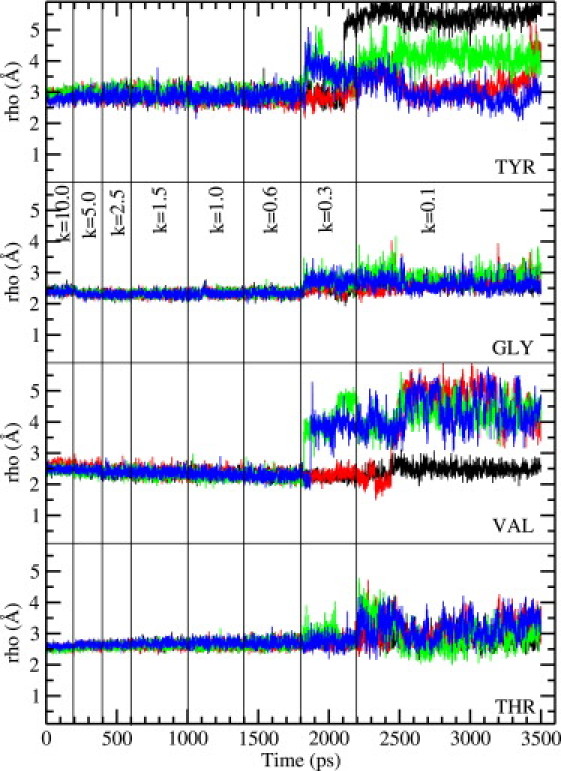
Behavior of the carbonyl oxygens in the selectivity filter of the Shaker potassium channel as the system is slowly relaxed to equilibrium. The results are obtained using the CHARMM22 force field without the CMAP correction. Distance of the carbonyl oxygens of Tyr, Gly, Val, and Thr residues from the filter axis are plotted as a function of time. The set of four oxygens for each residue is plotted separately for clarity. The value of the restraints used on the backbone atoms in each simulation window is indicated at the top (in kcal/mol/Å2). A sudden jump in the axial distance from the baseline of ∼2.5 Å indicates flipping of the carbonyl oxygen away from the filter axis.
Figure 3.
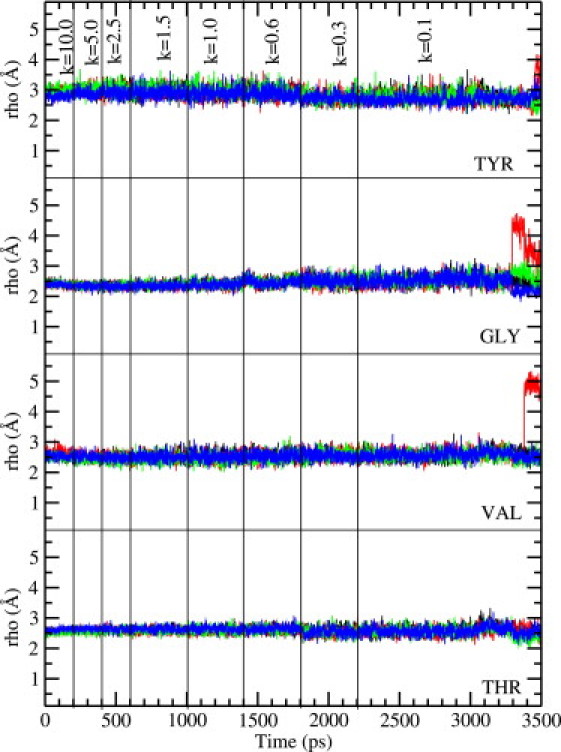
Same as Fig. 2, Shaker, but with the CMAP correction included in the CHARMM22 force field.
The results in Figs. 2 and 3 are obtained by placing a water molecule near the Tyr-377 residue, as observed in the KcsA structure. This water molecule does not form permanent hydrogen bonds with the filter atoms but remains in the vicinity of Tyr-377. When it does form a hydrogen bond for a period, it stabilizes the corresponding tyrosine carbonyl (e.g., the blue and red lines in the top panel of Fig. 2). To further assess its functional role, we have repeated the Shaker simulations described above without this water molecule (not shown). We find that the carbonyl orientation results shown in Figs. 2 and 3 are essentially replicated. Only the tyrosine carbonyls exhibit some difference—one more tyrosine-carbonyl flips, compared to Fig. 2 (without CMAP), and they exhibit larger fluctuations compared to the situation shown in Fig. 3 (with CMAP). Comparison of the two sets of simulations shows that the presence of a water molecule near Tyr-377 provides some help in stabilizing the tyrosine-carbonyl's orientation toward the filter axis, but as it is clear from Figs. 2 and 3, only the inclusion of the CMAP correction completely resolves the problem with the carbonyl orientations.
Many MD simulations of the KcsA channel have been performed using the CHARMM22 force field but so far none of them included the CMAP correction. In the light of the Shaker results discussed above, an interesting question here is how the stability of the carbonyl orientations are maintained in MD simulations of KcsA without including the CMAP correction. To address this question, we have repeated the equilibration simulations for the KcsA channel with and without the CMAP correction. One immediate difference from the Shaker simulations is that without CMAP, the results are more sensitive to equilibration times requiring longer simulations to settle down. Specifically, as the restraints are relaxed, the valine carbonyls tend to flip back, and some of the glycine and tyrosine carbonyls become unstable especially when the water molecule shown in Fig. 1 is removed from the system. The reason for the latter is traced back to the stability of the hydrogen bond between the amide group of Tyr-78 and the side chain of Glu-71. Regardless of how slowly the backbone atoms are relaxed and whether the CMAP correction is included or not, this hydrogen bond breaks in some of the monomers when the restraint is reduced from 0.6 to 0.3 kcal/mol/Å2. The Glu-71 side chain moves away from the filter, with the O-H hydrogen bond distance increasing from ∼2 Å to 4–5 Å. In simulations where the water molecule near Tyr-78 is removed, this leads to substantial fluctuations of the glycine and tyrosine carbonyls with some flipping back. When the water molecule is present, it makes occasional hydrogen bond with the amide group of Tyr-78, thus preserving the stability of the glycine and tyrosine carbonyl orientations to a large degree.
With the above caveats, we show in Figs. 4 and 5 the results of carbonyl orientations obtained from a 6.5-ns MD simulation of the KcsA system with the water molecule near Tyr-78 present. Inclusion of the CMAP correction in the CHARMM22 force field (Fig. 5) is again seen to suppress fluctuations of the carbonyl groups observed in MD simulations without CMAP (Fig. 4). However, when contrasted with the Shaker results (Figs. 2 and 3), the impact of the CMAP correction is seen to be somewhat less in KcsA because the reference results without CMAP are already in a better shape. All the tyrosine carbonyls are oriented toward the filter axis. As stressed above, this happens because the Glu-71 side chain and the water molecule near Tyr-78 help to stabilize the carbonyl orientations of tyrosine. There are some fluctuations in glycine and threonine carbonyls but none of them flip back, and only one or two valine carbonyls flip back. Flipping of the valine carbonyls have been noted in previous MD simulations of the KcsA channel. In our simulations also they are seen to flip back whether CMAP is included or not, though its presence is seen to sharpen the distinction between the two states with axial and flipped orientations (Fig. 5). Thus inclusion of the CMAP correction in KcsA simulations mainly helps in suppressing the fluctuations of the glycine and threonine carbonyls. Without CMAP the carbonyls are not as stable, exhibiting fairly large fluctuations as well as sizable deflections from the axial orientation. Overall, our results indicate that inclusion of the CMAP correction in CHARMM22 leads to a very robust filter structure in KcsA, but a reasonable structure can also be obtained in its absence.
Function of the selectivity filter
The selectivity of the potassium channels is controlled by the carbonyl groups in the filter, and hence, whether they are stably oriented toward the filter axis or wildly fluctuate, is expected to have a big impact on their selectivity. To study the functional implications of the CMAP correction on the selectivity of the Shaker and KcsA potassium channels, we have performed thermodynamic integration calculations with and without the CMAP correction and determined the free energy difference for binding of a K+ ion to the S1 site relative to a Na+ ion. A similar calculation is performed in bulk water, which is needed to determine the selectivity free energy.
In Fig. 6 we show the running averages of ΔGf(K+ → Na+) for the forward transformation (solid line) and −ΔGb(Na+ → K+) for the backward transformation (dashed line). Five sets of calculations are performed with the K+ ion in bulk water, at the S1 binding site in Shaker (with and without CMAP) and the same in KcsA. In all cases, there is minimal hysteresis between the forward and backward directions (≤1 kcal/mol), and the running averages remain almost flat, which ensure that the results have properly converged. The end point of each running average in Fig. 6 is listed in Table 1, from which the average free energy difference ΔGav (fourth column) and the final selectivity free energy ΔΔG (fifth column) are determined. The experimental values quoted in the last column are estimated from the permeability ratios obtained under bi-ionic conditions, which are given by PNa/PK < 0.03 for Shaker (27) and PNa/PK < 1/150 for KcsA (28).
Figure 6.
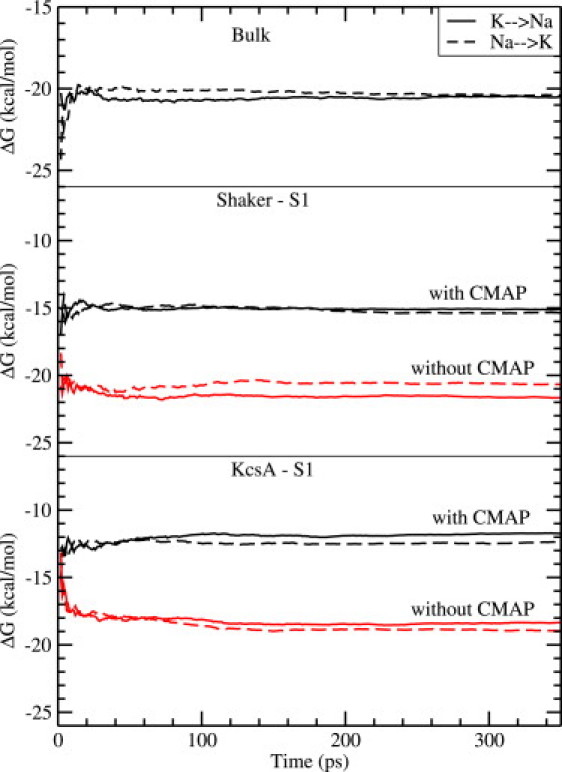
Running averages of the free energy differences for the forward ΔGf(K+ → Na+) (solid lines) and the backward −ΔGb(Na+ → K+) (dashed lines) transformations. The five sets of calculations shown correspond to bulk water (top panel), the S1 binding site in Shaker with and without CMAP (middle panel), and the same in KcsA (bottom panel). The end points of the free energy curves are listed in Table 1.
Table 1.
Free energy differences obtained using the thermodynamic integration method in bulk water (first row) and at the S1 binding site (second to fourth rows)
| ΔGf | −ΔGb | ΔGav | ΔΔG | Exp. | |
|---|---|---|---|---|---|
| Bulk | −20.6 | −20.4 | −20.5 | — | — |
| Shaker w/out CMAP | −21.7 | −20.7 | −21.2 | −0.7 | >2.1 |
| Shaker with CMAP | −15.2 | −15.4 | −15.3 | 5.2 | // |
| KcsA w/out CMAP | −18.3 | −19.0 | −18.7 | 1.8 | >2.9 |
| KcsA with CMAP | −11.7 | −12.4 | −12.1 | 8.4 | // |
The free energy differences for the forward (K+ → Na+) and negative of the backward (Na+ → K+) transformations are listed in the second and third columns. The fourth column gives the average of the two values (Eq. 2), and the fifth one gives the selectivity free energy obtained by subtracting the average bulk value from that of the binding site (Eq. 3). For comparison, the experimental lower-bounds for the selectivity free energies estimated from the permeability ratios are listed in the last column. The Shaker result is from Heginbotham and MacKinnon (27) and KcsA from LeMasurier et al. (28). All free energies are given in units of kcal/mol.
The most important result that emerges from this study is that, without the CMAP correction in the CHARMM force field, the S1 binding site in Shaker is not selective for K+ ions but slightly prefers Na+ ions with a free energy margin of 0.7 kcal/mol. Preferential binding of Na+ ions to S1 would lead to blocking of K+ permeation, which is not observed experimentally. Moreover the calculated selectivity free energy is 2.8 kcal/mol smaller than the experimental lower bound, which is a significant discrepancy when compared to the accuracy of the calculations (∼1 kcal/mol). Inclusion of the CMAP correction restores the K+ selectivity of the channel with a selectivity free energy of 5.2 kcal/mol. Thus, the CMAP correction has a substantial impact on the selectivity of Shaker, contributing almost 6 kcal/mol to the selectivity free energy and thereby restoring the agreement with the experimental observations. We note that new measurements of selectivity in Shaker is likely to improve this lower bound and thus sharpen the difference with KcsA and further strengthen the case for the necessity of the CMAP correction in MD simulations.
As one might surmise from the filter structure results, the situation is better in the KcsA channel with regard to selectivity. The S1 site retains K+ selectivity with a free energy margin of 1.8 kcal/mol even in the absence of CMAP. While this value is also smaller than the experimental lower bound, the margin of error in this case (1.1 kcal/mol) is nearly within the accuracy of the calculations. When the CMAP correction is included, the selectivity free energy increases to 8.4 kcal/mol, in comfortable agreement with the experimental lower bound. Again, inclusion of CMAP boosts the selectivity free energy of KcsA by >6 kcal/mol. Our result for the selectivity of the S1 site without CMAP is consistent with earlier calculations using the CHARMM force field, e.g., Noskov et al. found 2.6 kcal/mol for the selectivity free energy (26).
Discussion and Conclusions
Our main conclusion is that inclusion of the CMAP correction in the CHARMM22 force field is essential for maintaining the structural integrity and K+/Na+ selectivity of the Shaker channel. The situation in the KcsA channel in this regard is not as clear because it has a relatively more stable filter structure compared to Shaker, which helps KcsA to retain some selectivity even in the absence of the CMAP correction. A general conclusion that applies to both cases is that inclusion of the CMAP correction suppresses the fluctuations of the carbonyl groups in the filter and stabilizes their orientations toward the filter axis. As shown in free energy calculations, this extra stability of the carbonyl orientations leads to ∼6 kcal/mol increase in the selectivity free energy, turning the S1 site in Shaker from Na+-selective to K+-selective. It has been demonstrated previously that making the filter structure more rigid increases its selectivity free energy (26). The CMAP correction can be interpreted in the same light—it causes the filter structure to become more rigid, and thereby helps to increase the K+/Na+ selectivity free energy.
The KcsA potassium channel is the first biological ion channel to be solved, and therefore it has played a central role in computational studies of not only potassium but many other ion channels (see, for example (11–15),). Because the selectivity filter sequence is conserved in most potassium channels, it has been implicitly assumed that selectivity properties obtained from studies of KcsA would equally apply to other potassium channels. For example, a recent debate about the nature of the selectivity in potassium channels has mostly focused on the KcsA structure (26,29–32). The possibility that different results may be obtained in other potassium channels was considered only in a quantum mechanical study of the filter environment (33). Indeed, comparison of the structure and function of the selectivity filter in the KcsA and Shaker channels in this study has revealed significant differences between the two channels, which may have implications in selectivity. The most important difference is the replacement of the Glu-71 residue in KcsA with the nonpolar Val-370 residue in Shaker. An immediate question is: How common is this glutamate residue in other potassium channels? A glutamate residue in an equivalent position to Glu-71 in KcsA is found in all inward rectifier Kir channels. Thus, the KcsA filter is likely to be a good model for the Kir channels. On the other hand, for the main class of voltage-gated potassium channels, a completely different picture emerges. Of the 676 voltage-gated potassium channel sequences we have considered, a majority (35%) has a valine residue at the equivalent position to Glu-71 just like Shaker, followed by isoleucine (20%) and other nonpolar residues, but none has a glutamate residue. Thus, the Shaker channel, rather than KcsA, provides a more appropriate model for studying the selectivity property in voltage-gated potassium channels.
There have been very few computational studies of the Shaker pore domain so far (16,34,35). One reason for this paucity has been the difficulty of obtaining stable structures in MD simulations of Shaker in the absence of the CMAP correction in the CHARMM force field (e.g., the CMAP correction was incorporated in the popular NAMD code in version 2.6, in September, 2006). As demonstrated here, stable Shaker structures are feasible in MD simulations once the CMAP correction is included. More work is needed to study the permeation properties of the Shaker channel and especially the energetics of K+ ion conduction. More accurate measurements of the Na+/K+ permeability ratios in various potassium channels would also be very valuable in assessing and interpreting the computational results. Only through such work can we find out the functional consequences of the structural differences between the inward rectifier and voltage-gated potassium channels.
Acknowledgments
We thank Jamie Vandenberg for discussions of the structure of selectivity filter in various potassium channels. Calculations were carried out using the SGI Altix clusters at the Supercomputer Facility of the Australian National University (Canberra) and the Australian Center for Advanced Computing and Communications (Sydney), and ULAKBIM cluster of TUBITAK (Ankara).
This work was supported by grants from the Australian Research Council and Turkish Scientific and Technical Research Council (TUBITAK).
References
- 1.MacKerell A.D., Jr., Bashford D., Bellott M., Dunbrack R.L., Jr., Evanseck J.D. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 2.Feig A., MacKerell A.D., Jr., Brooks C.L. Force field influence on the observation of π-helical protein structures in molecular dynamics simulations. J. Phys. Chem. B. 2003;107:2831–2836. [Google Scholar]
- 3.MacKerell A.D., Feig M., Brooks C.L. Improved treatment of the protein backbone in empirical force fields. J. Am. Chem. Soc. 2004;126:698–699. doi: 10.1021/ja036959e. [DOI] [PubMed] [Google Scholar]
- 4.MacKerell A.D., Feig M., Brooks C.L. Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 5.MacKerell A.D. Empirical force fields for biological macromolecules: overview and issues. J. Comput. Chem. 2004;25:1584–1606. doi: 10.1002/jcc.20082. [DOI] [PubMed] [Google Scholar]
- 6.Buck M., Bouguet-Bonnet S., Friesner R.W., MacKerell A.D., Jr. Importance of the CMAP correction to the CHARMM22 protein force field: dynamics of hen lysozyme. Biophys. J. 2006;90:L36–L38. doi: 10.1529/biophysj.105.078154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li X.F., Hassan S.A., Mehler E.L. Long dynamics simulations of proteins using atomistic force fields and a continuum representation of solvent effects: calculation of structural and dynamic properties. Proteins. 2005;60:464–484. doi: 10.1002/prot.20470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steinbach P.J. Exploring peptide energy landscapes: a test of force fields and implicit solvent models. Proteins. 2004;57:665–677. doi: 10.1002/prot.20247. [DOI] [PubMed] [Google Scholar]
- 9.Hornak V., Abel R., Okur A., Strockbine B., Roitberg A. Comparison of multiple AMBER force fields and development of improved protein backbone parameters. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou Y., Morais-Cabral J.H., Kaufman A., MacKinnon R. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 Å resolution. Nature. 2001;414:43–48. doi: 10.1038/35102009. [DOI] [PubMed] [Google Scholar]
- 11.Kuyucak S., Andersen O.S., Chung S.H. Models of permeation in ion channels. Rep. Prog. Phys. 2001;64:1427–1472. [Google Scholar]
- 12.Tieleman D.P., Biggin P.C., Smith G.R., Sansom M.S.P. Simulation approaches to ion channel structure-function relationships. Q. Rev. Biophys. 2001;34:473–561. doi: 10.1017/s0033583501003729. [DOI] [PubMed] [Google Scholar]
- 13.Roux B., Allen T., Bernèche S., Im W. Theoretical and computational models of biological ion channels. Q. Rev. Biophys. 2004;37:15–103. doi: 10.1017/s0033583504003968. [DOI] [PubMed] [Google Scholar]
- 14.Roux B. Ion conduction and selectivity in K+ channels. Annu. Rev. Biophys. Biomol. Struct. 2005;34:153–171. doi: 10.1146/annurev.biophys.34.040204.144655. [DOI] [PubMed] [Google Scholar]
- 15.Domene C. Molecular dynamics simulations of potassium channels. Cent. Eur. J. Chem. 2007;5:653–671. [Google Scholar]
- 16.Khalili-Araghi F., Tajkhorshid E., Schulten K. Dynamics of K+ ion conduction through Kv1.2. Biophys. J. 2006;91:2–4. doi: 10.1529/biophysj.106.091926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Long S.B., Campbell E.B., MacKinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309:897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- 18.Luzhkov V.B., Aqvist J. A computational study of ion binding and protonation states in the KcsA potassium channel. Biochim. Biophys. Acta. 2000;1481:360–370. doi: 10.1016/s0167-4838(00)00183-7. [DOI] [PubMed] [Google Scholar]
- 19.Berneche S., Roux B. The ionization state and the conformation of Glu-71 in the KcsA K+ channel. Biophys. J. 2001;82:772–780. doi: 10.1016/S0006-3495(02)75439-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Humphrey W., Dalke A., Schulten K. VMD—visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 21.Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feller S., Zhang Y., Pastor R., Brooks B. Constant pressure molecular dynamics: the Langevin piston method. J. Chem. Phys. 1995;103:4613–4621. [Google Scholar]
- 23.Beveridge D.L., DiCapua F.M. Free energy via molecular simulation: applications to chemical and biomolecular systems. Annu. Rev. Biophys. Biophys. Chem. 1989;18:431–492. doi: 10.1146/annurev.bb.18.060189.002243. [DOI] [PubMed] [Google Scholar]
- 24.Bastug T., Kuyucak S. Energetics of ion permeation, rejection, binding and block in gramicidin A from free energy simulations. Biophys. J. 2006;90:3941–3950. doi: 10.1529/biophysj.105.074633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berneche S., Roux B. A gate in the selectivity filter of potassium channels. Structure. 2005;13:591–600. doi: 10.1016/j.str.2004.12.019. [DOI] [PubMed] [Google Scholar]
- 26.Noskov S.Y., Berneche S., Roux B. Control of ion selectivity in potassium channels by electrostatic and dynamic properties of carbonyl ligands. Nature. 2004;431:830–834. doi: 10.1038/nature02943. [DOI] [PubMed] [Google Scholar]
- 27.Heginbotham L., MacKinnon R. Conduction properties of the cloned Shaker K+ channel. Biophys. J. 1993;65:2089–2096. doi: 10.1016/S0006-3495(93)81244-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.LeMasurier M., Heginbotham L., Miller C. KscA: it's a potassium channel. J. Gen. Physiol. 2001;118:303–313. doi: 10.1085/jgp.118.3.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Noskov S.Y., Roux B. Ion selectivity in potassium channels. Biophys. Chem. 2006;124:279–291. doi: 10.1016/j.bpc.2006.05.033. [DOI] [PubMed] [Google Scholar]
- 30.Asthagiri D., Pratt L.R., Paulaitis M.E. Role of fluctuations in a snug-fit mechanism of KcsA channel selectivity. J. Chem. Phys. 2006;125:024701. doi: 10.1063/1.2205853. [DOI] [PubMed] [Google Scholar]
- 31.Bostick D.L., Brooks C.L. Selectivity in K+ channels is due to topological control of the permeant ion's coordinated state. Proc. Natl. Acad. Sci. USA. 2007;104:9260–9265. doi: 10.1073/pnas.0700554104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thomas M., Jayatilaka D., Corry B. The predominant role of coordination number in potassium channel selectivity. Biophys. J. 2007;93:2635–2643. doi: 10.1529/biophysj.107.108167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Varma S., Rempe S.B. Tuning ion coordination architectures to enable selective partitioning. Biophys. J. 2007;93:1093–1099. doi: 10.1529/biophysj.107.107482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Treptow W., Tarek M. Molecular restraints in the permeation pathway of ion channels. Biophys. J. 2006;91:26–28. doi: 10.1529/biophysj.106.087437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Treptow W., Tarek M. K+ conduction in the selectivity filter of potassium channels is monitored by the charge distribution along their sequence. Biophys. J. 2006;91:81–83. doi: 10.1529/biophysj.106.095992. [DOI] [PMC free article] [PubMed] [Google Scholar]