

Abstract
Sepsis is a generalized inflammatory disease, caused by the hyperinflammatory response of the host, rather than by invading organisms. Endothelial cells play a crucial role in the pathogenesis of sepsis. In this study, we investigated the effects of interleukin-8 (IL-8), a known neutrophil chemoattractant, on lipopolysaccharide (LPS) -induced reactive oxygen species (ROS) production by endothelial cells, and its significance in the pathogenesis of LPS-mediated sepsis. The results revealed that IL-8 directly induced ROS production in human umbilical vein endothelial cells (HUVECs), and also mediated LPS-induced ROS production by HUVECs. Stimulation of HUVECs by LPS strongly enhanced tissue factor expression, a hallmark of severe sepsis, which was suppressed by IL-8 knockdown. We further discovered that NADPH oxidase (Nox) 1 expression in LPS-stimulated HUVECs was markedly repressed by IL-8 knockdown, and Nox1 knockdown reduced tissue factor expression, suggesting that the LPS/IL-8 signalling in endothelial cells was predominantly mediated by Nox1. In conclusion, LPS stimulation of endothelial cells causes activation of the IL-8–Nox1 axis, enhances the production of ROS, and ultimately contributes to the progression of severe sepsis.
Keywords: endothelial cells, interleukin-8, lipopolysaccharide, NADPH oxidase 1, sepsis
Introduction
Sepsis is a serious medical condition characterized by a generalized inflammatory state caused by an invading organism, often referred to as systemic inflammatory response syndrome. The severe clinical manifestations of sepsis, such as hypotension, hypoperfusion to one or more organs, coagulopathy, and multiple organ failure, are caused by the hyperinflammatory, predominantly cytokine-mediated, response of the host, rather than by invading organisms.1 In the pathogenesis of sepsis, the crucial hallmarks are endothelial activation and dysfunction, in which components of the bacterial cell wall, such as lipopolysaccharide (LPS), activate pattern-recognition receptors on the endothelial surface, and subsequently trigger a hyperactivated systemic response with the generation of host-derived inflammatory mediators.2,3 Under normal conditions, the biological activity of inflammatory mediators is under stringent control, which is disrupted in sepsis, resulting in the dysregulated production of mediators. The inappropriate systemic inflammation further leads to the activation of coagulation with concomitant inhibition of anticoagulation systems. Whereas, under normal conditions, the endothelium exhibits antithrombotic properties by expressing tissue factor (TF) pathway inhibitors, thrombomodulin, nitric oxide and prostacyclin, it functions as a prothrombotic surface, releasing TF during the pathogenesis of sepsis.4,5
Endothelial cells respond to LPS through toll-like receptor4,6,7 followed by the release of pro-inflammatory cytokines such as interleukin-6 (IL-6), IL-8, and monocyte chemoattractant protein-1, and increased expression of adhesion molecules including intracellular adhesion molecule and vascular cell adhesion molecule.8–10 Interleukin-8 plays a crucial role in the recruitment of neutrophils, whereas monocyte chemoattractant protein-1 is involved in monocyte chemotaxis.11 The expression levels of cytokines and cell adhesion molecules are regulated by nuclear factor-κB, a key transcription factor in inflammation.12 Recently, similar to tumour necrosis factor-α (TNF-α),13–16 LPS has been demonstrated to induce the generation of reactive oxygen species (ROS) in an NADPH oxidase-dependent manner in endothelial cells.17–19 However, the importance of IL-8 in the LPS-induced ROS production by endothelial cells has not yet been studied. In this study, we examined the effects of IL-8 on ROS production using human umbilical vein endothelial cells (HUVECs), and the involvement of IL-8 in the pathogenesis of LPS-mediated sepsis.
Materials and methods
Cell culture
The HUVECs (KURABO, Osaka, Japan) were grown to confluence in endothelial growth medium (HuMedia-EG2; KURABO, Osaka, Japan) at 37° in a 5% CO2 humidified incubator. Upon reaching 90–100% confluence, cells were split using trypsin/EDTA (KURABO). All experiments were conducted using HUVECs at only one or two passages. Throughout the experiments, cell cultures remained free of endotoxin (data not shown).
Measurement of intracellular ROS production
Intracellular ROS generation in HUVECs was examined by flow cytometry using 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA; Molecular Probes, Eugene, OR), a ROS-sensitive dye. DCFH-DA is de-esterified to 2,7-dichlorodihydrofluorescein (DCFH), and further oxidized by ROS, yielding a fluorescent compound, 2,7-dichlorofluorescein (DCF), which can be detected by flow cytometry. The HUVECs (1 × 105) were cultured in six-well plates until 90–100% confluence in a serum-starved condition for the last 2 hr. They were pre-incubated for 30 min with or without 10 μm diphenyleneiodonium (DPI; Sigma Chemicals, St Louis, MO), an NADPH oxidase inhibitor, and then stimulated with 1 μg/ml LPS for 1 or 4 hr, or 100 nm IL-8 for 1 or 2 hr in the presence of 2 μm DCFH-DA. Subsequently, the cells were washed twice and suspended in phosphate-buffered saline (PBS). The amount of intracellular ROS was detected using the FACSCalibur flow cytometer (Becton Dickinson, Mountain View, CA). The data were analysed using CELLQuest software (Becton Dickinson).
Reverse transcription–polymerase chain reaction for messenger RNA expression of NADPH oxidase family
Total RNA was isolated from HUVECs treated with 1 μg/ml LPS or 100 nm IL-8 for 8 hr using RNAiso reagent (Takara Bio, Shiga, Japan) according to the manufacturer's instructions. For reverse transcription, 2 μg total RNA was subjected to first-strand complementary DNA (cDNA) synthesis using random primers (SuperScript III RT Kit; Invitrogen, Carlsbad, CA). Polymerase chain reaction (PCR) was performed in a 20-μl reaction volume and cDNAs were amplified with rTaq polymerase (Takara Bio). The PCR amplification of NADPH oxidase (Nox) 1–5 messenger RNA (mRNA) was carried out with the following primers: Nox1: forward, 5′-GCAATTGGATCACAACCTCA-3′, reverse, 5′-CTGGAGAGAATGGAGGCAAG-3′, Nox2: forward, 5′-GCTGTTCAATGCTTGTGGCT-3′, reverse, 5′-TCAGATTGGTGGCGTTATTG-3′, Nox3: forward, 5′-ATGAACACCTCTTGGGGTCAGCTGA-3′, reverse, 5′-GGATCGGAGTCACTCCCTTCGCTG-3′, Nox4: forward, 5′-CTCAGCGGAATCAATCAGCTGTG-3′, reverse, 5′-AGAGGAACACGACAATCAGCCTTAG-3′, Nox5: forward, 5′-ATCGAACATCCCCACCATT-3′, reverse, 5′-TTGTCACACACTCCTCGACAGC-3′) (Sigma-Aldrich Japan, Kyoto, Japan). The expression of β-actin mRNA served as the loading control. The resulting PCR fragments (10 μl) were electrophoresed on a 1·5% agarose gel.
Transfection of small interfering RNA for IL-8 or Nox1
RNA fragments of 21 nucleotide residues long (CAAAUCUACUUCAACACUUTT) specific to the human IL-8 cDNA [IL-8 small interfering (si)RNA], 19 nucleotide residues long (GAAAUCCAUCUGGUACAAA) specific to the human Nox1 cDNA (Nox1 siRNA), and scramble RNA (control siRNA) were purchased from Takara Bio. The HUVECs were transiently transfected with IL-8, Nox1 or control siRNA using the HUVEC Nucleofection® Kit (Amaxa Biosystems, Koeln, Germany) following the manufacturer's instructions. After 24 hr of transfection with siRNA, HUVECs were used for the subsequent experiments.
Quantitative real-time PCR for mRNA expression of IL-8, TF, Nox1 or Nox4
To examine mRNA expression of IL-8, TF, Nox1 or Nox4, HUVECs (2 × 106) were treated with LPS for 4 hr (IL-8), and 8 hr or 12 hr (TF, Nox1 and Nox4), after 24 hr transfection with appropriate siRNAs. After that, total RNA was isolated from HUVECs (2 × 106), using RNAiso reagent (Takara Bio) according to the manufacturer's instructions. For quantitative real-time PCR, first-strand cDNA was synthesized from 500 ng total RNA as described above. Real-time PCR was performed using SYBR Premix Ex Taq (Perfect Real Time; Takara Bio) and an ABI 7300 real-time instrument with Applied Biosystems Sequence Detection System, Version 1.2. (Applied Biosystems Japan, Tokyo, Japan). The primers for IL-8 (forward, 5′-ACACTGCGCCAACACAGAAATTA-3′; reverse, 5′-TTTGCTTGAAGTTTCACTGGCATC-3′), TF (forward, 5′-CACCG-ACGAGATTGTGAAGGAT-3′; reverse, 5′-CCCTGCGGGTAGGAGAA-3′), Nox1 (forward, 5′-CACTGACATCGTGACAGGTCTGAA-3′; reverse, 5′-CAAAGTCCGAGGGC-CACATAA-3′), Nox4 (forward, 5′-ATGTCATGGAAATCCGAATGGTC-3′; reverse, 5′-CCCAAATGTTGCTTTCGTTTCAG-3′), and β-actin (forward, 5′-ATTGCCGAC-AGGATGCAGA-3′; reverse, 5′-GAGTACTTGCGCTCAGGAGGA-3′) (Takara Bio) were used. The relative mRNA expression was normalized by β-actin mRNA.
Determination of IL-8 protein levels
To confirm the inhibition of IL-8 production by siRNA, HUVECs (2 × 106), transfected as described above, were treated with LPS (1 μg/ml) for 4 hr. After that, the medium was collected. Interleukin-8 protein levels were determined using an enzyme-linked immunosorbent assay kit according to the manufacturer's instructions (R&D Systems, Minneapolis, MN).
Measurement of TF expression on HUVECs by flow cytometry
Expression of TF on HUVECs was examined by flow cytometry. Briefly, HUVECs pre-treated with or without an antioxidant, N-acetylcysteine (20 mm), were stimulated with LPS (1 μg/ml) for 4 hr. After the stimulation, the cells were collected using trypsin and EDTA. The cells were washed twice and suspended in PBS. The suspension was incubated with anti-human TF antibody (Calbiochem, Gibbstown, NJ) at a saturated concentration at 4° for 30 min, and, subsequently, phycoerythrin-conjugated goat anti-mouse antibody (DAKO Japan, Tokyo, Japan) was added to the cells at 4° for 30 min. After two washings with PBS, the cells were subjected to flow cytometry, as described above.
Western blotting for TF expression
Expression of TF protein was analysed by Western blotting. Briefly, HUVECs treated with siRNA were collected and dissolved in lysis buffer, followed by centrifugation (20 000 g, 15 min). The protein was added to the same volume of 2× sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS–PAGE) sample buffer and boiled for 5 min. The protein concentration of lysates was measured by the Bradford method. The 40 μg of protein was separated by 10% SDS–PAGE and transferred to polyvinylidene-difluoride membranes (Immobilon-P; Millipore, Bedford, MA). At 1 : 1000 dilution, mouse anti-human tissue factor monoclonal antibody (Calbiochem, Gibbstown, NJ) and anti-mouse immunoglobulin G horseradish peroxidase-linked antibody (Amersham Biosciences, Zurich, Switzeland) were used as the first and secondary antibodies, respectively. Finally, signal was developed using enhanced chemiluminescence reagent (Amersham Biosciences). β-Actin (Chimicon, Temecula, CA) was used as an internal control.
Statistical analysis
Data are expressed as the means ± standard deviation (SD). P < 0·05 by Student's t-test was considered significant.
Results
Endothelial cells stimulated with IL-8 produce ROS
First, we examined ROS production in HUVECs exposed to 1 μg/ml LPS using flow cytometry. In agreement with previous observations, LPS induced ROS production (Fig. 1a), which was suppressed by the pre-treatment of HUVECs with DPI, an inhibitor of Nox (Fig. 1b). Although a small amount of ROS production was observed 1 hr after LPS stimulation, it was remarkable 4 hr after the simulation (Fig. 1c). Endothelial cells stimulated with LPS are known to generate IL-8, which subsequently recruits and activates neutrophils to degranulate. The activated neutrophils undergo a burst of oxygen consumption that is caused by Nox, ultimately leading to the formation of various ROS. However, the direct effects of IL-8 on ROS production by endothelial cells have not been adequately elucidated. Flow cytometric analysis revealed significant levels of ROS production in HUVECs stimulated with IL-8 (Fig. 1d). The ROS production was then suppressed by the pre-treatment of HUVECs with DPI (Fig. 1e). In contrast to LPS stimulation, the kinetic study demonstrated that IL-8-induced ROS generation was already notable 1 hr after the stimulation, followed by an undetectable level of ROS at 2 hr, suggesting that IL-8 is located down the LPS in this pathway (Fig. 1f). We further analysed mRNA expression of the Nox family by RT-PCR. Nox1 expression was induced by both LPS and IL-8, whereas Nox2 and Nox4 were constitutively expressed in HUVECs, irrespective of the presence of the stimuli (Fig. 2). These results suggest that IL-8-induced ROS production in endothelial cells is NADPH oxidase-related.
Figure 1.
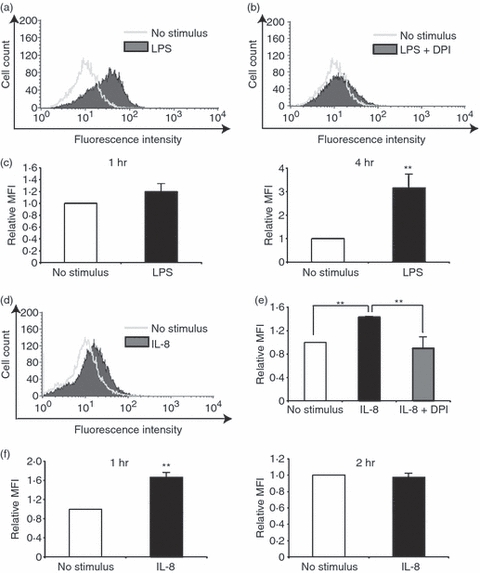
Reactive oxygen species (ROS) production in human umbilical vein endothelial cells (HUVECs) stimulated with lipopolysaccharide (LPS) or interleukin-8 (IL-8). ROS production was assessed by flow cytometry using DCFH-DA in HUVECs stimulated with LPS (a–c) or IL-8 (d–f). (a) Representative data of ROS production in LPS-stimulated HUVECs. (b) Representative data of ROS production in LPS-stimulated HUVECs pre-treated with DPI, an inhibitor of NADPH-oxidase (Nox). (c) Quantitative analysis of ROS production in HUVECs 1 hr (left) or 4 hr (right) after LPS stimulation using relative mean fluorescence intensity (MFI). (d) Representative data of ROS production in IL-8-stimulated HUVECs. (e) Quantitative analysis of ROS production in IL-8-stimulated HUVECs pre-treated with DPI. (f) Quantitative analysis of ROS production in HUVECs 1 hr (left) or 2 hr (right) after IL-8 stimulation. In (d–f) the data represent mean values ± SD (n = 4, **P < 0·01, Student's t-test).
Figure 2.
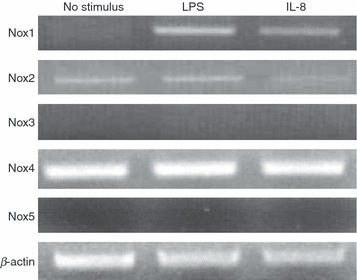
Reverse transcription–polymerase chain reaction analysis of total RNA from human umbilical vein endothelial cells stimulated with lipopolysaccharide (LPS) or interleukin-8 (IL-8). The messenger RNA expression of NADPH oxidase (Nox) 1–5 was analysed. β-Actin served as a loading control. The experiments were performed at least three times, and representative data are shown.
Interleukin-8 is a key mediator of LPS-induced ROS production in endothelial cells
We next examined the role of IL-8 in LPS-induced ROS production in endothelial cells. The HUVECs were subjected to transient transfection with siRNA specific for IL-8 (IL-8 siRNA). Quantitative real-time PCR revealed that the level of IL-8 mRNA was reduced to approximately 30% in HUVECs transfected with IL-8 siRNA compared with control cells transfected with scramble RNA (control siRNA)(Fig. 3a). The IL-8 protein level was also inhibited in HUVECs transfected with IL-8 siRNA, as revealed by enzyme-linked immunosorbent assay (Fig. 3b). Flow cytometric analysis revealed that LPS-induced ROS formation was significantly, although not completely, suppressed by the IL-8 siRNA treatment (Fig. 3c,d). To further substantiate the importance of IL-8 in LPS-induced ROS production, LPS-stimulated HUVECs were treated with anti-CXC chemokine receptor 1 (CXCR1) antibody, anti-CXCR2 antibody, or both. The CXCR1 and CXCR2 are specific receptors for IL-8. Either anti-CXCR1 or anti-CXCR2 antibody treatment inhibited the ROS production, suggesting that LPS-induced ROS production is mediated by both CXCR1 and CXCR2 (Fig. 4a,b,d). A synergistic effect was not observed by the simultaneous treatment of HUVECs with anti-CXCR1 and anti-CXCR2 antibodies (Fig. 4c,d). Moreover, similar effects, though not outstanding, of anti-CXCR1/CXCR2 antibodies on ROS production were observed in IL-8-stimulated HUVECs (Fig. 4e). These results indicate an important role for IL-8 in ROS production by endothelial cells.
Figure 3.
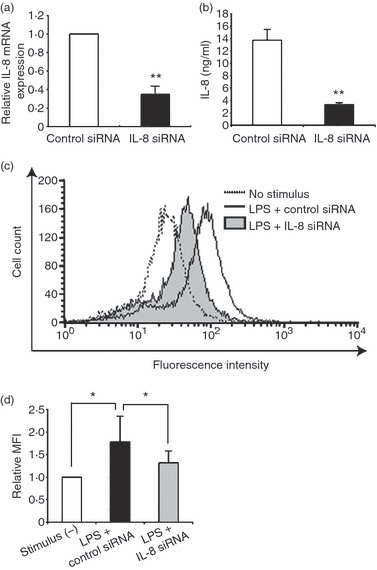
Effect of interleukin-8 (IL-8) knockdown on reactive oxygen species (ROS) production in lipopolysaccharide (LPS) -stimulated human umbilical vein endothelial cells (HUVECs). (a) The relative messenger RNA expression of IL-8 in LPS-stimulated HUVECs treated with IL-8 small interfering (si) RNA as determined by real-time polymerase chain reaction. The expression level is shown relative to that in LPS-stimulated HUVECs treated with control siRNA. The data represent mean values ± SD (n = 3, **P < 0·01, Student's t-test). (b) IL-8 protein expression in LPS-stimulated HUVECs treated with IL-8 siRNA by enzyme-linked immunosorbent assay. The data represent mean values ± SD (n = 4, **P < 0·01, Student's t-test). (c) Representative data of ROS production assessed by flow cytometry using DCFH-DA in LPS-stimulated HUVECs. The cells were treated with IL-8 siRNA or control siRNA. (d) Quantitative analysis of ROS production in LPS-stimulated HUVECs with control siRNA or IL-8 siRNA treatment. The data represent mean values ± SD (n = 4, *P < 0·05, Student's t-test).
Figure 4.
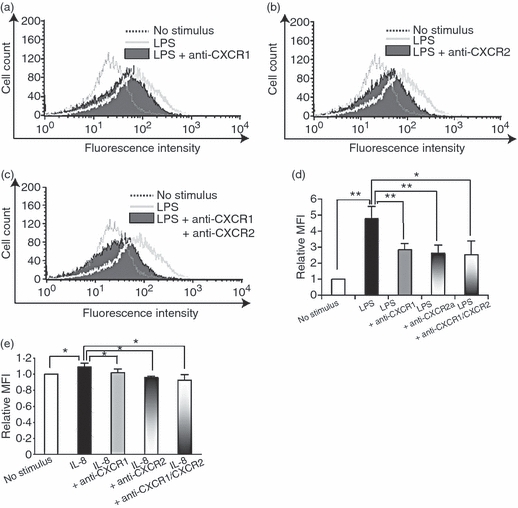
Effect of interleukin-8 (IL-8) receptor blockade on reactive oxygen species (ROS) production in human umbilical vein endothelial cells (HUVECs) stimulated with lipopolysaccharide (LPS) (a–d) or IL-8 (e). ROS production was assessed by flow cytometry using DCFH-DA. (a) Representative data of ROS production in LPS-stimulated HUVECs treated with anti-CXCR1 antibody. (b) Representative data of ROS production in LPS-stimulated HUVECs treated with anti-CXCR2 antibody. (c) Representative data of ROS production in LPS-stimulated HUVECs treated with both anti-CXCR1 and anti-CXCR2 antibodies. (d) Quantitative analysis of ROS production in LPS-stimulated HUVECs treated with anti-CXCR1 antibody, anti-CXCR2 antibody, or both. The data represent mean values ± SD (n = 4, **P < 0·01, *P < 0·05, Student's t-test). (e) Quantitative analysis of ROS production in IL-8-stimulated HUVECs treated with anti-CXCR1 antibody, anti-CXCR2 antibody, or both. The data represent mean values ± SD (n = 4, *P < 0·05, Student's t-test).
Interleukin-8 induces TF expression via Nox1 in endothelial cells
As shown in Fig. 2, Nox1 mRNA expression was dramatically induced by both LPS and IL-8 stimuli, indicating the relevance of Nox1 to the LPS/IL-8 cascade. We therefore examined whether Nox1 affects LPS-induced ROS production in HUVECs. Nox1 siRNA treatment significantly reduced ROS production in LPS-stimulated HUVECs (Fig. 5a). However, Nox4 expression, the predominant Nox enzyme in ROS production in LPS-stimulated endothelial cells,18 was not affected by Nox1 siRNA treatment (Fig. 5b), suggesting that the effect of Nox1 is not mediated by Nox4.
Figure 5.
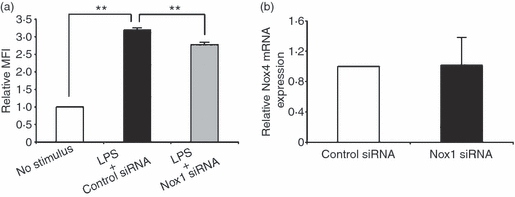
Effect of NADPH oxidase 1 (Nox1) knockdown on reactive oxygen species (ROS) production in lipopolysaccharide (LPS) -stimulated human umbilical vein endothelial cells (HUVECs). (a) Quantitative analysis of ROS production in LPS-stimulated HUVECs with control small interfering (si) siRNA or Nox1 siRNA treatment. The relative mean fluorescence intensity (MFI) is shown on the y-axis. The data represent mean values ± SD (n = 4, **P < 0·01, Student's t-test). (b) The relative messenger RNA expression of Nox4 in HUVECs treated with Nox1 siRNA was determined by real-time polymerase chain reaction. The expression level is shown relative to that in HUVECs treated with control siRNA. The data represent mean values ± SD (n = 4).
A previous study reported that TF expression and its release were induced in endothelial cells stimulated with cytokines such as TNF-α, and the induction was inhibited by antioxidative treatment, indicative of the relevance of ROS in TF expression.20 TF, the primary physiological initiator of coagulation, induces thrombus formation, and is implicated in various vascular disease processes including sepsis. We therefore investigated whether LPS-induced or IL-8-induced ROS formation affects TF expression in endothelial cells. Flow cytometric analysis showed that HUVECs stimulated with LPS induced TF expression, which was suppressed by the treatment with an antioxidant, N-acetylcysteine (Fig. 6a,b). Moreover, quantitative real-time PCR analysis showed that the increased TF expression in LPS-stimulated HUVECs was markedly inhibited by IL-8 siRNA treatment 12 hr after the stimulation (Fig. 7b), whereas the suppression was not observed 8 hr after the stimulation (Fig. 7a). Taken together, these results suggest that LPS-stimulated endothelial cells secrete IL-8, thereby generating ROS, contributing to a high level of TF expression. Next, we examined TF mRNA expression by treating LPS-stimulated HUVECs with Nox1 siRNA. In the same way as IL-8 siRNA treatment, TF mRNA expression was markedly reduced by Nox1 siRNA treatment 12 hr after LPS stimulation (Fig. 7d), but not at 8 hr (Fig. 7c), suggesting that Nox1 is implicated in the pathogenesis of LPS/IL-8-mediated sepsis. Similar effects of IL-8/Nox1 siRNA treatment were observed on the TF protein level, as denoted by immunoblotting (Fig. 7e,f). To further substantiate the above observation, we examined Nox1 and Nox4 expression in LPS-stimulated HUVECs with IL-8 siRNA treatment. Nox1 expression was dramatically attenuated by IL-8 knockdown 8 hr after LPS stimulation (Fig. 8a), in sharp contrast to the unchanged expression level of Nox4 (Fig. 8c). At 12 hr, IL-8 siRNA treatment had little effect on Nox1 and Nox4 expression (Fig. 8b,d), indicating the sequence of LPS–IL-8–Nox1–TF. These results add further support to the notion that the LPS/IL-8-induced septic process in endothelial cells, including a high level of TF expression, is predominantly mediated by Nox1, although Nox4 should play a central role in LPS-induced sepsis, where IL-8 is not involved.
Figure 6.
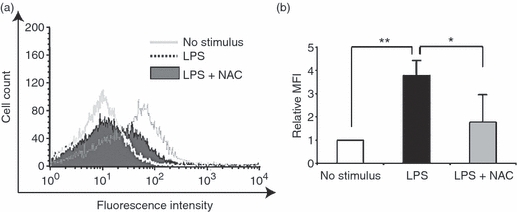
Antioxidative treatment suppresses tissue factor (TF) expression in lipopolysaccharide (LPS) -stimulated human umbilical vein endothelial cells (HUVECs). (a) Representative data of TF expression assessed by flow cytometry. Note that LPS-induced TF expression was suppressed by the treatment with an antioxidant, N-acetylcysteine. (b) Quantitative analysis of TF expression in LPS-stimulated HUVECs with antioxidant treatment. The relative mean fluorescence intensity (MFI) is shown on the y-axis. The data represent mean values ± SD (n = 4, **P < 0·01, *P < 0·05, Student's t-test).
Figure 7.
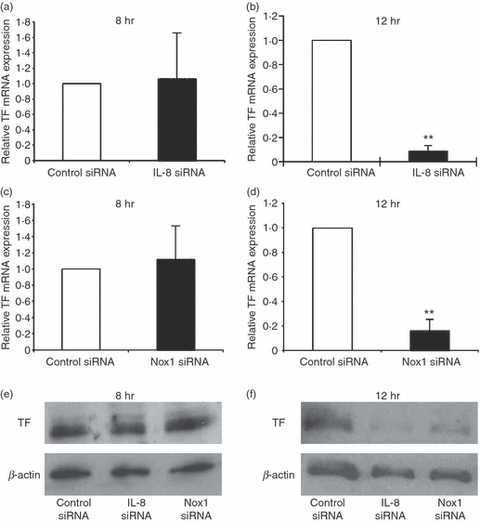
Effect of interleukin-8 (IL-8) or NADPH oxidase 1 (Nox1) knockdown on tissue factor (TF) expression in lipopolysaccharide (LPS) -stimulated human umbilical vein endothelial cells (HUVECs). (a, b) Effect of IL-8 knockdown on TF messenger (m) RNA expression in HUVECs 8 hr (a) or 12 hr (b) after LPS stimulation. The relative mRNA expression of TF was determined by real-time polymerase chain reaction (PCR). The expression level is shown relative to that in LPS-stimulated HUVECs treated with control small interfering (si) RNA. The data represent mean values ± SD (n = 4, **P < 0·01, Student's t-test). (c, d) Effect of Nox1 knockdown on TF mRNA expression in HUVECs 8 hr (c) or 12 hr (d) after LPS stimulation. The relative mRNA expression of TF was determined by real-time PCR. The expression level is shown relative to that in LPS-stimulated HUVECs treated with control siRNA. The data represent mean values ± SD (n = 4, **P < 0·01, Student's t-test). (e, f) Effect of IL-8 or Nox1 knockdown on TF protein expression in HUVECs 8 hr (e) or 12 hr (f) after LPS stimulation. The TF protein expression was determined by immunoblotting. Note that LPS-induced TF expression was markedly suppressed by IL-8 siRNA or Nox1 siRNA treatment at 12 hr.
Figure 8.
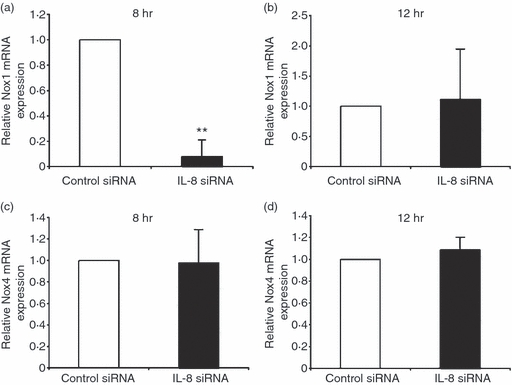
Effect of interleukin-8 (IL-8) knockdown on NADPH oxidase (Nox) 1 (a, b) or Nox4 (c, d) expression in lipopolysaccharide (LPS) -stimulated human umbilical vein endothelial cells (HUVECs). (a, b) Effect of IL-8 knockdown on Nox1 messenger (m) RNA expression in HUVECs 8 hr (a) or 12 hr (b) after LPS stimulation. The relative mRNA expression of Nox1 was determined by real-time polymerase chain reaction (PCR). The expression level is shown relative to that in LPS-stimulated HUVECs treated with control small interfering (si) RNA. The data represent mean values ± SD (n = 4, **P < 0·01, Student's t-test). (c, d) Effect of IL-8 knockdown on Nox4 mRNA expression in HUVECs 8 hr (c) or 12 hr (d) after LPS stimulation. The relative mRNA expression of Nox4 was determined by real-time PCR. The expression level is shown relative to that in LPS-stimulated HUVECs treated with control siRNA. The data represent mean values ± SD (n = 4).
Discussion
In the present study, we showed that IL-8 directly induced ROS production in HUVECs, and mediated LPS-induced ROS production from HUVECs, ultimately leading to a high level of TF expression. We also discovered that LPS/IL-8 signalling in endothelial cells was mainly mediated by Nox1. To our knowledge, this is the first report to demonstrate the importance of IL-8 via Nox1 in ROS production by endothelial cells.
Interleukin-8 is a member of the C-X-C family of chemokines that shows high-affinity binding to CXCR1 and CXCR2. The administration of IL-8 to HUVECs induced ROS production, indicative of the involvement of IL-8 in endothelial ROS production in a paracrine manner. Moreover, the blockage of IL-8 activity by anti-CXCR1, anti-CXCR2 neutralizing antibody, or IL-8 siRNA suppressed ROS production in LPS-stimulated HUVECs, suggesting that IL-8 also functions as an autocrine chemokine mediating LPS-induced ROS production by endothelial cells. Earlier reports have demonstrated that endothelial cells are a major source of IL-8, which is significantly enhanced during inflammation, infection and stress.21–23 Besides the promotion of neutrophil chemotaxis and degranulation, IL-8 signalling modulates endothelial proliferation, survival and migration, and regulates angiogenesis in both a paracrine and autocrine fashion.24–26 These observations are indicative of a crucial role for IL-8 in endothelial functions. We observed a novel role for IL-8 in regulating ROS formation in endothelial cells. However, interestingly, this function could be specific to endothelial cells, as we previously observed that neutrophils stimulated with IL-8 failed to release superoxide (O2−).27 It would be interesting to examine the relevance of IL-8 in ROS production in other cell types such as macrophages.
Furthermore, we found that IL-8-induced ROS production in endothelial cells was Nox-dependent. It has been reported that Nox4 is abundantly expressed in endothelial cells, and plays a central role in ROS production in LPS-stimulated endothelial cells, in contrast to very low expression levels of Nox1 and Nox2.18,28 In our study, Nox1, but not Nox4, expression was strongly induced in endothelial cells by both LPS and IL-8. We further demonstrated that Nox1 is a key mediator of the LPS/IL-8 signalling pathway in endothelial cells, as revealed by knockdown studies. Nox1 expression is normally highest in the colon, and LPS of Helicobacter pylori markedly enhanced Nox1 expression in guinea-pig gastric mucosal cells.29 However, so far as we know, the functions of Nox1 in endothelial cells are not clear. Our study provided a new insight into the function of Nox1 in endothelial cells, and the importance of Nox1 in the pathogenesis of sepsis.
The endothelium plays a pivotal role in orchestrating the host response in sepsis. Following the activation of pattern-recognition receptors on the surface of the endothelium by components of the bacterial wall such as LPS, a myriad of host-derived factors activate endothelial cells. The net phenotype of the endothelium in severe sepsis is pro-inflammation and pro-coagulation. Several clinical trials have been conducted to target LPS or inflammatory mediators such as TNF-α that activate endothelial cells. However, the anti-mediator therapies failed to improve survival in patients with severe sepsis because of the complexity of this pathological condition.30 One way to overcome this situation is to develop broadly based targeting strategies, in which multiple components are attenuated at the same time, for example, the inflammatory and coagulation cascades, because single-component targeting may not only be ineffective, but also may give rise to undesired, deleterious consequences. Phase 3 clinical trials with tissue factor pathway inhibitor demonstrated no beneficial effects, even with an increase in the risk of bleeding, in patients with severe sepsis, despite the promising results of pre-clinical studies.31 In the current study, we revealed that the blockage of IL-8 activity markedly suppressed TF expression induced by LPS stimulation of endothelial cells. We speculate that a therapeutic approach that inhibits both the TF pathway and IL-8 activity at the same time might have synergistic effects to improve survival in patients with severe sepsis.
Acknowledgments
This work was supported by grants from the Ministry of Education, Science, Sports, and Culture of Japan and the Japan Society for the Promotion of Science.
Glossary
Abbreviations:
- CXCR
CXC chemokine receptor
- DCFH-DA
2,7-dichlorodihydrofluorescein diacetate
- DPI
diphenyleneiodonium
- HUVEC
human umbilical vein endothelial cells
- IL
interleukin
- LPS
lipopolysaccharide
- Nox
NADPH oxidase
- ROS
reactive oxygen species
- RT-PCR
reverse transcription–polymerase chain reaction
- siRNA
small interfering RNA
- TF
tissue factor
- TNF-α
tumour necrosis factor-α
Disclosures
We have no competing financial or commercial interests.
References
- 1.Schouten M, Wiersinga WJ, Levi M, Van der Poll T. Inflammation, endothelium, and coagulation in sepsis. J Leukoc Biol. 2008;83:536–45. doi: 10.1189/jlb.0607373. [DOI] [PubMed] [Google Scholar]
- 2.Aird WC. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood. 2003;101:3765–77. doi: 10.1182/blood-2002-06-1887. [DOI] [PubMed] [Google Scholar]
- 3.Henneke P, Golenbock DT. Innate immune recognition of lipopolysaccharide by endothelial cells. Crit Care Med. 2002;30:S207–13. doi: 10.1097/00003246-200205001-00006. [DOI] [PubMed] [Google Scholar]
- 4.Esmon CT. The interactions between inflammation and coagulation. Br J Haematol. 2005;131:417–30. doi: 10.1111/j.1365-2141.2005.05753.x. [DOI] [PubMed] [Google Scholar]
- 5.Levi M, Van der Poll T, Buller HR. Bidirectional relation between inflammation and coagulation. Circulation. 2004;109:2698–704. doi: 10.1161/01.CIR.0000131660.51520.9A. [DOI] [PubMed] [Google Scholar]
- 6.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–76. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 7.Mollen KP, Anand RJ, Tsung A, Prince JM, Levy RM, Billiar TR. Emerging paradigm: Toll-like receptor 4-sentinel for the detection of tissue damage. Shock. 2006;26:430–7. doi: 10.1097/01.shk.0000228797.41044.08. [DOI] [PubMed] [Google Scholar]
- 8.Zhao B, Bowden RA, Stavchansky SA, Bowman PD. Human endothelial cell response to gram-negative lipopolysaccharide assessed with cDNA microarrays. Am J Physiol Cell Physiol. 2001;281:C1587–95. doi: 10.1152/ajpcell.2001.281.5.C1587. [DOI] [PubMed] [Google Scholar]
- 9.Zeuke S, Ulmer AJ, Kusumoto S, Katus HA, Heine H. TLR4-mediated inflammatory activation of human coronary artery endothelial cells by LPS. Cardiovasc Res. 2002;56:126–34. doi: 10.1016/s0008-6363(02)00512-6. [DOI] [PubMed] [Google Scholar]
- 10.Pober JS. Endothelial activation: intracellular signaling pathways. Arthritis Res. 2002;3:S109–16. doi: 10.1186/ar576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rollins BJ. Chemokines and atherosclerosis: what Adam Smith has to say about vascular disease. J Clin Invest. 2001;108:1269–71. doi: 10.1172/JCI14273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baltathakis I, Alcantara O, Boldt DH. Expression of different NF-κB pathway genes in dendritic cells (DC) or macrophages assessed by gene expression profiling. J Cell Biochem. 2001;83:281–90. doi: 10.1002/jcb.1231. [DOI] [PubMed] [Google Scholar]
- 13.Fan J, Frey RS, Rahman A, Malik AB. Role of neutrophil NADPH oxidase in the mechanism of tumor necrosis factor-α-induced NF-κB activation and intercellular adhesion molecule-1 expression in endothelial cells. J Biol Chem. 2002;277:3404–11. doi: 10.1074/jbc.M110054200. [DOI] [PubMed] [Google Scholar]
- 14.Frey RS, Rahman A, Kefer JC, Minshall RD, Malik AB. PKCζ regulates TNF-α-induced activation of NADPH oxidase in endothelial cells. Circ Res. 2002;90:1012–9. doi: 10.1161/01.res.0000017631.28815.8e. [DOI] [PubMed] [Google Scholar]
- 15.Li JM, Fan LM, Christie MR, Shah AM. Acute tumor necrosis factor alpha signaling via NADPH oxidase in microvascular endothelial cells: role of p47phox phosphorylation and binding to TRAF4. Mol Cell Biol. 2005;25:2320–30. doi: 10.1128/MCB.25.6.2320-2330.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frey RS, Gao X, Javaid K, Siddiqui SS, Rahman A, Malik AB. Phosphatidylinositol 3-kinase gamma signaling through protein kinase Czeta induces NADPH oxidase-mediated oxidant generation and NF-κB activation in endothelial cells. J Biol Chem. 2006;281:16128–38. doi: 10.1074/jbc.M508810200. [DOI] [PubMed] [Google Scholar]
- 17.Park HS, Jung HY, Park EY, Kim J, Lee WJ, Bae YS. Cutting edge: direct interaction of TLR4 with NAD(P)H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-κB. J Immunol. 2004;173:3589–93. doi: 10.4049/jimmunol.173.6.3589. [DOI] [PubMed] [Google Scholar]
- 18.Park HS, Chun JN, Jung HY, Choi C, Bae YS. Role of NADPH oxidase 4 in lipopolysaccharide-induced proinflammatory responses by human aortic endothelial cells. Cardiovasc Res. 2006;72:447–55. doi: 10.1016/j.cardiores.2006.09.012. [DOI] [PubMed] [Google Scholar]
- 19.Al Ghouleh I, Magder S. Nicotinamide adenine dinucleotide phosphate (reduced form) oxidase is important for LPS-induced endothelial cell activation. Shock. 2008;29:553–9. doi: 10.1097/SHK.0b013e318157ebc8. [DOI] [PubMed] [Google Scholar]
- 20.Szotowski B, Antoniak S, Goldin-Lang P, et al. Antioxidative treatment inhibits the release of thrombogenic tissue factor from irradiation- and cytokine-induced endothelial cells. Cardiovasc Res. 2007;73:806–12. doi: 10.1016/j.cardiores.2006.12.018. [DOI] [PubMed] [Google Scholar]
- 21.Oude Nijhuis CS, Vellenga E, Daenen SM, Kamps WA, De Bont ES. Endothelial cells are main producers of interleukin 8 through toll-like receptor 2 and 4 signaling during bacterial infection in leukopenic cancer patients. Clin Diagn Lab Immunol. 2003;10:558–63. doi: 10.1128/CDLI.10.4.558-563.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vadeboncoeur N, Segura M, Al-Numani D, Vanier G, Gottschalk M. Pro-inflammatory cytokine and chemokine release by human brain microvascular endothelial cells stimulated by Streptococcus suis serotype 2. FEMS Immunol Med Microbiol. 2003;35:49–58. doi: 10.1111/j.1574-695X.2003.tb00648.x. [DOI] [PubMed] [Google Scholar]
- 23.Huber AR, Kunkel SL, Todd RF, III, Weiss SJ. Regulation of transendothelial neutrophil migration by endogenous interleukin-8. Science. 1991;254:99–102. doi: 10.1126/science.1718038. [DOI] [PubMed] [Google Scholar]
- 24.Koch AE, Polverini PJ, Kunkel SL, Harlow LA, DiPietro LA, Elner VM, Elner SG, Strieter RM. Interleukin-8 as a macrophage-derived mediator of angiogenesis. Science. 1992;258:1798–801. doi: 10.1126/science.1281554. [DOI] [PubMed] [Google Scholar]
- 25.Li A, Dubey S, Varney ML, Dave BJ, Singh RK. IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. J Immunol. 2003;170:3369–76. doi: 10.4049/jimmunol.170.6.3369. [DOI] [PubMed] [Google Scholar]
- 26.Li A, Varney ML, Valasek J, Godfrey M, Dave BJ, Singh RK. Autocrine role of interleukin-8 in induction of endothelial cell proliferation, survival, migration and MMP-2 production and angiogenesis. Angiogenesis. 2005;8:63–71. doi: 10.1007/s10456-005-5208-4. [DOI] [PubMed] [Google Scholar]
- 27.Asagoe K, Yamamoto K, Takahashi A, et al. Down-regulation of CXCR2 expression on human polymorphonuclear leukocytes by TNF-α. J Immunol. 1998;160:4518–25. [PubMed] [Google Scholar]
- 28.Ago T, Kitazono T, Ooboshi H, et al. Nox4 as the major catalytic component of an endothelial NAD(P)H oxidase. Circulation. 2004;109:227–33. doi: 10.1161/01.CIR.0000105680.92873.70. [DOI] [PubMed] [Google Scholar]
- 29.Kawahara T, Kohjima M, Kuwano Y, et al. Helicobacter pylori lipopolysaccharide activates Rac1 and transcription of NADPH oxidase Nox1 and its organizer NOXO1 in guinea pig gastric mucosal cells. Am J Physiol Cell Physiol. 2005;288:C450–7. doi: 10.1152/ajpcell.00319.2004. [DOI] [PubMed] [Google Scholar]
- 30.Marshall JC. Sepsis: rethinking the approach to clinical research. J Leukoc Biol. 2008;83:471–82. doi: 10.1189/jlb.0607380. [DOI] [PubMed] [Google Scholar]
- 31.Abraham E, Reinhart K, Opal S, et al. Efficacy and safety of tifacogin (recombinant tissue factor inhibitor) in severe sepsis: a randomized controlled trial. JAMA. 2003;290:238–47. doi: 10.1001/jama.290.2.238. [DOI] [PubMed] [Google Scholar]