

Abstract
The amiloride-sensitive epithelial sodium channel (ENaC) plays a prominent role in sodium uptake from alveolar fluid, and is the major component in alveolar fluid clearance in normal and diseased lungs. The lectin-like domain of TNF-α has been shown to activate amiloride-sensitive sodium uptake in type II alveolar epithelial cells. Therefore, several synthetic peptides that mimic the lectin-like domain of TNF-α (TIP) were synthesised and their ability to enhance sodium current through ENaC was studied in A549 cells with the patch clamp technique. Our data suggest that a free positively-charged N-terminal amino group on residue 1 and/or a free negatively-charged carboxyl group on residue 17 of the TIP peptide is essential for the ENaC-activating effect. Ventilation strategies apart, no standard treatment exists for pulmonary permeability oedema. Therefore, novel therapies activating sodium uptake from the alveolar fluid via ENaC could improve clinical outcome.
Introduction
Fluid balance in the healthy adult mammalian lung depends on the regulation of reabsorption of fluid and solutes by the alveolar and distal epithelia on the one hand, and passive secretion of fluid, driven by hydrostatic pressure, from the vascular space into the alveolar lumen, on the other.1, 2 Excessive accumulation of fluid in the alveolar spaces can be accompanied by reduction of alveolar liquid clearance (ALC) capacity, an epithelial and endothelial hyperpermeability, or a disruption of the epithelial and endothelial barriers caused by increased apoptosis or necrosis.3 Pulmonary oedema is a major complication of acute lung injury (ALI), severe pneumonia and acute respiratory distress syndrome (ARDS), where failure of lungs to rapidly clear oedema fluid is associated with higher morbidity and mortality.4, 5 Resolution of alveolar oedema depends on the active removal of salt and water from the distal air spaces of the lung across the distal lung epithelial barrier.1, 6 Evidence from clinical studies shows that increased ALC leads to better clinical outcome in cases of pulmonary oedema and ALI/ARDS.6 Apart from ventilation strategies, no specific treatment exists for pulmonary oedema and novel therapies need to be developed to improve clinical outcome.7
The involvement of Na+ transporters in alveolar fluid clearance in the mammalian lung has been well established in recent years.2, 8–10 Transport via the amiloride-sensitive epithelial sodium channel (ENaC) in particular is one of the major pathways for Na+ entry across alveolar and distal epithelial cells.11 Although the primary site of oedema fluid clearance appears to be in the alveolar epithelium, distal airway epithelial cells can also absorb Na+.8 The current understanding of alveolar fluid clearance is that Na+ ions passively enter alveolar epithelial cells through the apically-located ENaC and are extruded into the interstitium by basally located Na+/K+-ATPase accompanied by transport of water in the same direction, following the osmotic gradient generated.9, 12 The large variety of metabolites and intricate signalling pathways involved in regulation of ENaC activity in lung epithelia illustrate the central role of regulation of lung Na+ absorption and ENaC activity in lung fluid balance and function.2, 3 For this reason, enhancement of ENaC activity in pathological conditions such as ALI and ARDS has been the focus of intense research.
Tumor necrosis factor α (TNF-α) has been shown to produce an amiloride-sensitive increase in ALC in Pseudomonas aeruginosa-induced pneumonia in rats and to stimulate amiloride-sensitive Na+ uptake by type II pneumocytes.13, 14 Furthermore, TNF-α has been shown to stimulate ALC during intestinal ischaemia-reperfusion and during bronchial asthma in rats.15, 16 The activating effect of TNF-α on ALC is mediated by its lectin-like domain.3, 16–20
The original TIP peptide, 1, designed some 15 years ago, mimics the lectin-like domain of TNF-α (TIP), corresponding to residues C101-E116 of wild type human TNF-α (TIP) (Lucas et al, 1994). Peptide 1 contains a disulphide bridge which effectively restrains the sequence of amino acid residues representing the lectin-like domain into a cyclic structure (Figures 1 and 2). However, the disulphide bridge is an unstable element in peptide 1 and undesirable in a compound intended for therapeutic use. The aim of the present study was to design TIP peptides lacking the disulphide bridge which should be more stable than the original TIP peptide and better suited for use as human medicine. The present work investigated the effect of the original TIP peptide 1, and a series of novel cyclic peptides that also mimic the lectin-like domain of TNF-α, on amiloride-sensitive sodium current in A549 cells. The TIP peptides have been designed and synthesised by solid-phase methods to retain the native conformation of the lectin-like domain as much as possible whilst at the same time exploring alternative linking solutions to bring about cyclisation of the linear sequence. The study was also aimed at elucidation of structural features which are important for the ENaC-activating effect in order to achieve an improved TIP peptide for therapeutic use. The continuous human ATII alveolar cell line, A549 was used to test the activity of the peptides in an electrophysiological assay. Furthermore, the selectivity of the activation effect of Na+ over K+ was investigated.
Figure 1. Human TNF-α around TIP or lectin-like domain (C101-E116) according to structure PDB ID:1A8M, and models for the TIP peptides 1, 2, 3 and 12 based on the 1A8M template.

Side chains are shown only for those residues relevant to the discussion and referred to in the text. Numbers adjacent to lines with double arrowheads refer to Cα-Cα distances in Å. i) Human TNF-α molecule, A chain, showing parts of polypeptide chain, F64-L76 (blue), K98-E116 (green), disulphide bridge between residues C69 and C101 and residues known from previous studies to be essential for trypanolytic and ENaC-activating effects: T105, E107 and E110. ii) 3, the TIP peptide which most resembles wild-type TNF-α, showing isopeptide link between side chains of K1 (K98 in TNF-α) and E19 (E116 in TNF-α) and amino acid change G4 (C101 in TNF-α). iii) 1, showing disulphide bond between C1 (P100 in TNF-α) and C17 (E116 in TNF-α) and amino acid change G2 (C101 in TNF-α); residues critical for ENaC-activating activity are T6, E8 and E11. iv) 2, showing disulphide bond between C1 (C101 in TNF-α) and C15; sequence C1-K12 is the same as C101-K112 in TNF-α, but residues corresponding to P113, W114 and Y115 are missing. v) 12, showing disulphide bond between C1 (P100 in TNF-α) and C17 (E116 in TNF-α) and amino acid change G2 (C101 in TNF-α), as well as mutated critical residues A6, A8 and A11 (T105, E107 and E110 in wild-type human TNF-α and T6, E8, E11 in 1).
Figure 2. TIP peptide 1, 2D chemical structure.
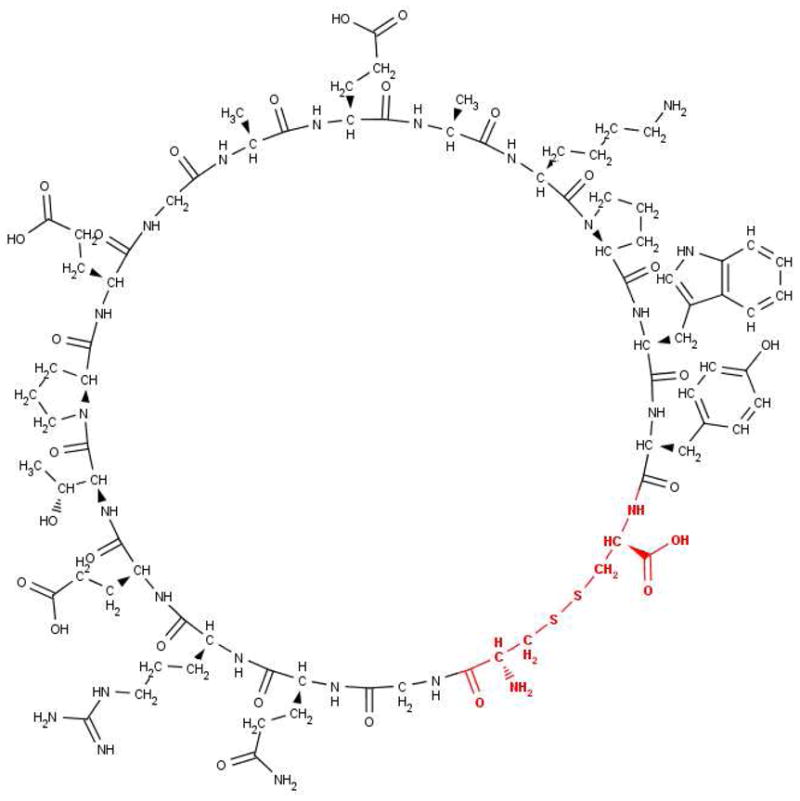
Atoms replaced in the derived TIP peptides 4, 5, 6, 7, 8, 9, 10 and 11 are shown in bold and coloured red. This part of the molecule refers to the cross linking structures, which are listed in Table 1.
Results
Peptide Design
In the present study, the lectin-like domain of human TNF-α (C101 to E116), previously shown to be implicated in sodium uptake activation in A549 cells (Fukuda et al, 2001),14 was used as a template for the design of a series of novel peptides, in which cyclisation was achieved, in all cases but one, by amide bond formation between an amino group of an N-terminal lysine or non-protein amino acid and a carboxyl group of a C-terminal α-amino acid residue. The 3D structural model for human TNF-α, PDB ID:1A8M was used as a template to build 3D models of the peptides using molecular graphics software and the models provided estimates of atomic distances in the cyclic molecules. Cyclic peptides were generally designed such that the distance between the N-terminal and C-terminal α-carbon atoms or atoms in positions equivalent to these, approximated that in 1 (C1-C17 Cα-Cα distance 7.15 Å) or in human TNF-α (P100-E116 Cα-Cα distance 7.23 Å in PDB ID:1A8M A chain) (Figure 1).
The 19-residue cysteine-free, cyclic peptide (3) comprises the sequence of human TNF-α from K98 to E116, with C101 replaced by glycine and cyclisation achieved by isopeptide bond formation between the ε-amino group of K98 and the γ-carboxyl group of E116 (Figure 1).
The cysteine-containing peptide (2) has the wild-type sequence of TNF-α from C101 to K112 to which two glycine residues have been added followed by a C-terminal cysteine residue. Disulphide bond formation between the two terminal cysteine residues brings about cyclisation in the 15-peptide 2. Thus 2 lacks P113 and bulky hydrophobic residues W114 and Y115 of TNF-α (P14, W15 and Y16 of 1), whilst retaining the residues T105, E107 and E110 (T6, E8 and E11 of 1) (Figure 1), shown to be essential for the ENaC-activating effect of TNF-α and 1.14, 17–19
A mutant TIP peptide (12)17 having the same sequence as 1, but with T6, E8 and E11 each replaced by alanine, was included in the study for comparative purposes (Figure 1).
These novel peptides were compared to the original human TIP analogue, CGQRETPEGAEAKPWYC (1), which contains the sequence from P100 to E116 of human TNF-α, but with P100 and E116 replaced by cysteine residues and C101 replaced by glycine, so that cyclisation of the linear sequence was achieved by disulphide bond formation between the two terminal cysteine residues.17 The disulphide bond in analogue 1 may be susceptible to reduction and scission leading to complications in medicinal use. Therefore, the focus of the new peptide design described in this manuscript was replacement of the disulphide bridge of the original TIP analogue, 1, with other molecular arrangements, whilst preserving the sequence of residues T105-E110 (Figure 2, Table 1), shown to be essential for the ENaC-activating effect of TNF-α.14, 17–19
Table 1.
Cross-linking solutions to replace the disulphide bridge of 1 in TIP peptide analogues
| Peptide | Cross-linking structure (positions 1 and 17) | Characteristics of amide bond in cross-linking structure |
|---|---|---|
| 4 | Side chain amino group of lysine (K1) and carboxyl group of glycine (G17) | |
| 5 | 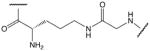 |
Side chain amino group of ornithine and carboxyl group of glycine (G17) |
| 6 | Amino group of 5-aminopentanoic acid and carboxyl group of glycine (G17) | |
| 7 | 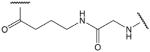 |
Amino group of 4-aminobutanoic acid and carboxyl group of glycine (G17) |
| 8 | 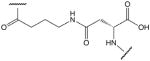 |
Amino group of 4-aminobutanoic acid and side chain carboxyl group of aspartic acid (D17) |
| 9 | 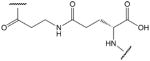 |
Amino group of β-alanine (3-aminopropanoic acid) and side chain carboxyl group of glutamic acid (E17) |
| 10 | Amino group of 7-aminoheptanoic acid and carboxyl group of tyrosine (Y16) | |
| 11 | Amino group of 6-aminohexanoic acid and carboxyl group of glycine (G17) |
4–11- have the same sequence as 1 from G2 to Y16, with natural and non-protein amino acids and amino carboxylic acids replacing C1 and C17; 10 is a 16-peptide in which 7-aminoheptanoic acid replaces C1. In each peptide the cross-linking structure replaces the atoms shown in bold and red in Figure 2.
These cysteine-free cyclic peptides contain the amino acid sequence P100 to Y115 of human TNF-α, with P100 substituted by lysine, ornithine or an aliphatic amino acid and with the C101 replaced by glycine. Replacement of the N-terminal lysine or ornithine by aliphatic amino acid homologues, which lack the α-amino group, afforded a comparison of the effects of positive charge distribution on the ability of the peptides to activate ENaC in A549 cells. Similarly, the effect of negative charge is revealed by replacement of C-terminal aspartic or glutamic acid residues with glycine. In peptides with N-terminal lysine or ornithine, cyclisation has been achieved via the ε-amino or δ-amino group, respectively. In peptides with C-terminal aspartic or glutamic acid, cyclisation has been achieved via the β-carboxyl or γ-carboxyl group of aspartic acid or glutamic acid, respectively. All cysteine-free peptides have in common the 15-residue sequence -GQRETPEGAEAKPWY-.
Structure-activity relationship
TNF-α has been shown to activate ENaC in alveolar epithelium.14 Therefore, synthetic peptides that mimic the lectin-like domain of TNF-α (TIP) were synthesised and evaluated for their ability to enhance the amiloride-sensitive sodium current through ENaC in A549 cells. Currents were recorded by means of the patch clamp technique in the whole-cell mode. To prove the specificity of the test compounds on the amiloride-sensitive Na+ current, firstly, amiloride was added in control experiments to identify the registered current as the amiloride-sensitive Na+ current. Secondly, after a control period and addition of the tested TIP peptide, amiloride was added after a steady-state effect has been reached in order to prove whether the peptide-induced increase in current is due to the amiloride-sensitive Na+ current. In this experimental setting it could be shown that, in a similar manner to TNF-α, TIP peptides 1, 3, 4, 5, 8 and 9 induced an amiloride-sensitive sodium ion flux through ENaC at all tested membrane holding potentials ranging from −100 to +100 mV (Figure 3). At a membrane holding potential of Eh= −100 mV, a maximal response to TNF-α and each ENaC-activating peptide was reached at a current level of 1073 ± 15 pA, whereas TIP peptides 2, 6, 7, 10, 11 and 12 showed no effect on ENaC at concentrations up to 480 nM.
Figure 3. Effect of 1 on current-voltage (IV) relationship.

Representative original whole-cell recordings from a cell clamped at holding potentials ranging from −100 to +100 mV (in 20 mV increments) are shown during control (A) and in presence of 120 nM 1 (B). At this concentration the ENaC-activating effect was maximal. In C the IV-curves are illustrated for control (open circles, n=5), in presence of 60 nM 1 (filled squares, note: in panel C concentration of 1 is 60 nM that is the concentration close to the EC50-value of 1, n=5). Open triangles represent obtained after addition of 10 mM amiloride, which blocks the sodium current in control (n=5) as well as in presence of 1 (n=5). Mean values ± S.E. are given for 5 experiments.
For comparison of activity, the concentration for half maximal response (EC50) was estimated at Eh= −100 mV for TNF-α and all tested TIP peptides. TNF-α as the reference compound increased Na+ current with an EC50 of 8.2 ± 0.1 nM (n=5). The active TIP peptides showed 0.14- (TIP peptide 3) to 0.43-fold (TIP peptide 9) the activity of TNF-α. TIP peptide 3 with an estimated EC50 of 56.0 ± 0.8 nM (n=4) increased the Na+ current to the same extent as compound 1 (Table 2, 4, 5, 8 and 9Figure 4). All other active TIP peptides, , were significantly (P<0.05) more effective in increasing the Na+ current than TIP peptide 1 (Table 2, Figure 4).
Table 2.
Potency of ENaC-activating TIP peptides and selectivity of effect.
| Active test compound | ENaC EC50 (nM) | n | Selectivity | n | |
|---|---|---|---|---|---|
| ENaC (%) | Potassium current (%) | ||||
| TNF-α | 8.2 ± 0.1 | 5 | 93.4 ± 1.9 | 6.9 ± 1.8 | 4 |
| 1 | 54.3 ± 0.8 | 5 | 99.4 ± 0.1 | 0.6 ± 0.1 | 3 |
| 3 | 56.0 ± 0.8 | 4 | 93.4 ± 0.6 | 6.1 ± 0.9 | 3 |
| 4 | 38.3 ± 1.7 | 3 | 99.4 ± 0.2 | 0.6 ± 0.1 | 3 |
| 5 | 45.5 ± 0.6 | 3 | 98.9 ± 0.3 | 1.1 ± 0.3 | 3 |
| 8 | 24.8 ± 0.5 | 5 | 91.2 ± 0.2 | 8.6 ± 0.1 | 3 |
| 9 | 19.9 ± 0.7 | 4 | 89.6 ± 1.2 | 10.4 ± 1.1 | 3 |
Figure 4. Concentration-response curves.

For all ENaC-activating TIP peptides a maximal effect could be observed at 120 nM. TIP peptides 4, 5, 8 and 9 were more active than reference compound 1, but less active than TNF-α, whereas 3 was as active as 1. Estimated values for the Hill slope of the concentration-response curves were smaller for TIP peptides than for TNF-α and 1, indicating attenuated steepness of the curves. Mean values ± S.E. for 3–5 experiments are given.
These data imply that either a free positively-charged N-terminal amino group on residue 1 (4 and 5) or a free negatively-charged carboxyl group on residue 17 (8 and 9) is essential for the ENaC-activating effect. The presence of both types of charged group on the terminal residues, as in compounds 1 and 3, results in an ENaC-activating effect, which is less pronounced than when either charged group is present alone. This structure-activity relationship becomes evident, since the TIP peptides 6, 7, 10 and 11, which lack a free positively-charged N-terminal amino group on residue 1 as well as a free negatively-charged carboxyl group on residue 17, showed no effect on ENaC at concentrations up to 240 nM. TIP peptides 2 and 12 were also ineffective.
Selectivity of ENaC-activating effect
All TIP peptides showing an activating effect were highly selective for amiloride-sensitive sodium current. The percentage increase in whole-cell current, which could be attributed to ENaC ranged between 89.6% in the case of 9 and 99.4% for 1 and 4. Specifically, TIP peptides 1, 4 and 5 have virtually no effect on potassium channels whereas 3, 8 and 9 do have a slight effect (Table 2, Figure 5). The small current which was induced by these latter peptides, when ENaC was blocked by amiloride, could be reversed by the K+ channel blocker tetraethylammonium chloride (TEA). These findings obtained with the whole-cell recording mode were verified with single channel experiments. Effects of peptides 3, 8 and 9 on amiloride-sensitive Na+ current with a conductivity of 9.4 ± 0.1 pS (n=17) as well as on TEA-sensitive K+ current with a conductance of 261 ± 20 pS (n=6) were observed. This estimated conductance is very close to the value of 242 ± 33 pS reported for the TEA-sensitive Ca2+-activated K+ channel in A549 cells.20
Figure 5. TEA-selective K+ current.

Current traces induced by clamping the cell membrane to a holding potential of −100 mV are presented. The original whole-cell recordings from two cells are illustrated during control, and after addition of 100 μM amiloride, which blocks the amiloride-sensitive Na+ current through ENaC. Addition of 1 and 8 to the bathing solution induced virtually no (1, right panel) or only a small (8, left panel) current, respectively. This peptide-induced current was completely blocked by TEA indicating that this current is a K+ current. The pulse protocol and calibration bar is illustrated in the inset.
With regard to this point, it is interesting to note that 4 and 5 have a free positively-charged, N-terminal amino group on residue 1 and no free negatively-charged carboxyl group on residue 17, whereas 8 and 9 have no free positively-charged amino group, but they do have a free negatively-charged carboxyl group on residue 17. Both 1 and 3 have a free amino group on residue 1, but in 3, residue 1 is equivalent to K98 in native TNF-α, whereas in 1 residue 1 is equivalent to P100, thus the N-terminal residues in these peptides represent structurally different parts of the native TNF-α molecule.
Discussion and Conclusions
Both alveolar type I (ATI) cells (which cover at least 95% of the internal surface area of the lung) and type II (ATII) cells (which cover about 2–5% of the internal surface area of the lung) have several different sodium-permeable channels in their apical membranes that play a role in normal lung physiology and pathophysiology.2 ENaC appears to be expressed in both ATI and ATII cells.12 The channel plays a prominent role in sodium uptake from alveolar fluid, and its involvement in alveolar clearance in normal and diseased lungs has been widely demonstrated.1, 6, 9, 21 K+ channels are also expressed in ATI and ATII cells12, 22, and they can participate in transepithelial Na+ and Cl− exchange22, as well as alveolar fluid clearance.23 Apart from ENaC, several other ion channels such as acid-sensing ion channels or ion pumps and transporters like the sodium-calcium exchanger, Na+/K+-ATPase or the Na+/K+/Cl− co-transporter contribute to the sodium homeostasis of a cell. Previous work showed an effect of TNF-α on ENaC14. ENaC is located at the apical side of the cell, and in addition there is evidence that TIP peptides rather work from the apical than basolateral side.19 To our knowledge A549 cells express ENaC24, potassium channels25 and a cyclic nucleotide-gated ion channel26, but no co-expression of ASIC and ENaC in the A549 cell line has been reported. Therefore, based on these previous findings, we chose A549 cells for our structure-activity relationship studies on ENaC. However, we are aware that the TIP peptides could affect directly or indirectly also other channels than ENaC and also ion pumps and transporters, which are responsible for sodium homeostasis.
TNF-α has been shown to activate amiloride-sensitive sodium currents in alveolar epithelium.14 In accordance, several in situ, ex vivo and in vivo studies have indicated that the lectin-like domain of TNF-α promotes amiloride-sensitive oedema reabsorption and alveolar fluid clearance.3, 19, 27–30 The seemingly contradictory roles of TNF-α in pulmonary oedema can be explained by the spatial separation of the different functions of TNF-α, with its lectin-like domain which activates, and its receptor-binding sites which inhibit oedema reabsorption,31, 32 being located in different domains in the native molecule.3, 18, 19, 29, 30 Native murine TNF-α (mTNF) has been shown to produce a similar amiloride-sensitive increase in ALC in mice expressing TNF receptors as in mice genetically deficient for the TNF receptors, indicating a TNF receptor-independent mechanism for amiloride sensitive ALC.28 A mutant mouse TNF-α with amino acid changes T104A, E106A and E109A in the lectin-like domain, has no effect on Na+ uptake in flooded rat lungs in situ, unlike the wild type mTNF, which increases uptake.14 Furthermore, wild-type mTNF, but not mutant, stimulates the amiloride-sensitive Na+ uptake in A549 cells in vitro.14 The mouse TIP peptide also has been shown to improve alveolar fluid balance in injured isolated rabbit lungs.30 More recently it has been demonstrated that human TIP peptide improved rat lung function after transplantation, reducing ROS generation and neutrophil infiltration.19 The latter study also provided evidence for the role of ENaC as the primary site of action of the TIP peptide in monolayers of rat type II alveolar epithelial cells, as the peptide activated transepithelial current only when introduced to the apical side of the cells.19 Taken together, these observations indicate that the receptor-binding sites of TNF-α inhibit, whereas its lectin-like domain activates oedema reabsorption and support a therapeutic role for TIP peptides in treatment of pulmonary oedema and ALI/ARDS.
Native TNF-α is a homotrimer with the lectin-like domain of each subunit (in human TNF-α the sequence of amino acid residues C101 to E116) located close together at the tip of the structure, whereas the TNF receptor-binding sites are located in the basolateral regions of the molecule. Initially characterised for its trypanolytic effect17, the lectin-like domain of TNF-α has been shown to activate amiloride-sensitive Na+ uptake in lung microvascular endothelial cells18 and in the alveolar epithelial cell line A549.14 Therefore, synthetic peptides that mimic the lectin-like domain of TNF-α were synthesized. Replacement of C101 by a glycine residue (G2) and addition of N-terminal (C1) and C-terminal cysteine (C17) residues, linked by a disulphide bridge, resulted in the 17-residue cyclic peptide, 1. The amino acid residues T105, E107 and E110 of human TNF-α corresponding to T6, E8 and E11 of 1, were shown to be essential for the ENaC-activating effect of TNF-α and the analogue 1 peptide.17, 18 From the TIP-domain of TNF-α, a series of synthetic TIP peptides, 3, 4, 5, 6, 7, 8, 9, 10 and 11 was derived.
A comparison of the ENaC-activating effect of all the TIP peptides allows tentative conclusions to be drawn regarding the influence of three main structural features of the TIP-domain and 1 on ENaC. These features are firstly, the T105, E107, E110 triad of residues at the tip of the TNF-α molecule previously shown to be necessary for the trypanolytic, chitobiose-binding and ENaC-activating effects of TNF-α and 1,14, 17–19, 33 secondly, a hydrophobic region introduced by P113 and bulky, hydrophobic side chains of adjacent residues W114 and Y115, and thirdly, the presence of either a positively-charged N-terminal amino group, or a negatively-charged C-terminal carboxyl group or both of these.
The importance of residues T105, E107 and E110 (corresponding to T6, E8 and E11 in 1) is substantiated in the present study as the mutant TIP peptide (12), in which these residues are replaced by alanine, has no effect on the sodium current in A549 cells, whereas 1 does have an effect (EC50 = 54.3 ± 0.8 nM). All novel peptides contain T6, E8 and E11, which have been shown to be essential for the ENaC-activating effect of 1. The inactive TIP peptide 2 lacks residues equivalent to P113, W114 and Y115 of human TNF-α, and therefore the bulky hydrophobic region of 1. Furthermore, studying the ENaC-activating effect of these TIP peptides, it became obvious that a free positively-charged N-terminal amino group on residue 1 and/or a free negatively-charged carboxyl group on residue 17 is essential for activity. The most active peptides have either a free positively-charged amino group at the N-terminal in the position of residue 1 – this is the case for 4 and 5 – or they have a free negatively-charged carboxyl group at the C-terminal in the position of residue 17, as is the case for 8 and 9. TIP peptides which lack a free positively-charged N-terminal amino group on residue 1 as well as a free negatively-charged carboxyl group on residue 17 (6, 7, 21, 22), did not affect ENaC. The importance of the charged groups in the linker part of the peptides for activity can be better appreciated by comparison of the activity of pairs of peptides differing only in the presence or absence of a charged group on the linker portion of the molecule. Specifically, peptide 4 and peptide 11 differ solely in that 4 has a charged amino group on residue 1, 4 has an EC50 of 38.3 nM, whereas 11 is inactive. Peptide 5 and peptide 6 also differ solely in the presence of a charged amino group on residue 1 in 5 and its absence in 6; 5 has an EC50 of 45.4 nM, whereas 6 is inactive (Table 2). Similar comparison concerning the presence of a free carboxyl group show that peptide 8, which has a free carboxyl group on residue 17, has an EC50 of 24.8 nM, whereas 7, which lacks a carboxyl group is inactive. TIP peptides 1 and 3, which are approximately identical in their ability to activate amiloride-sensitive Na+ current, (EC50 of 54.3 ± 0.8 and 56.0 ± 0.8 nM, respectively), each have both a positively-charged, N-terminal amino group and a negatively-charged, C-terminal carboxyl group. Therefore, it seems that the presence of charge, positive on the N-terminal residue 1 or negative on the C-terminal residue 17 (or 19 in the case of 3) is essential for the ENaC-activating effect of the TIP peptides. All active TIP peptides were highly selective for ENaC with a tendency towards less selectivity in molecules without a free positively-charged N-terminal amino group.
To conclude, the results of the present study show that TIP peptides which have an ENaC-activating effect in A549 cells possess i) the triad of residues equivalent to T105, E107 and E110 of human TNF-α, ii) the group of adjacent hydrophobic residues equivalent to P113, W114 and Y115 of human TNF-α, and iii) either a positively-charged N-terminal amino group or a negatively-charged C-terminal carboxyl group or both of these, whereas peptides which lack one or more of these features do not exert such an effect.
Experimental Section
The analysis for the determination of purity of the test compounds was done by HPLC. The purity of tested TIP peptides was ≥95%, except for peptide 3 (see peptide description).
Peptide Synthesis
All peptides in this study were synthesised by solid-phase peptide synthesis according to the fluorenylmethyloxycarbonyl/t-butyl protection strategy on 2-chlorotritylchloride resin. Diisopropyl carbodiimide and N-hydroxybenzotriazole were used as coupling reagents. All coupling steps were carried out in N-N-dimethyl formamide. Protected amino acids were coupled in succession to the peptide chain, starting with the C-terminal amino acid. Deprotection of fluorenylmethoxycarbonyl was carried out in 20% piperidine in N-N-dimethyl formamide. Cleavage of the completed, partially-protected peptide from the resin was carried out in a 1:1 mixture of acetic acid and dichloromethane. In the case of cysteine-containing peptides, after cleavage from the resin, side-chain deprotection in 95% trifluoroacetic acid, 5% water, was carried out followed by cyclisation by oxidation of terminal cysteine residues, achieved by aeration of the crude linear peptide at pH 8.5 for 90 hours. Crude peptide product was purified by reverse phase medium pressure liquid chromatography (RP-MPLC) on an RP-C18-silica gel column with a gradient of 5% – 40% acetonitrile. Finally, the trifluoracetate counter-ion was replaced by acetate on a Lewatit MP64 column (acetate form). Following a final wash in water, the purified peptide as acetate salt was lyophilised and obtained as a white to off-white powder. In the case of cysteine-free peptides, the cyclisation step was carried out on the partially-protected linear peptide following cleavage from the 2-chlorotritylchloride resin. After selective cyclisation of the cysteine-free peptides, side-chain deprotection in trifluoroacetic acid followed by preparative RP-MPLC, replacement of the trifluoroacetate ion by acetate and lyophilisation of the acetate form of the peptide was carried out as for cysteine-containing peptides. In the case of peptide 8, in which cyclisation involved amide bond formation through the side chain carboxyl group of aspartic acid, selective cyclisaton was achieved by starting the synthesis using the C-terminal aspartic acid N-protected with the fluorenylmethyloxycarbonyl group and with the C-α carboxyl protected with a tertiary butyl (OtBu) group. Synthesis proceeded by linkage of the C-terminal aspartic acid residue to the trityl resin through the side chain carboxyl group, followed by stepwise addition of the protected amino acid residues to the peptide chain. After deprotection of the amino group of N-terminal 4-aminobutanoic acid and cleavage of the side-chain protected peptide from the resin, cyclisation was carried out through the free side-chain carboxyl group and the amino group of N-terminal 4-aminobutanoic acid. Finally, side chain protecting groups were removed with trifluoracetic acid and the peptide purified by RP-MPLC as for the other peptides. The synthesis of peptide 9, followed the same principles as that of 8, except that the C-terminal amino acid was glutamic acid with the Cα carboxyl group protected with OtBu, and with 3-aminopropanoic acid as the N-terminal amino carboxylic acid.
The molecular masses of the peptides were confirmed by electrospray ionisation mass spectrometry or MALDI-TOF-MS and their purity was determined by analytical high performance liquid chromatography.
Peptide Description
Cyclo(CGQRETPEGAEAKPWYC) (1)
Cyclisation in peptide 1 was achieved by oxidation of the terminal cysteine residues to form a disulphide bridge. The purity of the peptide was 96.3%. m/z (ESI) 1924.2 (M++1); theoretical average molecular mass 1923.1.
Cyclo(CQRETPEGAEAKGGC) (2)
Cyclisation in peptide 2 was achieved by oxidation of the terminal cysteine residues to form a disulphide bridge. The purity of the peptide was 97.0%. m/z (MALDI) 1534.8 (M++1); theoretical average molecular mass 1533.6.
Cyclo(KSPGQRETPEGAEAKPWYE) (3)
Cyclisation in peptide 3 was achieved by formation of an amide bond between the amino group attached to the ε-carbon of the N-terminal lysine residue and the side chain carboxyl group attached to the γ-carbon of the C-terminal glutamic acid residue. The purity of the peptide was 89%. m/z (MALDI-TOF) 2142.7 (M++1); theoretical average molecular mass 2142.3.
Cyclo(KGQRETPEGAEAKPWYG) (4)
Cyclisation in peptide 4 was achieved by creating an amide bond between the amino group attached to the ε-carbon of the side chain of the N-terminal lysine residue and the carboxyl group of the C-terminal glycine residue. The purity of the peptide was 98.8%. m/z (ESI) 1888.2 (M++1); theoretical average molecular mass 1886.0.
Cyclo(ornithine-GQRETPEGAEAKPWYG) (5)
Cyclisation in peptide 5 was achieved by creating an amide bond between the amino group attached to the δ-carbon of the side chain of the N-terminal ornithine residue and the carboxyl group of the C-terminal glycine residue. The purity of the peptide was 97.4%. m/z (ESI) 1873.4 (M++1); theoretical average molecular mass 1872.0.
Cyclo(5-aminopentanoic acid-GQRETPEGAEAKPWYG) (6)
Cyclisation in peptide 6 was achieved by creating an amide bond between between the amino group of N-terminal 5-aminopentanoic acid and the carboxyl group of the C-terminal glycine residue. The purity of the peptide was 100%. m/z (MALDI-TOF) 1857.9 (M++1); theoretical average molecular mass 1857.0.
Cyclo(4-aminobutanoic acid-GQRETPEGAEAKPWYG) (7)
Cyclisation in peptide 7 was achieved by creating an amide bond between the amino group of the N-terminal 4-aminobutanoic acid and the carboxyl group of the C-terminal glycine residue. The purity of the peptide was 100%. m/z (MALDI-TOF) 1843.3 (M++1); theoretical average molecular mass 1843.0.
Cyclo(4-aminobutanoic acid-GQRETPEGAEAKPWYD) (8)
Cyclisation in peptide 8 was achieved by creating an amide bond between the amino group of 4-aminobutanoic acid and the side chain carboxyl group attached to the β-carbon of the C-terminal aspartic acid residue. The purity of the peptide was 100%. m/z (MALDI-TOF) 1901.6 (M++1); theoretical average molecular mass 1901.0.
Cyclo(β-alanine-GQRETPEGAEAKPWYE) (9)
Cyclisation in peptide 9 was achieved by creating an amide bond between the amino group of the N-terminal β-alanine (3-aminopropanoic acid) and the side chain carboxyl group attached to the γ-carbon of the C-terminal glutamic acid residue. The purity of the peptide was 100%. m/z (MALDI-TOF) 1902.7 (M++1); theoretical average molecular mass 1901.0.
Cyclo(7-aminoheptanoic acid-GQRETPEGAEAKPWY) (10)
Cyclisation in peptide 10 was achieved by creating an amide bond between the amino group of the N-terminal 7-aminoheptanoic acid and the carboxyl group of the C-terminal tyrosine residue. The purity of the peptide was 97.2%. m/z (MALDI-TOF) 1828.9 (M++1); theoretical average molecular mass 1828.0.
Cyclo(6-aminohexanoic acid-GQRETPEGAEAKPWYG) (11)
Cyclisation in peptide 11 was achieved by creating an amide bond between the amino group of 6-aminohexanoic acid and the carboxyl group of the C-terminal glycine residue. The purity of the peptide was 100%. m/z (MALDI-TOF) 1872.6 (M++1); theoretical average molecular mass 1871.0.
Cyclo(CGQREAPAGAAAKPWYC) (12)
Cyclisation in peptide 12 was achieved by oxidation of the terminal cysteine residues to form a disulphide bridge. The purity of the peptide was 98.2%. m/z (ESI) 1778.2 (M++1); theoretical average molecular mass 1777.0.
Structural Modelling
Using 3D structural model for human TNF-α, PDB ID:1A8M as a template, 3D models of the peptides were built in Accelrys DS Visualizer, version 2.0.1 (http://accelrys.com), Avogadro, version 1.0.0 (http://avogadro.openmolecules.net/wiki/Main_Page) or DeepView/Swiss-PdbViewer, version 4.0.1 (http://www.expasy.org/spdbv/) molecular graphics and editing programs.34 For all peptide models the part of the structure immediately surrounding the lectin-like domain in the A chain, between residues K98 and P117 was taken as a template for modelling. A model for 1 was built in DeepView by mutating the P100 and E116 residues to cysteine and the C101 to glycine, followed by removal of extra residues and energy minimisation. Models for the remaining peptides were built in DeepView, Accelrys DS and Avogadro by mutation of residues, replacement of elements and insertion or deletion of bonds to achieve the desired molecular arrangement, followed by energy minimisation. Images of the peptide models were made in PyMol, version 0.99 (http://www.pymol.org/) or RasMol, version 2.7.4.2 (http://rasmol.org/).
Energy minimisation computations were performed with the GROMOS96 implementation of Swiss-PdbViewer. Computations were performed in vacuo with the Gromos96 43B1 parameter set without reaction field35 (http://www.igc.ethz.ch/GROMOS).
Two-dimensional chemical structures were drawn using MarvinSketch version 5.3.1 (http://www.chemaxon.com/) and BioDraw Ultra, version 11.01 (http://cambridgesoft.com).
Cell line
Experiments were carried out on the human epithelial cell line A549 (ATTAC Nr. CCL-185) in passages 80–90. Cells were grown in Dulbecco’s modified Eagle’s medium/nutrient mixture F12 Ham, supplemented with 10% fetal bovine serum and containing 1% penicillin-streptomycin. All culture media were purchased from Sigma-Aldrich GmbH (St. Louis, MO).
Electrophysiological Recordings
Effects on ENaC were studied on A549 cells at room temperature (19–22°C) 24 to 48 h after plating. Currents were recorded with the patch clamp method in the whole-cell mode. Glass cover slips with the cultured cells were transferred to a chamber of 1 ml capacity, mounted on the stage of an inverted microscope (Zeiss, Axiovert 100). The chamber contained 1 ml of the bath solution of the following composition (in mM): 145 NaCl, 2.7 KCl, 1.8 CaCl2, 2 MgCl2, 5.5 glucose and 10 HEPES, adjusted to pH 7.4 with 1 M NaOH solution. Micropipettes were pulled from thin-walled borosilicate glass capillaries (World Precision Instruments, Inc., FL, USA) with a Flaming Brown micropipette puller (P87, Sutter Instruments, CA, USA) and polished on a microforge (Narishige, Tokyo, Japan) to obtain electrode resistances ranging from 2.0 to 3.5 MΩ. The pipette solution contained (in mM): 135 potassium methane sulphonate, 10 KCl, 6 NaCl, 1 Mg2ATP, 2 Na3ATP, 10 HEPES and 0.5 EGTA, adjusted to pH 7.2 with 1 M KOH solution. Chemicals for pipette and bathing solutions were supplied by Sigma-Aldrich (Vienna, Austria). Electrophysiological measurements were carried out with an Axopatch 200B patch clamp amplifier (Axon Instruments, CA, USA). Capacity transients were cancelled, and series resistance was compensated. Whole cell currents were filtered at 5 kHz and sampled at 10 kHz. Data acquisition and storage were processed directly to a PC equipped with pCLAMP 10.0 software (Axon Instruments, CA, USA).
After GΩ-seal formation, the equilibration period of 5 min was followed by recordings at holding potentials (Eh) between −100 and +100 mV in 20 mV increments for 1 min at each Eh. Amiloride was added in control experiments in order to identify if the registered current is the amiloride-sensitive Na+ current. In the series of experiments with the test compounds, after a control period of 5 min aliquots of a stock solution, which was prepared with distilled water, were cumulatively added into the bathing solution, resulting in concentrations ranging from 1.75 to 30 nM TNF-α, and from 3.5 to 480 nM TIP peptide, respectively. The wash-in phase lasted about 1 min. After steady-state had been reached, the same clamp protocol was applied during control recordings and for each concentration of the peptide. At the end of the experiments amiloride was added in order to prove whether the peptide-induced increase in current is due to the amiloride-sensitive Na+ current, so that the specificity of these compounds on the amiloride-sensitive Na+ current could be proven. Concentration-response curves and EC50-values were fitted and estimated for currents recorded at Eh of −100 mV with SigmaPlot 9.0. Differences in EC50 were calculated for statistical significance (P<0.05) with the Student’s t-test.
For evaluation of ion selectivity, ENaC was blocked by 10 to 100 μM amiloride hydrochloride hydrate before the addition of peptide. Subsequent addition of 10 mM tetraethylammonium chloride (TEA) indicated whether any observed increases in the current were due to potassium current. These experiments were also carried out at Eh = −100 mV.
Acknowledgments
P. H. received financial support by the Austrian Research Promotion Agency (FFG).
Abbreviations
amino acids and designation of peptides according to the IUPAC-IUB Joint Commission on Biochemical Nomenclature Recommendations 1983 (IUPAC-IUB, 1984).
- ALC
alveolar liquid clearance capacity
- ALI
acute lung injury
- ARDS
acute respiratory distress syndrome
- ENaC
epithelial sodium channel
- TIP
lectin-like domain of TNF-α
References
- 1.Matthay MA, Folkesson HG, Clerici C. Lung epithelial fluid transport and the resolution of pulmonary edema. Physiol Rev. 2002;82:569–600. doi: 10.1152/physrev.00003.2002. [DOI] [PubMed] [Google Scholar]
- 2.Eaton DC, Helms MN, Koval M, Bao HF, Jain L. The contribution of epithelial sodium channels to alveolar function in health and disease. Annu Rev Physiol. 2009;71:403–423. doi: 10.1146/annurev.physiol.010908.163250. [DOI] [PubMed] [Google Scholar]
- 3.Lucas R, Verin AD, Black SM, Catravas JD. Regulators of endothelial and epithelial barrier integrity and function in acute lung injury. Biochem Pharmacol. 2009;77:1763–1772. doi: 10.1016/j.bcp.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Verghese GM, Ware LB, Matthay BA, Matthay MA. Alveolar epithelial fluid transport and the resolution of clinically severe hydrostatic pulmonary edema. J Appl Physiol. 1999;87:1301–1312. doi: 10.1152/jappl.1999.87.4.1301. [DOI] [PubMed] [Google Scholar]
- 5.Ware LB, Matthay MA. Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. American journal of respiratory and critical care medicine. 2001;163:1376–1383. doi: 10.1164/ajrccm.163.6.2004035. [DOI] [PubMed] [Google Scholar]
- 6.Sartori C, Matthay MA. Alveolar epithelial fluid transport in acute lung injury: new insights. Eur Respir J. 2002;20:1299–1313. doi: 10.1183/09031936.02.00401602. [DOI] [PubMed] [Google Scholar]
- 7.Johnson ER, Matthay MA. Acute Lung Injury: Epidemiology, Pathogenesis, and Treatment. J Aerosol Med Pulm Drug Deliv. doi: 10.1089/jamp.2009.0775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berthiaume Y, Folkesson HG, Matthay MA. Lung edema clearance: 20 years of progress: invited review: alveolar edema fluid clearance in the injured lung. J Appl Physiol. 2002;93:2207–2213. doi: 10.1152/japplphysiol.01201.2001. [DOI] [PubMed] [Google Scholar]
- 9.Berthiaume Y, Matthay MA. Alveolar edema fluid clearance and acute lung injury. Respir Physiol Neurobiol. 2007;159:350–359. doi: 10.1016/j.resp.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hummler E, Planes C. Importance of ENaC-mediated sodium transport in alveolar fluid clearance using genetically-engineered mice. Cell Physiol Biochem. 25:63–70. doi: 10.1159/000272051. [DOI] [PubMed] [Google Scholar]
- 11.Folkesson HG, Matthay MA. Alveolar epithelial ion and fluid transport: recent progress. Am J Respir Cell Mol Biol. 2006;35:10–19. doi: 10.1165/rcmb.2006-0080SF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guidot DM, Folkesson HG, Jain L, Sznajder JI, Pittet JF, Matthay MA. Integrating acute lung injury and regulation of alveolar fluid clearance. Am J Physiol Lung Cell Mol Physiol. 2006;291:L301–306. doi: 10.1152/ajplung.00153.2006. [DOI] [PubMed] [Google Scholar]
- 13.Rezaiguia S, Garat C, Delclaux C, Meignan M, Fleury J, Legrand P, Matthay MA, Jayr C. Acute bacterial pneumonia in rats increases alveolar epithelial fluid clearance by a tumor necrosis factor-alpha-dependent mechanism. J Clin Invest. 1997;99:325–335. doi: 10.1172/JCI119161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fukuda N, Jayr C, Lazrak A, Wang Y, Lucas R, Matalon S, Matthay MA. Mechanisms of TNF-alpha stimulation of amiloride-sensitive sodium transport across alveolar epithelium. Am J Physiol Lung Cell Mol Physiol. 2001;280:L1258–1265. doi: 10.1152/ajplung.2001.280.6.L1258. [DOI] [PubMed] [Google Scholar]
- 15.Borjesson A, Norlin A, Wang X, Andersson R, Folkesson HG. TNF-alpha stimulates alveolar liquid clearance during intestinal ischemia-reperfusion in rats. Am J Physiol Lung Cell Mol Physiol. 2000;278:L3–12. doi: 10.1152/ajplung.2000.278.1.L3. [DOI] [PubMed] [Google Scholar]
- 16.Tillie-Leblond I, Guery BP, Janin A, Leberre R, Just N, Pittet JF, Tonnel AB, Gosset P. Chronic bronchial allergic inflammation increases alveolar liquid clearance by TNF-alpha -dependent mechanism. Am J Physiol Lung Cell Mol Physiol. 2002;283:L1303–1309. doi: 10.1152/ajplung.00147.2002. [DOI] [PubMed] [Google Scholar]
- 17.Lucas R, Magez S, De Leys R, Fransen L, Scheerlinck JP, Rampelberg M, Sablon E, De Baetselier P. Mapping the lectin-like activity of tumor necrosis factor. Science. 1994;263:814–817. doi: 10.1126/science.8303299. [DOI] [PubMed] [Google Scholar]
- 18.Hribar M, Bloc A, van der Goot FG, Fransen L, De Baetselier P, Grau GE, Bluethmann H, Matthay MA, Dunant Y, Pugin J, Lucas R. The lectin-like domain of tumor necrosis factor-alpha increases membrane conductance in microvascular endothelial cells and peritoneal macrophages. Eur J Immunol. 1999;29:3105–3111. doi: 10.1002/(SICI)1521-4141(199910)29:10<3105::AID-IMMU3105>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 19.Hamacher J, Stammberger U, Roux J, Kumar S, Yang G, Xiong C, Schmid RA, Fakin RM, Chakraborty T, Hossain HM, Pittet JF, Wendel A, Black SM, Lucas R. The lectin-like domain of tumor necrosis factor improves lung function after rat lung transplantation--potential role for a reduction in reactive oxygen species generation. Crit Care Med. 38:871–878. doi: 10.1097/CCM.0b013e3181cdf725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ridge FP, Duszyk M, French AS. A large conductance, Ca2+-activated K+ channel in a human lung epithelial cell line (A549) Biochim Biophys Acta. 1997;1327:249–258. doi: 10.1016/s0005-2736(97)00073-4. [DOI] [PubMed] [Google Scholar]
- 21.Lazrak A, Samanta A, Venetsanou K, Barbry P, Matalon S. Modification of biophysical properties of lung epithelial Na(+) channels by dexamethasone. Am J Physiol Cell Physiol. 2000;279:C762–770. doi: 10.1152/ajpcell.2000.279.3.C762. [DOI] [PubMed] [Google Scholar]
- 22.Leroy C, Dagenais A, Berthiaume Y, Brochiero E. Molecular identity and function in transepithelial transport of K(ATP) channels in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2004;286:L1027–1037. doi: 10.1152/ajplung.00249.2003. [DOI] [PubMed] [Google Scholar]
- 23.Sakuma T, Takahashi K, Ohya N, Nakada T, Matthay MA. Effects of ATP-sensitive potassium channel opener on potassium transport and alveolar fluid clearance in the resected human lung. Pharmacol Toxicol. 1998;83:16–22. doi: 10.1111/j.1600-0773.1998.tb01436.x. [DOI] [PubMed] [Google Scholar]
- 24.Lazrak A, Samanta A, Matalon S. Biophysical properties and molecular characterization of amiloride-sensitive sodium channels in A549 cells. Am J Physiol Lung Cell Mol Physiol. 2000;278:L848–857. doi: 10.1152/ajplung.2000.278.4.L848. [DOI] [PubMed] [Google Scholar]
- 25.Karle C, Gehrig T, Wodopia R, Hoschele S, Kreye VA, Katus HA, Bartsch P, Mairbaurl H. Hypoxia-induced inhibition of whole cell membrane currents and ion transport of A549 cells. Am J Physiol Lung Cell Mol Physiol. 2004;286:L1154–1160. doi: 10.1152/ajplung.00403.2002. [DOI] [PubMed] [Google Scholar]
- 26.Xu W, Leung S, Wright J, Guggino SE. Expression of cyclic nucleotide-gated cation channels in airway epithelial cells. J Membr Biol. 1999;171:117–126. doi: 10.1007/s002329900564. [DOI] [PubMed] [Google Scholar]
- 27.Berthiaume Y. Tumor necrosis factor and lung edema clearance: the tip of the iceberg? Am J Respir Crit Care Med. 2003;168:1022–1023. doi: 10.1164/rccm.2308003. [DOI] [PubMed] [Google Scholar]
- 28.Elia N, Tapponnier M, Matthay MA, Hamacher J, Pache JC, Brundler MA, Totsch M, De Baetselier P, Fransen L, Fukuda N, Morel DR, Lucas R. Functional identification of the alveolar edema reabsorption activity of murine tumor necrosis factor-alpha. Am J Respir Crit Care Med. 2003;168:1043–1050. doi: 10.1164/rccm.200206-618OC. [DOI] [PubMed] [Google Scholar]
- 29.Braun C, Hamacher J, Morel DR, Wendel A, Lucas R. Dichotomal role of TNF in experimental pulmonary edema reabsorption. J Immunol. 2005;175:3402–3408. doi: 10.4049/jimmunol.175.5.3402. [DOI] [PubMed] [Google Scholar]
- 30.Vadasz I, Schermuly RT, Ghofrani HA, Rummel S, Wehner S, Muhldorfer I, Schafer KP, Seeger W, Morty RE, Grimminger F, Weissmann N. The lectin-like domain of tumor necrosis factor-alpha improves alveolar fluid balance in injured isolated rabbit lungs. Crit Care Med. 2008;36:1543–1550. doi: 10.1097/CCM.0b013e31816f485e. [DOI] [PubMed] [Google Scholar]
- 31.Dagenais A, Frechette R, Yamagata Y, Yamagata T, Carmel JF, Clermont ME, Brochiero E, Masse C, Berthiaume Y. Downregulation of ENaC activity and expression by TNF-alpha in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2004;286:L301–311. doi: 10.1152/ajplung.00326.2002. [DOI] [PubMed] [Google Scholar]
- 32.Bao HF, Zhang ZR, Liang YY, Ma JJ, Eaton DC, Ma HP. Ceramide mediates inhibition of the renal epithelial sodium channel by tumor necrosis factor-alpha through protein kinase C. Am J Physiol Renal Physiol. 2007;293:F1178–1186. doi: 10.1152/ajprenal.00153.2007. [DOI] [PubMed] [Google Scholar]
- 33.Marquardt A, Bernevic B, Przybylski M. Identification, affinity characterisation and biological interactions of lectin-like peptide-carbohydrate complexes derived from human TNF-alpha using high-resolution mass spectrometry. J Pept Sci. 2007;13:803–810. doi: 10.1002/psc.902. [DOI] [PubMed] [Google Scholar]
- 34.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 35.van Gunsteren WF, Billeter SR, Eising AA, Hünenberger PH, Krüger P, Mark AE, Scott WRP, Tironi IG. Biomolecular Simulation: The GROMOS96 Manual and User Guide 1996. Vdf Hochschulverlag AG an der ETH Zürich, Zürich; Switzerland: pp. 1–1042. [Google Scholar]