

Abstract
Macrophages (Mϕ) are professional antigen-presenting cells, but when they accumulate at sites of inflammation, they can inhibit T-cell proliferation. In experimental autoimmune uveoretinitis, this limits the expansion of T cells within the target organ. To define requirements for the elaboration of this outcome, we have generated populations of Mϕin vitro that could also regulate T-cell responses; stimulating CD4+ T-cell activation and cytokine production, but simultaneously suppressing T-cell proliferation. When T cells are removed from the influence of such cells, normal T-cell responses are restored. We show that tumour necrosis factor 1 (TNFR1) signalling is a critical checkpoint in the development of such Mϕ, as TNFR1−/− Mϕ are unable to suppress T-cell proliferation. This deficit in antigen-presenting cells results in a lack of production of prostaglandin E2 (PGE2) and nitric oxide, which are critical effector mechanisms that inhibit T-cell division. However, TNFR1 signalling is not required for the inhibitory function of Mϕ because we could circumvent the requirement for this receptor, by maturing Mϕ in the presence of exogenous interferon-γ and PGE2. This produced TNFR1−/− Mϕ that inhibited T-cell proliferation and indicates that TNFR1 delivers a signal that is necessary for the development but not the execution of this function.
Keywords: autoimmunity, experimental autoimmune uveoretinitis, macrophage, nitric oxide, prostaglandin E2
Introduction
Organ-specific autoimmune diseases, such as multiple sclerosis and inflammatory eye disease, are co-ordinated by the activation of autoantigen-specific T cells, which are recruited specifically to sites of disease.1,2. The release of inflammatory mediators leads to a leucocyte influx that consists of a complex mixture of cell types.3,4 For example, at the peak of experimental autoimmune uveoretinitis (EAU), the murine model of human inflammatory eye disease, we observe a heterogeneous population of cells including CD11b+ cells, which form the largest fraction of the immune cells present, with significant numbers of CD4+ T cells and smaller numbers of CD8+ T cells also detected.5–7 In this environment, the large majority of CD11b+ cells are usually described as macrophages (Mϕ); they release inflammatory mediators and act as professional antigen-presenting cells (APCs).8–10 They can stimulate autoantigen-specific CD4+ T cells, by presenting MHC class II-restricted peptides and we have recently reported that Mϕ derived from the inflamed retina of mice with EAU can act as myeloid regulatory cells, inhibiting T-cell proliferation while allowing normal antigen-specific T-cell cytokine production.10
One important cytokine produced by activated Mϕ is tumour necrosis factor-α (TNF-α) and the expression of one of its receptors, TNFR1, is necessary for the normal development of organ-specific autoimmunity.11,12 Blocking signals through this receptor produces a number of important changes in Mϕ function, including the abrogation of nitric oxide (NO) release following interferon-γ (IFN-γ) stimulation,11 with a concomitant reduction in tissue damage. In murine EAU, the loss of TNFR1 signalling is also associated with a dramatic reduction in CD11b+ cell trafficking to the target organ, but an increase in the relative proportion of CD4+ cells within the target organ,10 suggesting that the control of T-cell proliferation by myeloid CD11b+ cells in EAU may be dependent on TNFR1 signalling. Furthermore, CD11b+ cells from the eyes of TNFR1−/− mice immunized for EAU were able to stimulate T-cell proliferation, unlike similar cells from TNFR1 replete mice.10 This TNFR1-dependent correlation between myeloid regulatory function and NO production is consistent with previous findings in the mouse and rat that have associated NO with the inhibition of T-cell proliferation.13–15 However, it was not established whether the abrogation of NO production, consequent to the absence of TNFR1, was sufficient to explain the relative increase in CD4+ T cells in EAU and the loss of regulatory function by target organ-infiltrating Mϕ. In addition, Mϕ-like cells, called MDSC,16 isolated under other chronic inflammatory situations, particularly from tumours, and that suppress anti-tumour immune responses, have been described previously.17–19 These cells exhibit a range of characteristics that are unlikely to be controlled directly by TNFR1 signalling.
Here we describe populations of Mϕ, generated in vitro, which can regulate T-cell responses. We show that the critical requirement for TNFR1 expression and signalling relates to the development of a regulatory myeloid cell phenotype, rather than being required for this regulatory function. Therefore, restoring signals downstream of TNF-α signalling leads to the generation of TNFR1-deficient cells that are competent to inhibit T-cell proliferation. We identify two independent processes that result from TNFR1 signalling that together play a critical role in the control of T-cell responses by Mϕ, which are mediated by IFN-γ and prostaglandin E2 (PGE2) respectively. In cells where the TNFR1−/− signalling pathway is inhibited, the absence of these signals prevents Mϕ differentiation into myeloid cells that regulate T-cell proliferation.
Materials and methods
Mice and reagents
C57BL/6 mice were originally obtained from Harlan UK Limited (Oxford, UK), C57BL/6 TNFR1(p55)-deficient mice (TNFR1−/−) were obtained from The Jackson Laboratory (Bar Harbor, ME), and C57BL/6 OT-II transgenic mice20 expressing the T-cell receptor-specific for chicken ovalbumin323–339 (OVA) and I-Ab were a kind gift of Dr Steve Anderton (Department of Biological Sciences, University of Edinburgh, UK). All mice were housed under specific pathogen-free conditions with food and water continuously available. In all experiments, female mice aged between 6 and 8 weeks were used. Treatment of animals conformed to the regulations for animal research as set down by the Home Office, UK and also to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research.
The OVA323–339 (ISQAVHAAHAEINEAGR) peptide (Sigma, Poole, UK) was at least 95% pure as determined by HPLC. Complete tissue culture medium was Dulbecco's modified Eagle's minimum essential medium (without phenol red) supplemented with 10% volume/volume (v/v) fetal calf serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 2mm l-glutamine, 1 mm sodium pyruvate and 5 μm 2-mercaptoethanol (all from Invitrogen, Paisley, UK). Anti-IFN-γ neutralizing monoclonal antibody (mAb) was purified from culture supernatant of XMG1.2 cells, using protein G columns according to standard protocols. Soluble TNFR1 fusion protein (sTNFR1-Ig) was a kind gift from Geoff Hale (Therapeutic Antibody Group, University of Oxford, UK). All fluorochrome-conjugated anti-mouse mAbs and secondary detection reagents used were purchased from BD Biosciences (Oxford, UK). Biotinylated anti-CD3ζ was from Upstate (Watford, UK), and purified polyclonal rabbit anti-mouse EP1, EP2, EP3 and EP4 were from Cayman Chemicals (Ann Arbor, MI).
Preparation of cells
Bone marrow (BM) Mϕ were generated using a method adapted from Munder et al.21 Briefly, bone marrow cells were resuspended at 5 × 105 cells/ml in complete media supplemented with 5% v/v horse serum (Invitrogen), and 50 pg/ml macrophage colony-stimulating factor. The cell suspension was transferred to hydrophobic PTFE-coated tissue culture bags (supplied by Dr M. Munder, University of Heidelberg, Heidelberg, Germany) and incubated for 8 days at 37° in 5% v/v CO2.
Single-cell splenocyte suspensions were generated by grinding spleens through a 70-μm cell strainer (BD Biosciences) with a syringe plunger. When used as APCs, splenocytes were irradiated with 3000 Rads using a caesuim-137 source (Gravatom, Hants, UK).
The OT-II CD4+ T cells were prepared by enriching CD4+ cells from single cell suspensions of C57BL/6 OT-II splenocytes, using anti-CD4 microbeads (Miltenyi Biotech, Bisley, UK) according to the manufacturer's instructions. B cells were prepared from spleens using anti-B220 microbeads (Miltenyi Biotech). Dendritic cells were generated from cultures of bone marrow cells as previously described.22
Cell culture
The 1 × 105 APCs were co-cultured with CD4+ T cells at ratio of 1 : 1 in round-bottom 96-well plates in complete media. The OVA peptide was added at the indicated concentrations. To some cultures the arginine analogue, l-NG-monomethyl arginine, the NO donor S-nitroso-N-acetyl-l,l-penicillamine, or the cyclo-oxygenase (COX) inhibitor indomethacin (all from Sigma) was added. In some experiments, recombinant IFN-γ (Peprotech, London, UK), or PGE2 (Sigma) was added. Cells were cultured in a humidified environment at 37°, 5% v/v CO2. Proliferation was measured by pulsing with 18·5 kBq [3H]thymidine (GE Healthcare, Bucks, UK) per well for the final 8 hr of culture and determining thymidine uptake [measured in counts per minute (c.p.m.)]. Accumulated NO production was measured after 64 hr in culture supernatants using Griess reagent (Sigma) as previously described.23 Production of IFN-γ was assessed using a murine T helper type 1 (Th1)/Th2 Flow cytomix 10plex kit (Bender Medsystems, Vienna, Austria) according to the manufacturer's instructions. Concentration of PGE2 was measured using an enzyme immunoassay competition enzyme-linked immunosorbent assay kit (Caymen Chemical, Ann Arbor, MI) according to the manufacturer's instructions.
Flow cytometry
Cell suspensions were incubated with 24G2 cell supernatant for 5 min at room temperature before incubation with primary mAb at 4° for 20 min. Intracellular staining was carried out using a cytofix/cytoperm kit according to the manufacturer's instructions (BD Biosciences). Cell suspensions were acquired with an LSR-II flow cytometer (BD Cytometry Systems). Analysis was carried out using FlowJo software (TreeStar, San Carlos, CA).
Statistical analyses
Using Prism 4 software (GraphPad Software Inc., San Diego, CA), comparisons of statistical significance between groups were assessed using the Mann–Whitney U-test.
Results
In inflammatory environments, recruited leucocytes may have emergent properties that are dependent on multiple local interactions with many different soluble signalling molecules. In EAU, accumulating Mϕ, derived from BM cells, infiltrate inflammatory sites in large numbers and perform as professional APCs. They interact with T cells, both enhancing and regulating immunity. We have demonstrated that the Mϕ that accumulate in the target organ modify T cell responses, suppressing T cell proliferation but preserving cytokine secretion.10 These Mϕ express cell surface markers such as Gr1 and CD31 that are associated with immune regulation, and to investigate the function of such cells, we generated Mϕin vitro from BM cells cultured in an inert environment (hydrophobic PTFE-coated tissue culture bags). We compared the ability of these cells to present antigen with other APCs. The OVA323–339-specific TCR transgenic OT-II CD4+ T cells were co-cultured with different populations of professional APCs in the presence or absence of cognate OVA peptide. Wild-type (WT) splenocytes, B cells and dendritic cells stimulated peptide-specific T-cell proliferation, but BM-Mϕ did not (Fig. 1a).
Figure 1.
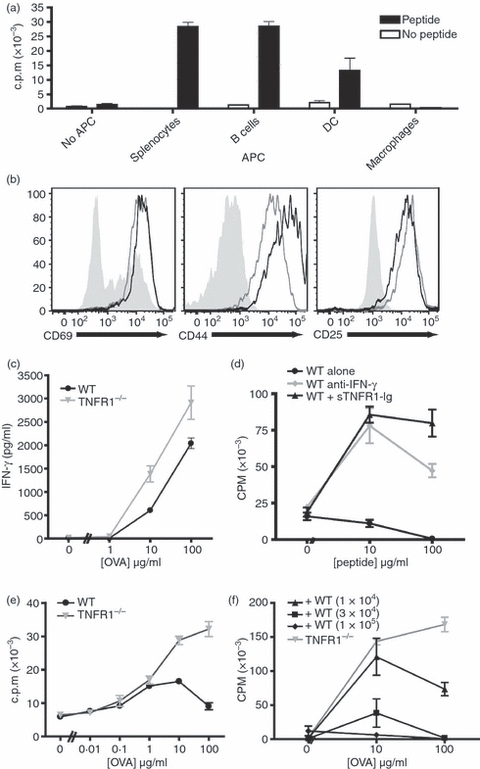
Antigen presentation by macrophages activates both T cells and macrophages, and inhibits T cell proliferation in a tumour necrosis factor-α (TNF-α) dependent manner. Ovalbumin (OVA) -specific OT-II CD4+ T cells were purified and co-cultured at a 1 : 1 ratio with a variety of syngeneic antigen-presenting cells (APCs) for 72 hr in the presence or absence of 10 μg/ml OVA peptide. The APCs tested were whole splenocytes, purified B cells, and dendritic cells (DCs) and macrophages (Mϕ) derived ex vivo from bone marrow cells. T-cell activation assessed by proliferation (a). The activation of CD4+ cells co-cultured with Mϕ (black lines), or with splenocytes (grey lines) is similar compared with naive CD4+ cells (filled grey) (b). Wild-type (WT) or tumour necrosis factor receptor 1 deficient (TNFR1−/−) Mϕ elicit similar levels of interferon-γ (IFN-γ) production by T cells 72 hr after activation (c). Mϕ-dependent inhibition of T-cell proliferation is prevented by blocking with anti-IFN-γ neutralizing monoclonal antibody (10 μg/ml) (grey circles) or sTNFR1-immunoglobulin fusion protein (10 ng/ml) (black triangles) compared with untreated control cultures (black circles). (d). OT-II T cells were co-cultured with TNFR1−/− Mϕ or WT Mϕ across a range of peptide concentrations (e). OT-II T cells were co-cultured with TNFR1−/− Mϕ (1 × 105 cells/well) and WT Mϕ in increasing numbers as indicated (f). These data are representative of three independent experiments.
To address whether this was the result of a failure of Mϕ to interact with T cells, we analysed other markers of T-cell activation. Despite the lack of proliferation, we observed that, following co-culture with BM-Mϕ, OT-II T cells adopted an activated cell surface phenotype and expressed high levels of CD69, CD44 and CD25 (Fig. 1b). The OT-II T cells activated by Mϕ also produced high levels of IFN-γ, the production of which was shown to be independent of TNFR1 signalling as BM-Mϕ derived from TNFR1 knockout (TNFR1−/−) mice stimulated T cells to produce similar amounts of IFN-γ. Interferon-γ activates Mϕ, which in turn leads to autocrine TNF-α signalling that further mediates Mϕ activation.11 Blocking Mϕ activation by neutralizing IFN-γ or TNF-α by the addition of anti IFN-γ mAb or sTNFR1-immunoglobulin fusion protein restored peptide-dependent T-cell proliferation (Fig. 1d), supporting our previous data that the regulation of T-cell proliferation by myeloid cells in the target organ during autoimmunity is dependent on the activation of myeloid cells by IFN-γ and TNF-α.10 Consistent with these in vitro blocking studies, TNFR1−/− Mϕ stimulated T-cell proliferation across a range of peptide concentrations, whereas WT Mϕ stimulated little proliferation (Fig. 1e). Therefore, both WT and TNFR1−/− can activate T cells in a peptide-dependent manner, but WT Mϕ do not induce T-cell proliferation.
We then addressed whether WT Mϕ inhibition of T-cell proliferation was a dominant effect. Addition of increasing numbers of WT Mϕ to cultures where OT-II T cells were activated by TNFR1−/− Mϕ led to a dose-dependent inhibition of proliferation. Adding WT Mϕ at a ratio of 1 : 1 with the TNFR1−/− Mϕ, prevented the proliferation induced by TNFR1−/− Mϕ (Fig. 1f). This TNF-α-dependent suppression of T-cell proliferation by naive Mϕ is similar to that induced by Mϕ in autoimmunity and by populations of myeloid-derived suppressor cells (MDSC), which prevent T-cell responses in tumour sites.13,16
The Mϕ from sites of autoimmune inflammation and MDSC share phenotypic markers, including the expression of CD11b, Gr-1 and CD31, which have been useful in identifying myeloid cells that can inhibit T-cell proliferation. As a consequence, we examined the phenotype of in vitro-generated naive Mϕ and observed that, consistent with in vivo-generated Mϕ, they expressed CD11b, CD31 and F4/80, but not Gr-1 (Supplementary Fig. S1). The activation of BM-Mϕ with LPS or IFN-γ, in the absence of T cells, did not lead to the expression of Gr-1 (data not shown). However, when BM-Mϕ were activated by co-culture with T cells and cognate peptide, both WT and TNFR1−/− Mϕ up-regulated Gr-1 (Fig. 2a), indicating a requirement for signals supplied by T cells for Gr-1 expression. Naive Mϕ from either mouse strain expressed CD31, which was down-regulated to a greater extent on TNFR1−/− Mϕ compared with WT Mϕ following activation (Fig. 2a). Interestingly, the mechanism by which Mϕ acquire a suppressive Gr-1+ phenotype appears to require cell–cell contact with activated T cells, rather than resulting from stimulation by soluble factors (Supplementary Fig. S2).
Figure 2.
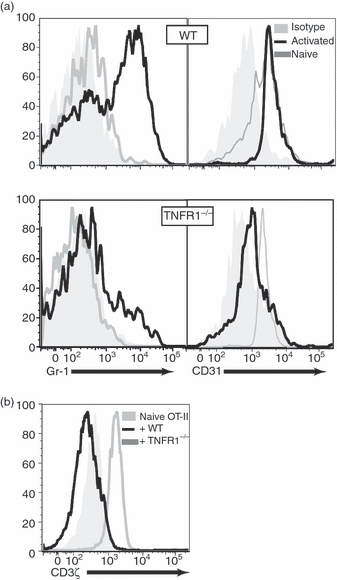
Tumour necrosis factor receptor 1 deficient (TNFR1−/−) macrophages have a less inhibitory phenotype than wild-type (WT) macrophages. WT or TNFR1−/− bone-marrow derived macrophages (BM-Mϕ) were co-cultured with OT-II T cells with 10 μg/ml ovalbumin (OVA) for 72 hr, then surface stained CD4, CD11b, CD31 and Gr-1 (a) and stained intracellularly with anti-TCRξ monoclonal antibody (b). Gr-1 up-regulation was greater with WT Mϕ while CD3ξ expression was reduced compared with naive OT-II CD4+ T cells (grey filled) and OT-II CD4+ T cells co-cultured for 72 hr with TNFR1−/− Mϕ (grey lines). These data are representative of three independent experiments.
The inhibition of T-cell proliferation in the presence of tumour-derived Mϕ has been associated with down-regulation of the ζ-chain of the CD3/TCR signal transduction complex.10,24 To determine the effects on the intracellular expression of CD3ζ by OT-II CD4+ cells, we examined cells stimulated by WT or TNFR1−/− BM-Mϕ. Compared with unstimulated T cells, activation with WT Mϕ led to lower levels of CD3ζ (Fig. 2b) consistent with T-cell inhibition,25 whereas activation with TNFR1−/− Mϕ led to CD3ζ up-regulation, consistent with normal activation26 (Fig. 2b).
Since Mϕ in the local environment stimulate lymphocyte cytokine production but block the proliferation of T cells, we wished to ascertain the fate of T cells that escape from their presence. To do this, we tested whether co-culture with inhibitory BM-Mϕ produced a long-term unresponsive state in the T cells. OT-II CD4+ T cells were combined with BM-Mϕ and OVA peptide for 24 hr and then the non-adherent lymphocytes were removed and the T cells were re-plated in fresh medium. Cell proliferation was then assessed by [3H]thymidine incorporation. OT-II T cells that had been stimulated by TNFR1−/− Mϕ were able to proliferate and this capacity was maintained when they were removed from the APCs. Continuous culture of T cells with WT Mϕ prevented proliferation, but in contrast, when the T cells were removed from the WT Mϕ they were able to proliferate without further antigenic stimulation (Fig. 3). These data show that antigen presentation by Mϕ to T cells for 24 hr produces a T cell that is poised to divide, but is held in check by factors in the local microenvironment.
Figure 3.
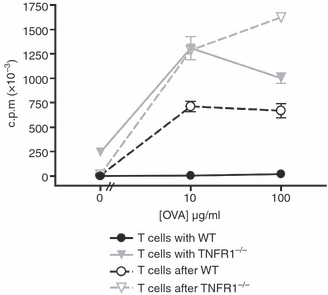
T cells regain the ability to proliferate when removed from macrophages. Wild-type (WT) or tumour necrosis factor receptor 1 deficient (TNFR1−/−) bone-marrow-derived macrophages (BM-Mϕ) were co-cultured with OT-II T cells in the presence of ovalbumin (OVA) peptide. After 24 hr, non-adherent cells were transferred to fresh plates (re-plated) and all cells were washed into fresh media. Proliferation was measured after a further 48 hrs of culture for control cells (solid lines) and re-plated cells (broken lines) for cultures initially containing either WT macrophages (black lines) or TNFR1−/− macrophages (grey lines). These data are representative of four independent experiments.
Inhibition of T-cell proliferation by tumour-derived MDSC and inflammatory monocytes in experimental autoimmune encephalomyelitis has been reported to be the result of the production of NO.27,28 Since TNFR1−/− BM-Mϕ do not produce NO in response to IFN-γin vitro, we wanted to test whether this deficiency was sufficient to explain the WT inhibition of T-cell proliferation, by restoring NO levels in the presence of TNFR1−/− BM-Mϕ. In cultures of OT-II T cells with either WT or TNFR1−/− Mϕ, we could significantly reduce NO production from WT BM-Mϕ with the inhibitor N(G)-mono-methyl-l-arginine (l-NMMA), or raise NO levels to concentrations above those produced by WT BM-Mϕ with the NO donor S-nitroso-N-acetyl-l,l-penicillamine (SNAP) (Fig. 4a). Co-cultures of OT-II T cells and WT Mϕ that were treated with a concentration of l-NMMA that reduced NO production to the levels observed in cultures with TNFR1−/− Mϕ (Fig. 4a and Supplementary Fig. S3) only partially restored proliferation (Fig. 4b). Furthermore, levels of NO that were associated with reduced T-cell proliferation in the context of WT BM-Mϕ, were not sufficient to inhibit the proliferation induced by TNFR1−/− BM-Mϕ (Fig. 4b and Supplementary Fig. S3). Therefore, although some T-cell suppression is the result of the presence of NO, NO alone is not sufficient to produce the complete spectrum of inhibitory effects induced by WT Mϕ.
Figure 4.
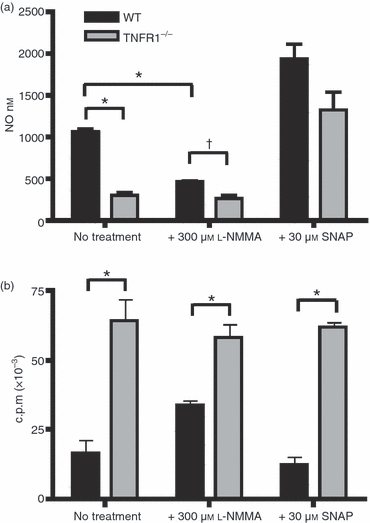
Regulation of T-cell proliferation is reversible and is partially mediated by nitric oxide (NO). Wild-type (WT) or tumour necrosis factor receptor 1 deficient (TNFR1−/−) bone-marrow-derived macrophages (BM-Mϕ) were co-cultured with OT-II T cells in the presence of 100 μg/ml ovalbumin (OVA) peptide for 72 hr. To some cultures, 300 μmN(G)-mono-methyl-l-arginine (l-NMMA), an arginine analogue, or 30 μmS-nitroso-N-acetyl-l,l-penicillamine (SNAP), a nitric oxide (NO) donor, was added. Plots show the effect of addition of l-NMMA or SNAP on NO production (a) and proliferation (b) on co-cultures containing WT (black bars) or TNFR1−/− (grey bars) Mϕ, as compared with control untreated co-cultures. These data are representative of four independent experiments. *P < 0·01; †not significantly different.
We then investigated other mechanisms by which Mϕ can regulate T-cell responses. The soluble factor PGE2 is produced by Mϕ in response to TNF-α29 and we found that culture of OT-II T cells with WT Mϕ in the presence of cognate peptide led to high levels of PGE2, whereas similar culture with TNFR1−/− Mϕ did not (Fig. 5a). As PGE2 has previously been associated with the differentiation of myeloid cells that inhibit T-cell responses in tumours,30 we examined whether its presence was a significant factor in the inhibition of T-cell proliferation by BM-Mϕ. We inhibited PGE2 production with COX inhibitors (SC-560, a COX-1 inhibitor, or indomethacin, a pan-COX inhibitor), which restored OT-II T-cell proliferation (Fig. 5b) to levels that were a third to a half as great as those induced by TNFR1−/− Mϕ. The addition of exogenous PGE2 led to a dose-dependent reduction in OT-II T-cell proliferation stimulated by TNFR1−/− Mϕ (Fig. 5c), and also inhibited WT NO production from WT Mϕ in co-culture.
Figure 5.
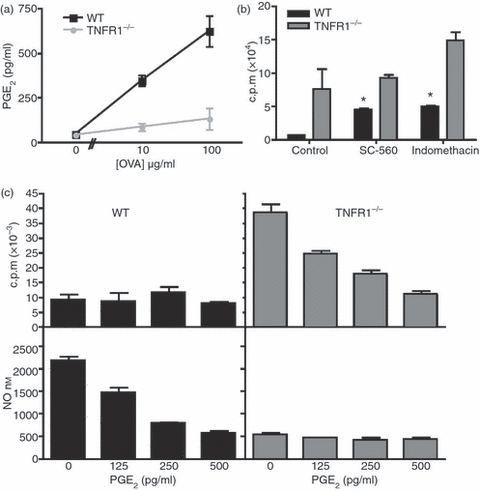
Signalling via tumour necrosis factor receptor 1 (TNFR1) enhances prostaglandin E2 (PGE2) production, which contributes to T-cell regulation. Wild-type (WT) or TNFR1−/− bone-marrow-derived macrophages (BM-Mϕ) were co-cultured with OT-II T cells and ovalbumin (OVA) peptide for 72 hr. In some experiments the pan-cyclo-oxygenase (COX) inhibitor indomethacin or the COX-2-specfic inhibitor SC-256 was added. In other experiments, PGE2 was added. PGE2 concentration (a) and proliferation (b) of co-cultures containing WT Mϕ (black bars) or TNFR1−/− Mϕ (grey lines) treated with COX inhibitors was measured. Proliferation (top) and nitric oxide (NO) production (bottom) were measured from co-cultures with WT Mϕ (black bars, left-hand graphs) or TNFR1−/− (grey bars, right-hand graph) with PGE2 added at a range of concentrations (c). These data are representative of two independent experiments. *Significantly different to control WT MϕP < 0·05.
The effects of PGE2 are mediated through one or more of the four E prostanoid (EP) receptors, EP1, EP2, EP3 and EP4.31 To determine which population of cells PGE2 is acting on, we examined the expression of EP receptors on OT-II T cells and Mϕ. Naive BM-Mϕ expressed low levels of EP2 receptor, but stimulation by IFN-γ led to rapid up-regulation of EP2 by both WT and TNFR1−/− Mϕ, although this up-regulation was greater on WT cells (Fig. 6a). A similar up-regulation was observed when WT or TNFR1−/− Mϕ were activated by co-culture with OT-II T cells and cognate peptide (Fig. 6b). In contrast, OT-II T cells expressed little or no EP2 receptor either when naive or when activated by cognate OVA peptide presented by either WT or TNFR1−/− Mϕ (Fig. 6c). Similar results were obtained for other EP receptors, EP1, EP3 and EP4 (data not shown). These data indicated that, unlike PGE2 (and NO) production, EP receptor up-regulation was independent of TNF-α signalling and that PGE2 in this system most likely acts through effects on Mϕ.
Figure 6.
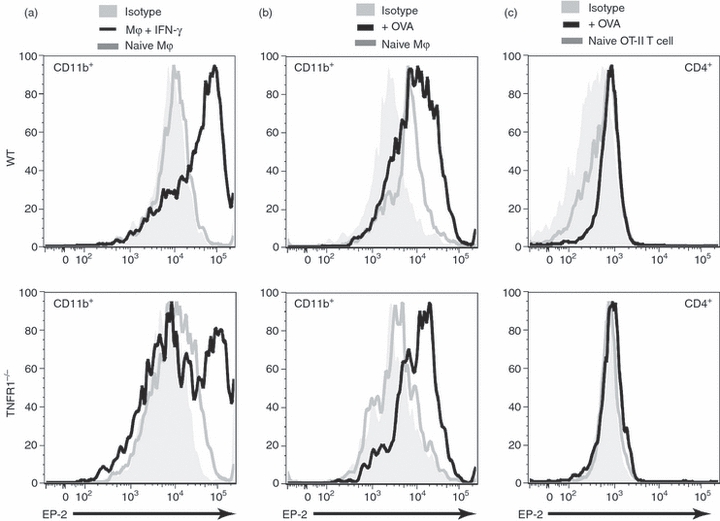
Interferon-γ (IFN-γ) induces E prostanoid (EP) receptor up-regulation on macrophages (Mϕ). Wild-type (WT) or tumour necrosis factor receptor 1 deficient (TNFR1−/−) Mϕ were treated with 100 U/ml IFN-γ for 24 hr, or cultured with OT-II T cells in the presence of 10 μg/ml ovalbumin (OVA) for 72 hr, and stained with monoclonal antibodies (mAbs) against CD11b and EP-2. EP-2 expression by IFN-γ-activated CD11b+ cells (a), naive OT-II CD4+ cells or cells activated by Mϕ (b) or CD11b+ cells from the same co-cultures (c) are shown. Isotype controls (grey-filled) and corresponding naive cells (grey lines) is shown. These data are representative of two independent experiments.
As EP receptor up-regulation was IFN-γ dependent, but TNFR1 independent (Fig. 6a), we reasoned that the up-regulation of these receptors might poise the Mϕ to receive an autocrine PGE2 signal the induction of which was TNFR1 dependent. If this were the case, and if TNFR1 signalling was critical in maturing inhibitory Mϕ but not needed for their function, then treatment with a combination of IFN-γ and PGE2 should circumvent the lack of TNFR1 signalling in TNFR1−/− Mϕ. To test this TNFR1−/− BM-Mϕ were pre-incubated for 72 hr with a combination of PGE2 and IFN-γ separately or together. These treatments did not result in an up-regulation of Gr-1. Nevertheless, the use of the combination of reagents, but not either reagent alone, produced a TNFR1−/− Mϕ that could both inhibit T-cell proliferation (Fig. 7a) and produce NO (Fig. 7b).
Figure 7.
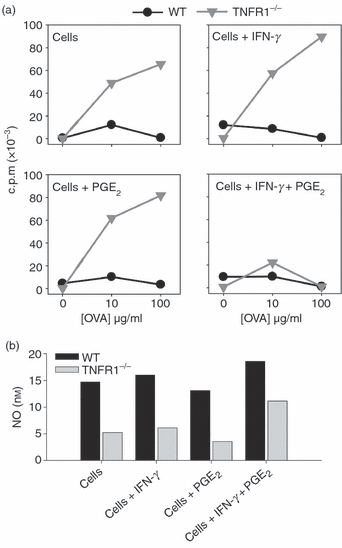
Treatment with interferon-γ/prostaglandin E2 (IFN-γ/PGE2) circumvents the requirement for tumour necrosis factor receptor 1 (TNFR1) signalling in the generation of regulatory macrophages. Wild-type (WT) or TNFR1−/− bone-marrow-derived macrophages (BM-Mϕ) were incubated in PTFE-coated plates for 72 hr in the presence of recombinant IFN-γ (100 U/ml), recombinant PGE2 (1 ng/ml), or both together. After 72 hr, washed cells were co-cultured with OT-II T cells in the presence of OVA peptide (100 μg/ml) for a further 72 hr when T-cell proliferation (a) and nitric oxide (NO) elicited (b) were measured. Pre-treatment with IFN-γ and PGE2 together produces TNFR1−/− Mϕ that suppress proliferation. These data are representative of three independent experiments.
Discussion
In this paper, we explore the role that TNFR1 signalling plays in inducing myeloid cells that can selectively limit T-cell growth. Cognate interactions between T cells and Mϕ that lack TNFR1 lead to activation marker up-regulation, cytokine production and T-cell proliferation, whereas the interaction between the similar T cells and WT BM-Mϕ results in activation marker up-regulation and cytokine production, but not in T-cell division. We have shown that peptide presentation by WT or TNFR1−/− Mϕ to OT-II T cells is sufficient to induce IFN-γ, and that IFN-γ alone can stimulate the up-regulation of EP receptors on WT and TNFR1−/− Mϕ (Figs 6 and 8). This up-regulation is induced more efficiently in the presence of T cells and, furthermore, cell–cell interactions are required for the up-regulation of Gr-1, which accompanies the differentiation to a suppressive phenotype in vivo (Fig. 8).
Figure 8.
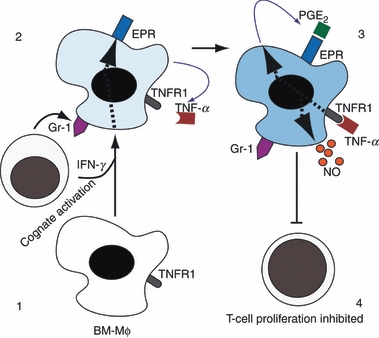
Maturation of bone-marrow-derived macrophages (BM-Mϕ) to produce a suppressive antigen-presenting cell (APC). BM-Mϕ that receive signals via cell contact and interferon-γ/ (IFN-γ) (1) up-regulate Gr-1 and E prostanoid receptor (EPR) and produce autocrine tumour necrosis factor-α (TNF-α) (2). TNF-α signalling stimulates prostaglandin E2 (PGE2) and nitric oxide (NO) production by the Mϕ (3) leading it to generate a local microenvironment that permits T-cell activation but inhibits T-cell proliferation (4).
Interferon-γ also drives the production of low levels of TNF-α that are sufficient to stimulate BM-Mϕ to produce PGE2 and NO in an autocrine manner (Fig. 8). Both of these effector molecules contribute to the subsequent cell cycle arrest in neighbouring T cells; an inhibition that is reversible if the T cells are removed from the vicinity of the Mϕ and washed (Fig. 3).
The ability of Mϕ to reduce T-cell responses has been documented for many years.32 In tumour models, this is thought to contribute to tumour escape from immunosurveillance, but it is unlikely that this represents a normal physiological expression of this process. In inflammation stimulated by infection, restricting T-cell proliferation within the tissue could have a role simply by sparing finite metabolic resources for other effector cells that are present. Rapid T-cell division is highly dependent on local glucose33 and activated Mϕ also consume glucose and other sources of metabolic energy at a high rate.34,35 Therefore, limiting proliferation may be a form of immune system triage at the site of inflammation. Another possibility is that restricting T-cell activation prevents the differentiation of antigen-specific T cells within tissues. Segregating the environment in which T cells differentiate, from that in which they exercise effector function, could reduce the generation of T-cell effector cells that can be activated by autoantigens. At a site of acute inflammation, Mϕ will be processing large amounts of damaged normal tissue that might lead to an increased risk of local autoimmunity.
It is not, however, the case that T-cell immunity is entirely shut down in this inflammatory microenvironment. Our demonstration that T cells removed from the presence of Mϕ can resume proliferation (Fig. 2) shows that T cells that traffic away from the inflammatory environment will still be able to contribute to the pool of circulating activated antigen-specific cells. This local immune response could still serve to amplify T-cell responses and support the production of immunological memory.
In terms of Mϕ function, our data suggest that a lack of TNFR1 signalling impedes the development of Mϕ with the capacity to inhibit T cells. This critical role for TNFR1 in the generation of these cells also suggests TNFR1 may be important to the generation of MDSC in tumours. Therefore, our study throws light on other previously unexplained findings: that in a model of metastasizing lung carcinoma, although tumours initially expand at normal rates, in TNFR1−/− mice, metastases regress after 21 days.36 Also in TNFR1−/− mice and mice treated with TNFR1−/− bone marrow,37 there is a reduced tumour burden in a model of colorectal carcinoma. We suggest that this may relate to a failure to generate functional MDSC. However, other factors also remain important, because the efficacy of TNF-α blockade, which has been used as a therapy in late-stage ovarian carcinoma, maps at least partially to a defect in TNFR1 signalling to T cells.38
The lack of TNFR1 was also associated with a lack of PGE2 production. It has been previously demonstrated that PGE2 is required for MDSC maturation in vivo.30,39 PGE2 can also modulate the function of dendritic cells as APCs, and this effect depends on expression of EP2 or EP4 by the dendritic cell.40 It is also important to note that certain aspects of Mϕ activation (specifically up-regulation of Gr-1) are dependent on cell–cell contact and not mediated by soluble factors (Fig. 2 and supplementary Fig. S2). Up-regulation of Gr-1 is not part of the maturation process demonstrated in Fig. 7, and although a role in limiting T-cell proliferation is not ruled out by this experiment, the soluble mediators NO and PGE2 together are sufficient to restrict T-cell proliferation.
Finally, it was striking that despite the strong phenotype of TNFR1−/− Mϕin vitro and in vivo, we could restore near normal inhibitory function by treating with a combination of soluble mediators (Fig. 7). This result illustrates on the one hand the emergent properties that multiple signals can have on cell function and on the other hand the many levels of redundancy that are inherent in Mϕ responses. This redundancy complicates the analysis of function in vitro and in vivo, but it is also likely to produce immune responses that in the wild are more robust and less susceptible to a single targeted attack by a pathogen.
Acknowledgments
This work was carried out with support from the National Eye Research Centre.
Glossary
Abbreviations:
- APC
antigen-presenting cell
- BM
bone marrow
- COX
cyclo-oxygenase
- c.p.m.
counts per minute
- EPR
E prostanoid receptor
- IFN
interferon
- L-NMMA
N(G)-mono-methyl-l-arginine
- Mϕ
macrophages
- mAb
monoclonal antibody
- MDSC
myeloid-derived suppressor cells
- NO
nitric oxide
- OVA
ovalbumin
- PG
prostaglandin
- SNAP
S-nitroso-N-acetyl-l,l-penicillamine
- TNFR1
tumour necrosis factor receptor 1
- v/v
volume/volume
Disclosures
The authors declare no competing interests.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Figure S1. Wild-type bone-marrow-derived macrophages (WT BM-Mϕ) were prepared as described in the Materials and methods. Cells were then stained with antibodies against CD11b, CD31, F4/80, and Gr-1. Plot A shows F4/80 and CD11b expression by naive BM-Mϕ. Plots B and C show CD31 and Gr-1 expression naive BM-Mϕ (black lines) compared with isotype controls (grey filled).
Figure S2. Wild-type (WT) or tumour necrosis factor receptor 1 deficient (TNFR1−/−) bone-marrow-derived macrophages (BM-Mϕ) were co-cultured with OT-II T cells in the lower chambers of 0.22 μm transwells in the presence of 100 μg/ml ovalbumin (OVA) peptide. Equal numbers of either WT or TNFR1−/− BM-Mϕ were added to the top chamber. After 72 hr, from the both chambers were harvested separately and stained with antibodies against CD11b and Gr-1 for flow cytometric analysis. Plots show Gr-1 expression of CD11b+ cells, with Mϕ from the top chamber (red lines) and those from the lower chamber (blue lines).
Figure S3. Wild-type (WT) or tumour necrosis factor receptor 1 deficient (TNFR1−/−) bone-marrow-derived macrophages (BM-Mϕ) were co-cultured with OT-II T cells in the presence of 100 μg/ml ovalbumin (OVA) peptide for 72 hr. N(G)-Mono-methyl-l-arginine (l-NMMA) or S-nitroso-N-acetyl-l,l-penicillamine (SNAP) was added at the indicated concentrations. T-cell proliferation was measured over the final 8 hr of culture. Nitric oxide (NO) production was measured in the culture supernatant. Plots show the effect of addition of l-NMMA or SNAP on NO production and proliferation on co-cultures containing WT (black lines) or TNFR1−/− (grey lines) Mϕ, as compared with control co-cultures in which Mϕ alone were cultured.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Liversidge J, Forrester JV. Experimental autoimmune uveitis (EAU): immunophenotypic analysis of inflammatory cells in chorio retinal lesions. Curr Eye Res. 1988;7:1231–41. doi: 10.3109/02713688809033227. [DOI] [PubMed] [Google Scholar]
- 2.Martin R, McFarland HF, McFarlin DE. Immunological aspects of demyelinating diseases. Annu Rev Immunol. 1992;10:153–87. doi: 10.1146/annurev.iy.10.040192.001101. [DOI] [PubMed] [Google Scholar]
- 3.Dick AD, Kreutzer B, Laliotou B, Forrester JV. Phenotypic analysis of retinal leukocyte infiltration during combined cyclosporin A and nasal antigen administration of retinal antigens: delay and inhibition of macrophage and granulocyte infiltration. Ocul Immunol Inflamm. 1997;5:129–40. doi: 10.3109/09273949709085061. [DOI] [PubMed] [Google Scholar]
- 4.Zeine R, Owens T. Direct demonstration of the infiltration of murine central nervous system by Pgp-1/CD44high CD45RBlow CD4+ T cells that induce experimental allergic encephalomyelitis. J Neuroimmunol. 1992;40:57–69. doi: 10.1016/0165-5728(92)90213-5. [DOI] [PubMed] [Google Scholar]
- 5.Raveney BJE, Richards CM, Aknin ML, et al. The B subunit of Escherichia coli heat-labile enterotoxin inhibits Th1 but not Th17 cell responses in established autoimmune uveoretinitis. Invest Ophthalmol Vis Sci. 2008;49:4008–17. doi: 10.1167/iovs.08-1848. [DOI] [PubMed] [Google Scholar]
- 6.Kerr EC, Raveney BJE, Copland DA, Dick AD, Nicholson LB. Analysis of retinal cellular infiltrate in experimental autoimmune uveoretinitis reveals multiple regulatory cell populations. J Autoimmun. 2008;31:354–61. doi: 10.1016/j.jaut.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 7.Copland DA, Wertheim MS, Raveney BJE, Armitage WJ, Nicholson LB, Dick AD. The clinical time-course of experimental autoimmune uveoretinitis using topical endoscopic fundal imaging with histological and cellular infiltrate correlation. Invest Ophthalmol Vis Sci. 2008;49:5458–65. doi: 10.1167/iovs.08-2348. [DOI] [PubMed] [Google Scholar]
- 8.Unanue ER. Antigen-presenting function of the macrophage. Annu Rev Immunol. 1984;2:395–428. doi: 10.1146/annurev.iy.02.040184.002143. [DOI] [PubMed] [Google Scholar]
- 9.Geppert TD, Lipsky PE. Antigen presentation at the inflammatory site. Crit Rev Immunol. 1989;9:313–62. [PubMed] [Google Scholar]
- 10.Raveney BJE, Copland DA, Dick AD, Nicholson LB. TNFR1-dependent regulation of myeloid cell function in experimental autoimmune uveoretinitis. J Immunol. 2009;183:2321–9. doi: 10.4049/jimmunol.0901340. [DOI] [PubMed] [Google Scholar]
- 11.Calder CJ, Nicholson LB, Dick AD. A selective role for the TNFp55 receptor in autocrine signalling following IFN-gamma stimulation in experimental autoimmune uveoretinitis. J Immunol. 2005;175:6286–93. doi: 10.4049/jimmunol.175.10.6286. [DOI] [PubMed] [Google Scholar]
- 12.Renno T, Krakowski M, Piccirillo C, Lin JY, Owens T. TNF-alpha expression by resident microglia and infiltrating leukocytes in the central nervous system of mice with experimental allergic encephalomyelitis. Regulation by Th1 cytokines. J Immunol. 1995;154:944–53. [PubMed] [Google Scholar]
- 13.Angulo I, Rullas J, Campillo JA, et al. Early myeloid cells are high producers of nitric oxide upon CD40 plus IFN-gamma stimulation through a mechanism dependent on endogenous TNF-alpha and IL-1alpha. Eur J Immunol. 2000;30:1263–71. doi: 10.1002/(SICI)1521-4141(200005)30:5<1263::AID-IMMU1263>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 14.van der Veen RC, Dietlin TA, Hofman FM. Tissue expression of inducible nitric oxide synthase requires IFN-gamma production by infiltrating splenic T cells: more evidence for immunosuppression by nitric oxide. J Neuroimmunol. 2003;145:86–90. doi: 10.1016/j.jneuroim.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 15.Broderick C, Hoek RM, Forrester JV, Liversidge J, Sedgwick JD, Dick AD. Constitutive retinal CD200 expression regulates resident microglia and activation state of inflammatory cells during experimental autoimmune uveoretinitis. Am J Pathol. 2002;161:1669–77. doi: 10.1016/S0002-9440(10)64444-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand-Rosenberg S, Schreiber H. The terminology issue for myeloid-derived suppressor cells. Cancer Res. 2007;67:425–6. doi: 10.1158/0008-5472.CAN-06-3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bronte V, Apolloni E, Cabrelle A, et al. Identification of a CD11b+/Gr-1+/CD31+ myeloid progenitor capable of activating or suppressing CD8+ T cells. Blood. 2000;96:3838–46. [PMC free article] [PubMed] [Google Scholar]
- 18.Gabrilovich DI, Velders MP, Sotomayor EM, Kast WM. Mechanism of immune dysfunction in cancer mediated by immature Gr-1+ myeloid cells. J Immunol. 2001;166:5398–406. doi: 10.4049/jimmunol.166.9.5398. [DOI] [PubMed] [Google Scholar]
- 19.Nicholson LB, Raveney BJE, Munder M. Monocyte dependent regulation of autoimmune inflammation. Curr Mol Med. 2009;9:23–9. doi: 10.2174/156652409787314499. [DOI] [PubMed] [Google Scholar]
- 20.Barnden MJ, Allison J, Heath WR, Carbone FR. Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol Cell Biol. 1998;76:34–40. doi: 10.1046/j.1440-1711.1998.00709.x. [DOI] [PubMed] [Google Scholar]
- 21.Munder M, Eichmann K, Moran JM, Centeno F, Soler G, Modolell M. Th1/Th2-regulated expression of arginase isoforms in murine macrophages and dendritic cells. J Immunol. 1999;163:3771–7. [PubMed] [Google Scholar]
- 22.Raveney BJE, Morgan DJ. Dynamic control of self-specific CD8+ T cell responses via a combination of signals mediated by dendritic cells. J Immunol. 2007;179:2870–9. doi: 10.4049/jimmunol.179.5.2870. [DOI] [PubMed] [Google Scholar]
- 23.Copland DA, Calder CJ, Raveney BJE, et al. Monoclonal antibody-mediated CD200 receptor signalling suppresses macrophage activation and tissue damage in experimental autoimmune uveoretinitis. Am J Pathol. 2007;171:580–8. doi: 10.2353/ajpath.2007.070272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rodriguez PC, Zea AH, Culotta KS, Zabaleta J, Ochoa JB, Ochoa AC. Regulation of T cell receptor CD3zeta chain expression by l-arginine. J Biol Chem. 2002;277:21123–9. doi: 10.1074/jbc.M110675200. [DOI] [PubMed] [Google Scholar]
- 25.Ezernitchi AV, Vaknin I, Cohen-Daniel L, et al. TCR zeta down-regulation under chronic inflammation is mediated by myeloid suppressor cells differentially distributed between various lymphatic organs. J Immunol. 2006;177:4763–72. doi: 10.4049/jimmunol.177.7.4763. [DOI] [PubMed] [Google Scholar]
- 26.Wang L, Bronstein N, Hsu V, Baniyash M. Transcriptional regulation of the murine TCR zeta gene. Int Immunol. 1995;7:1627–35. doi: 10.1093/intimm/7.10.1627. [DOI] [PubMed] [Google Scholar]
- 27.Brandao MM, Soares E, Salles TS, Saad ST. Expression of inducible nitric oxide synthase is increased in acute myeloid leukaemia. Acta Haematol. 2001;106:95–9. doi: 10.1159/000046596. [DOI] [PubMed] [Google Scholar]
- 28.van der Veen RC, Dietlin TA, Dixon GJ, Gilmore W. Macrophage-derived nitric oxide inhibits the proliferation of activated T helper cells and is induced during antigenic stimulation of resting T cells. Cell Immunol. 2000;199:43–9. doi: 10.1006/cimm.1999.1597. [DOI] [PubMed] [Google Scholar]
- 29.Engelmann H, Holtmann H, Brakebusch C, et al. Antibodies to a soluble form of a tumor necrosis factor (TNF) receptor have TNF-like activity. J Biol Chem. 1990;265:14497–504. [PubMed] [Google Scholar]
- 30.Sinha P, Clements VK, Fulton AM, Ostrand-Rosenberg S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res. 2007;67:4507–13. doi: 10.1158/0008-5472.CAN-06-4174. [DOI] [PubMed] [Google Scholar]
- 31.Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiol Rev. 1999;79:1193–226. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- 32.Allison AC. Mechanisms by which activated macrophages inhibit lymphocyte-responses. Immunol Rev. 1978;40:3–27. doi: 10.1111/j.1600-065x.1978.tb00399.x. [DOI] [PubMed] [Google Scholar]
- 33.Fox CJ, Hammerman PS, Thompson CB. Fuel feeds function: energy metabolism and the T-cell response. Nat Rev Immunol. 2005;5:844–52. doi: 10.1038/nri1710. [DOI] [PubMed] [Google Scholar]
- 34.Newsholme P, Newsholme EA. Rates of utilization of glucose, glutamine and oleate and formation of end-products by mouse peritoneal macrophages in culture. Biochem J. 1989;261:211–8. doi: 10.1042/bj2610211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Newsholme P, Rosa LFBP, Newsholme EA, Curi R. The importance of fuel metabolism to macrophage function. Cell Biochem Funct. 1996;14:1–10. doi: 10.1002/cbf.644. [DOI] [PubMed] [Google Scholar]
- 36.Tomita Y, Yang X, Ishida Y, et al. Spontaneous regression of lung metastasis in the absence of tumor necrosis factor receptor p55. Int J Cancer. 2004;112:927–33. doi: 10.1002/ijc.20493. [DOI] [PubMed] [Google Scholar]
- 37.Popivanova BK, Kitamura K, Wu Y, et al. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J Clin Invest. 2008;118:560–70. doi: 10.1172/JCI32453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Charles KA, Kulbe H, Soper R, et al. The tumor-promoting actions of TNF-alpha involve TNFR1 and IL-17 in ovarian cancer in mice and humans. J Clin Invest. 2009;119:3011–23. doi: 10.1172/JCI39065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rodriguez PC, Hernandez CP, Quiceno D, et al. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med. 2005;202:931–9. doi: 10.1084/jem.20050715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harizi H, Grosset C, Gualde N. Prostaglandin E2 modulates dendritic cell function via EP2 and EP4 receptor subtypes. J Leukoc Biol. 2003;73:756–63. doi: 10.1189/jlb.1002483. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.