

Abstract
Both erythropoietin (EPO) and haem oxygenase-1 (HO-1), an anti-oxidative stress protein, have proven protective roles in experimental autoimmune encephalomyelitis (EAE), a reliable animal model of multiple sclerosis. In this study, EPO delivered intraperitoneally could reduce disease severity in myelin oligodendrocyte glycoprotein (MOG)–EAE mice. To assess the effect of EPO on endogenous HO-1 in EAE, we investigated expression of HO-1 mRNA by real-time polymerase chain reaction (RT–PCR), protein expression centrally and peripherally by Western blot and immunohistochemistry and mean fluorescence intensity of splenic HO-1 by flow cytometry. A significantly higher expression of HO-1 in both the central nervous system (CNS) and spleen was shown in EPO-treated MOG–EAE mice than in controls. We further examined the immunomodulatory effect of EPO in EAE, and via RT–PCR demonstrated significantly lower expression of interferon-γ, interleukin (IL)-23, IL-6 and IL-17 mRNA, and significantly higher expression of IL-4 and IL-10 mRNA in CNS of EPO-treated MOG–EAE mice than in controls. Using flow cytometry, we also observed a significantly decreased ratio of both T helper type 1 (Th1) and Th17 lymphocyte subsets isolated from CNS and a significantly increased ratio of splenic regulatory CD4 T cells in EPO-treated MOG–EAE mice. In addition, we demonstrated that MOG-specific T cell proliferation was lower in the EPO-treated group than in controls and showed amelioration of EAE by adoptive transfer of splenocytes from EPO-treated MOG–EAE mice. Together, our data show that in EAE, EPO induction of endogenous HO-1 and modulation of adaptive immunity both centrally and peripherally may involve the repression of inflammatory responses.
Keywords: erythropoietin, experimental autoimmune encephalomyelitis, haem oxygenase-1, regulatory T cells, Th17
Introduction
Multiple sclerosis (MS) is a chronic disease of the central nervous system (CNS) affecting primarily young adults, of which the pathogenesis is characterized by inflammation, demyelination and axonal injury [1]. Experimental autoimmune encephalomyelitis (EAE) is the most widely recognized animal model of MS. The immunopathogenic mechanism of EAE is viewed widely as a T cell-mediated inflammatory disease of the CNS in which activated T cells recruit invading macrophages, resident astrocytes and microglia, leading to release of inflammatory mediators and toxic molecules including glutamate, nitric oxide (NO) and/or reactive oxygen species. These contribute to axonal damage, which is followed by complement activation or antibody-mediated phagocytosis of axons [2,3].
The potential neuroprotective effect of erythropoietin (EPO) against neuronal death induced by ischaemia and hypoxia has been studied extensively both in vitro and in vivo[4]. Bernaudin et al. first demonstrated a temporally and spatially regulated cellular expression of EPO and EPO receptor (Epo-R) with the progression of a cerebral infarct and showed a basal level of expression of EPO in ischaemic mice in a range of neural cells. In addition, they demonstrated a significant reduction in the degree of infarct in mice treated with intracerebral injection of EPO [5]. Sinor et al. showed that EPO reduced neuronal cell death from hypoxia and attenuated the neurotoxic effect of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) in cultured cortical neurones [6]. Fletcher et al. showed that intranasal administration of EPO plus insulin-like growth factor-I (IGF-I) reduced stroke volumes within 24 h and improved neurological function in a murine stroke model [7]. Nadam et al. provided evidence of the neuroprotective potential of EPO induced in astroglia displaying delayed neuronal death in the rat hippocampus after pilocarpine-induced status epilepticus [8]. Moreover, Zhang et al. showed the neuroprotective effect of EPO in murine EAE [9], and recently Ehrenreich et al. introduced EPO to treat patients with chronic MS and showed its effectiveness in a clinical trial [10].
Haem oxygenase-1 (HO-1) is a heat shock protein induced by oxidative stress that can be amplified significantly in lesions in animals with EAE [11]. HO-1 expression in active MS lesions has also been identified [12]. This increased endogenous HO-1 may have the defensive potential to minimize tissue damage in EAE [13]. Furthermore, Chora et al. showed that Hmox1–/–C57BL/6 HO-1 gene knock-out mice displayed more exacerbated EAE than Hmox1+/+ mice and that induction of HO-1 by cobalt protoporphyrin IX (CoPPIX) administration suppressed EAE progression, but that this protection of CoPPIX was abrogated in Hmox1–/– mice with EAE [14]. Thus, targeting induction of HO-1 over-expression could be considered a novel therapeutic strategy in the treatment of MS.
Park et al. have confirmed that T helper type 17 (Th17) cells [interleukin (IL)-17-producing CD4+ T cells] have a fundamental role in the immunopathogenesis of EAE [15]. Tzima et al. demonstrated that myeloid HO-1 deficiency exacerbated EAE in mice and enhanced infiltration of activated macrophages and Th17 cells to the CNS, thereby establishing HO-1 as a critical early mediator of the innate immune response in EAE [16]. Yuan et al. designed an elegant study that showed that the immunomodulation of EPO in EAE was mediated by promoting a large expansion in CD4+forkhead box P3 (FoxP3+) regulatory T cells (Tregs) to inhibit Th17 polarization and to abrogate proliferation of the antigen-presenting dendritic cells (DCs) either in the peripheral circulation or in the inflamed spinal cord [17].
Katavetin et al. reported that EPO induces HO-1 expression in cultured renal endothelial cells and that EPO-induced HO-1 expression is probably responsible for cytoprotection against oxidative stress in experimental renal injury in vivo[18]. Recently, Burgers et al. demonstrated that up-regulation of HO-1 expression contributes to EPO-mediated cytoprotection during myocardial ischaemia–reperfusion injuries [19]. However, the effect of EPO on endogenous HO-1 has not yet been studied in EAE. Here, we demonstrate that EPO can enhance endogenous HO-1 both peripherally and centrally in EAE and that it up-regulates splenic Tregs and Th2 cells and down-regulates Th1 and Th17 cells in EPO-treated myelin oligodendrocyte glycoprotein (MOG)–EAE mice in situ.
Materials and methods
Mice
C57BL/6 mice (6–8 weeks old) were purchased from the National Laboratory Animal Center, Taiwan. All animal protocols were approved by the Institutional Animal Care and Use Committees (IACUC) in Taiwan.
EAE induction and treatment protocol
MOG35–55 peptide (M-E-V-G-W-Y-R-S-P-F-S-R-O-V-H-L-Y-R-N-G-K) corresponding to the mouse sequence was synthesized by QCB Inc., a division of BioSource International (Hopkinton, MA, USA), and purified by high-performance liquid chromatography (HPLC). Peptide purity was greater than 95% after HPLC. C57BL/6 mice were immunized on day 0 with 100 µg per mouse of MOG in 100 µl of an emulsion in complete Freund's adjuvant (CFA) containing 400 µg of Mycobacterium tuberculosis H37Ra (Difco, Detroit, MI, USA). Each mouse also received 500 ng intraperitoneally of pertussis toxin (PTX) (List Biological Laboratories, Campbell, CA, USA) on days 0 and 2 after immunization.
EPO (Eprex®) was kindly supplied by Janssen Cilag (Taipei, Taiwan) in syringes with 2000 U/ml/vial and was stored at 4°C. Mice were administered EPO [100 U/100 µl/mouse/injection diluted in sterilized phosphate-buffered saline (PBS), equivalent to 5000 U/kg/mouse/injection] or an equal volume of PBS as control on days 1, 3, 5 and 7 post-immunization.
Clinical EAE score
The clinical EAE score was assessed using the following scale: 0 = no symptoms, 0·5 = distal weak or spastic tail, 1 = completely limp tail, 1·5 = limp tail and hindlimb weakness (feet slip through cage grill), 2·0 = unilateral partial hindlimb paralysis, 2·5 = bilateral partial hindlimb paralysis, 3·0 = complete bilateral hindlimb paralysis, 3·5 = complete hindlimb and unilateral partial forelimb paralysis, 4·0 = moribund and 5 = dead [20].
Real-time polymerase chain reaction (PCR)
The expression of mRNA for interferon (IFN)-γ, IL-6, IL-12p35, IL-17, IL-23p19, IL-27EBI3, transforming growth factor (TGF)-β, IL-4 and IL-10 in spinal cord or brain was analysed with specific primers from Applied Biosystems (Life Technologies, CA, USA). The expression was normalized to that of hypoxanthine–guanine phosphoribosyl transferase (HPRT).
Immunohistochemistry
For immunohistochemical (IHC) staining, mice were perfused with PBS, and organs were harvested, formaldehyde-fixed and paraffin-embedded. The endogenous peroxidase activity was quenched, and the sections were blocked with 1% (w/v) bovine serum albumin (BSA) in PBS for 1 h. The sections were then incubated with a 1:300 dilution of rabbit polyclonal anti-HO-1 antibody (Stressgen Biotechnologies, Victoria, BC, Canada) in PBS, followed by horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (Novus Biologicals, Littleton, CO, USA). The reaction products were visualized with a colour solution consisting of aminoethylcarbazole (AEC; Dako, Carpinteria, CA, USA), and the slides were counterstained with haematoxylin [21].
Isolation of lymphocytes from the spleens, brains and spinal cords
Mice were anaesthetized by intramuscular injection of 100 mg/kg ketamine (Imalgene 1000, Merial Laboratoire de Toulouse, France) and 20 mg/kg xylazine (Rompum, Bayer AG, Germany), and perfused transcardially through the left ventricle with ice-cold PBS. Spleens were harvested, placed in RPMI-1640 medium (Invitrogen Life Technologies, Gaithersburg, MD, USA) and minced, and erythrocytes were depleted with Tris-buffered ammonium chloride. The remaining cell pellets, representing the total splenic mononuclear cell population, were resuspended in RPMI-1640 medium. The non-adherent lymphocyte population was collected, washed and resuspended in PBS containing 1% (v/v) fetal bovine serum (FBS) (all supplements from Invitrogen Life Technologies).
Brains and spinal cords were dissociated by glass homogenization through a fine mesh screen using a syringe plunger and collected into 10 ml of Hanks' balanced salts solution (HBSS) containing 0·05% collagenase D (Boehringer Mannheim Biochemicals, Indianapolis, IN, USA), 0·1 µg/ml of the trypsin inhibitor N-α-tosyl-L-lysyl chloromethyl ketone (TLCK) (Sigma Chemical Co., St Louis, MO, USA), 10 µg/ml DNase I (Sigma) and 10 mM Hepes buffer pH 7·4. The resulting tissue slurry was mixed at room temperature for 60 min and allowed to settle at unit gravity for 30 min to deplete any undigested debris. The supernatant was collected, pelleted at 200 g for 5 min and resuspended in 10 ml Ca2+/Mg2+-free HBSS for each brain. Five millilitres of this suspension were layered carefully onto 10 ml of a modified density separation medium (a mixture of 75% RPMI-1640 containing 10% FBS, 10 mM HEPES and 50 µg gentamicin with 25% Ficoll-Paque) in a 50-ml centrifuge tube and centrifuged at 500 g for 30 min before the overlying media and tissue interface were removed. The entire 10 ml of gradient medium was diluted 10-fold with HBSS and centrifuged at 300 g for 10 min [20].
Sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS–PAGE) and Western blot
Proteins were extracted from brains and spinal cords, and separated by SDS–PAGE (12%). The gel was electroblotted onto a nitrocellulose membrane, incubated for 1 h in 20 ml of the blocking buffer [Tris-buffered saline (TBS), 5% skimmed milk], washed three times in TBS with 0·1% Tween-20 and incubated overnight at 4°C with rabbit anti-HO-1 antibody (Stressgen). The blots were washed three times and incubated for 1 h with HRP-conjugated goat anti-rabbit antibody (Novus) at room temperature. The membranes were washed three times, and the membrane-bound antibody was detected with Western Lightning Chemiluminescent Reagent Plus (PerkinElmer Life Sciences, Boston, MA, USA) and visualized on X-ray film. The molecular weight of HO-1 is 32 kDa and of β-actin 43 kDa.
Flow cytometry analysis
Prepared cells (1 × 106 cells in 0·1 ml of PBS) were incubated on ice and stained with the following marker-specific antibodies (0·5 µg of antibody/1 × 106 cells): fluorescein isothiocyanate (FITC)-conjugated anti-HO-1 antibody (eBioscience, San Diego, CA, USA) and allophycocyanin (APC)-conjugated anti-mouse CD4 antibody (eBioscience, San Diego, CA, USA). Either lymphocytes isolated from central nervous system (CNS) or erythrocyte-depleted splenocytes were stained with anti-mouse CD4 and stimulated with 20 ng/ml of phorbol myristate acetate (PMA) plus ionomycin for 4 h, the last 2 h in the presence of monensin [22]. After fixation in 4% formaldehyde for 20 min, cells were stained with fluorescein isothiocyanate (FITC)-conjugated monoclonal antibody (mAb) to IFN-γ, FITC-conjugated mAb to FoxP3, phycoerythrin (PE)-conjugated mAb to IL-17, PE-conjugated mAb to IL-4 and PE-conjugated mAb to CD25 or isotype control mAbs, according to the manufacturer's instructions (BD Biosciences, San Diego, CA, USA) in the presence of 0·5% saponin for permeabilization (eBioscience). Flow cytometric analysis was performed with a fluorescence activated cell sorter (FACS)Calibur (Becton Dickinson), and data were analysed with CellQuest software. Results were analysed using WinMDI software.
Antigen-specific proliferation
Splenocyte cell suspensions were isolated on day 21 from MOG35–55-immunized mice treated with or without EPO. Pooled splenocytes from six individual mice from the same group were plated in triplicate in 96-well round-bottomed plates at 2 × 105 cells/well in 200 µl of complete RPMI-1640 medium supplemented with 2 mM l-glutamine, 25 mM HEPES, 100 U/ml penicillin, 100 µg/ml streptomycin, 5·5 × 10–5 M 2-mercaptoethanol and 5% FBS (all supplements from Invitrogen Life Technologies) containing 0–10 µg/ml MOG35–55 (Enzo Life Sciences, PA, USA) and cultured at 37°C in 5% CO2. On day 3, 1 Ci/well [3H]-thymidine (Amersham Pharmacia Biotech, Piscataway, NJ, USA) was added, and the cells were cultured for an additional 6 h, after which cells were harvested and proliferation was assessed by [3H]-thymidine incorporation detected with a TopCount scintillation counter (Packard, PerkinElmer, Boston, MA, USA).
Adoptive transfer
Prepared splenocytes were isolated from donor mice treated or not treated with EPO 21 d after MOG35–55 immunization and 2 × 107 cells injected intravenously into recipients. The recipient mice were immunized with 50 µg of MOG35–55 in CFA and given 500 ng of PTX intraperitoneally on days 0 and 2.
Statistical analysis
Disease scores were analysed using repeated-measures analysis of variance (anova). A significant difference in disease progression between two groups corresponds to P < 0·05 for a two-sided significance test. Where significant differences were found, the Tukey–Kramer multiple comparisons test was used to identify differences between individual groups. The significance of the results from real-time PCR was determined using the Newman–Keul test.
Results
EPO ameliorates EAE and enhances endogenous HO-1 in situ
We first treated mice with intraperitoneal (i.p.) injection of EPO four times on days 1, 3, 5 and 7, respectively, after MOG35–55/CFA immunization to induce EAE. EPO was delivered in a dosage of 100 U/100 µl/mouse/injection (equivalent to 5000 U/kg/mouse/injection), and control mice received an i.p. injection of the same volume of PBS at the same times. We observed a significantly lower clinical score in EPO-treated MOG–EAE mice compared with control mice (Fig. 1a). In addition, we evaluated the incidence of disease and time of onset of EAE and observed a delayed onset and a lower incidence of EAE in EPO-treated MOG–EAE mice compared with controls (Fig. 1b). Furthermore, we extracted mRNA for quantitative comparison and protein for Western blotting from brains and spinal cords. We demonstrated a significantly elevated expression of endogenous HO-1 mRNA in brain and a trend to increased expression in spinal cord from EPO-treated MOG–EAE mice compared with controls. Similarly, we detected a significantly higher expression of HO-1 mRNA in lymphocytes isolated from CNS of EPO-treated MOG–EAE mice (Fig. 1c). Interestingly, we observed a higher level of HO-1 protein in spinal cords and only a trend to increased HO-1 protein in brains from EPO-treated MOG–EAE mice compared with controls (Fig. 1d). In addition, we observed enhanced immunohistochemical staining for HO-1 in the overlying inflammatory layers and in parenchyma of spinal cord from EPO-treated MOG–EAE mice compared with controls (Fig. 1e). This clinically protective effect of EPO on EAE was consistent with a previous report [9], where we first addressed the augmentation of endogenous HO-1 in situ in EPO-treated MOG–EAE mice.
Fig. 1.
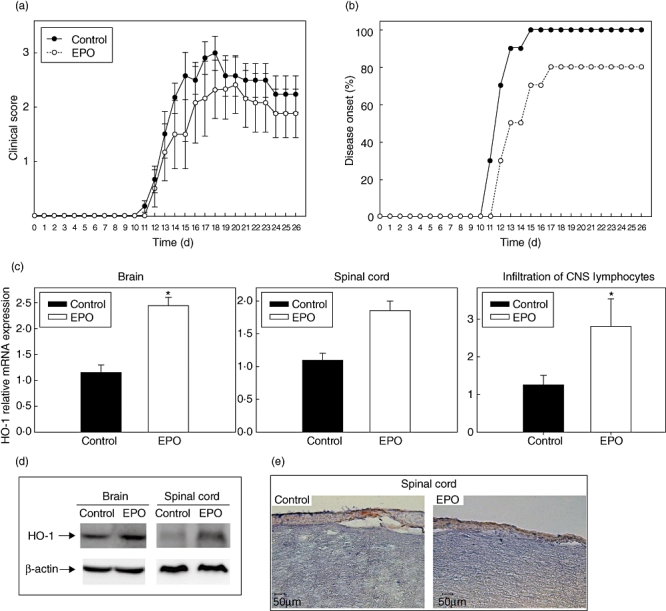
Erythropoietin (EPO) lessens experimental autoimmune encephalomyelitis (EAE) and enhances haem oxygenase-1 (HO-1) expression in situ. (a) Clinical score and (b) time to onset of EAE in C57BL/6 mice treated intraperitoneally either with phosphate-buffered saline (PBS) (100 µl/mouse/injection) or EPO (100 U/100 µl/mouse/injection diluted in sterilized PBS, equivalent to 5000 U/kg/mouse/injection) on days 1, 3, 5 and 7 after subcutaneous (s.c.) immunization with myelin oligodendrocyte glycoprotein (MOG)35–55/ complete Freund's adjuvant (CFA) on day 0 and intraperitoneal (i.p.) pertussis toxin (PTX) on day 0 and 2. Each group contained 10 mice. Data represent means ± standard error of the mean. (c) Expression of HO-1 mRNA in the brain, spinal cord or lymphocytes isolated from the central nervous system of EPO-treated and PBS-treated (as control) MOG–EAE mice determined by real time-polymerase chain reaction. Data in plots are expressed as mean ± standard error from three independent experiments. Significance P < 0·05. (d) Western blot of HO-1 in brain and spinal cord from EPO-treated and control mice on day 14 after MOG injection. (e) Immunohistochemical staining for HO-1 of spinal cord from EPO-treated mice (right) or controls (left) on day 14 after MOG injection. Images are at 200× magnification, and the length of the bar represents 50 µm.
EPO inhibits production of inflammatory cytokines in situ
The neuroprotective effects of EPO in EAE may not only be mediated by the induction of endogenous HO-1 expression, and we hypothesized that immunomodulation could play an important role in this protective effect of EPO in EAE. Therefore we tested the extracted mRNA of brains and spinal cords from mice on day 14 after MOG induction of EAE by quantitative PCR for the presence of mRNA for cytokines including IFN-γ, IL-6, IL-12p35, IL-17, IL-23p19, IL-27EBI3, TGF-β, IL-4 and IL-10 (Fig. 2). Predictably, we found significantly lower expression of IFN-γ, IL-12p35, IL-23p19, IL-6 and IL-17 mRNA in spinal cords from MOG–EAE mice treated with EPO compared with controls. However, the levels of these cytokines were not significantly different in the brains of the two groups. Interestingly, the expression of mRNA for anti-inflammatory cytokines IL-4 and IL-10 was significantly higher in brains from the EPO-treated group than from controls. However, IL-4 mRNA was undetectable in spinal cords from both groups, and only a mild trend towards an increase in IL-10 mRNA in spinal cords was shown in the EPO-treated group compared with controls. We also investigated the expression of IL-27EBI3 and TGF-β mRNA in the CNS in EAE. However, the expression of IL-27EBI3 and TGF-β mRNA did not differ between the EPO-treated group and controls in either brains or spinal cords, implying a minor effect of EPO on IL-27 and TGF-β in EAE.
Fig. 2.
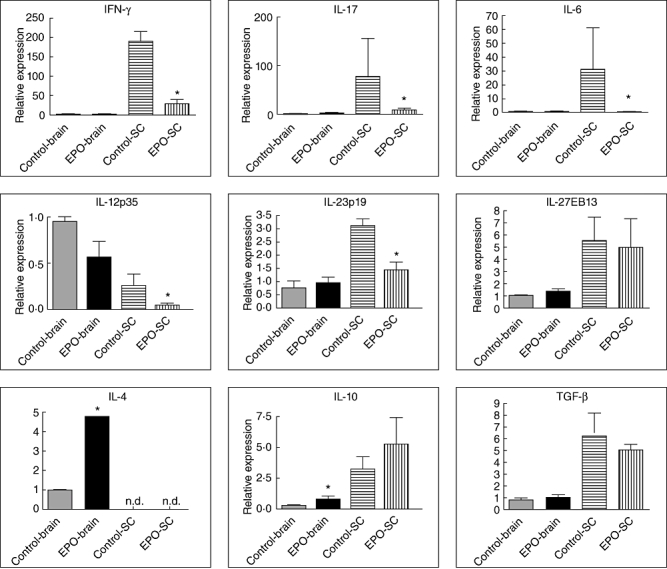
mRNA expression of relevant cytokines in central nervous system (CNS). Cytoplasmic RNA was prepared from the spinal cord and brain of autoimmune encephalomyelitis (EAE) mice treated with erythropoietin (EPO) and from controls, and the levels of cytokine mRNA expression relative to hypoxanthine–guanine phosphoribosyl transferase (HPRT) were determined by quantitative real-time polymerase chain reaction (n = 3).
EPO counteracts encephalitogenic Th1 and Th17 subsets
Encephalitogenic Th17 cells are the essential mediators of the pathogenic effects of EAE [23]. Th1 cells facilitate the invasion of Th17 cells to the CNS during EAE [24], and CNS-derived Th2 cytokine IL-4 is crucial to regulate inflammation in EAE [25]. To investigate the effect of EPO treatment on Th lineages, we isolated mononuclear cells from the CNS of MOG–EAE mice treated with EPO and from controls. We used flow cytometry to detect directly intracellular IFN-γ-, IL-4- and IL-17-producing CD4+ T cells (representing Th1, Th2 and Th17 cells, respectively) on day 21, and their proportion as a percentage of total CD4+ cells was also analysed for comparison (Fig. 3a). Interestingly, we showed a significantly lower proportion of both Th1 and Th17 CD4+ cells in the encephalitogenic CD4+ cells from EPO-treated MOG–EAE mice compared with controls and only a trend to mildly increased encephalitogenic Th2 CD4+ cells in the EPO-treated group (Fig. 3b). This suggests that EPO preserves, at least in part, the capacity to counteract encephalitogenic Th1 and Th17 cells in situ and protects neuronal cells during EAE.
Fig. 3.
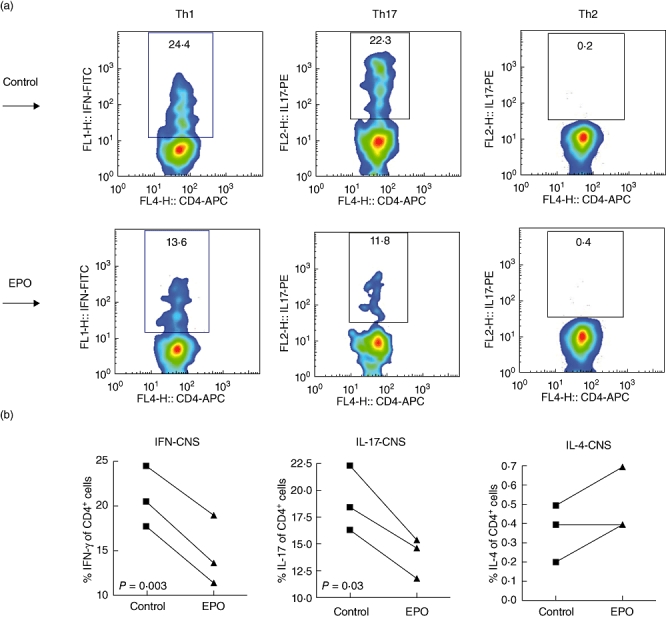
Distribution of T helper (Th) lymphocyte subsets in situ. (a) Flow cytometric analysis of intracellular cytokines in CD4+ T cells isolated from central nervous system (CNS) of mice treated with either erythropoietin (EPO) or phosphate-buffered saline. CD4 T lymphocytes isolated from CNS from either EPO-treated mice or from controls on day 21 at peak disease stage were stained intracellularly with interleukin (IL)-4, interferon (IFN)-γ and IL-17 by flow cytometry. (b) Percentage of IFN-γ-producing CD4+ T cells (Th1), IL-17-producing CD4+ T cells (Th17) and IL-4-producing CD4+ T cells (Th2) are presented. Data are representative of three experiments.
EPO up-regulates splenic HO-1
Recently, in our co-laboratory Wu et al. demonstrated the therapeutic effect of induction of HO-1 in ameliorating experimental murine membranous nephropathy via anti-oxidative, anti-apoptotic and immunomodulatory effects [21]. To study further the potential effects of EPO on peripheral HO-1 expression, we also examined splenic HO-1 expression by IHC and Western blotting analysis, and used flow cytometry for evaluation of the level of HO-1 protein in splenic lymphocytes on day 14 post-EAE induction. We demonstrated a more extended area of HO-1-positive staining in splenocytes from EPO-treated MOG–EAE mice than in those from controls (Fig. 4a), and a marked increase in the splenic HO-1 protein level in EPO-treated mice with EAE was revealed by Western blotting (Fig. 4b). To address whether EPO affected the cellular intensity of HO-1 expression in splenocytes, using flow cytometry we further observed a significantly augmented mean fluorescence intensity (MFI) of HO-1 staining in the same number of splenic lymphocytes from EPO-treated MOG–EAE mice compared with controls (Fig. 4c). Taken together, we demonstrated that EPO not only causes central up-regulation of HO-1 in the CNS but is also a potent inducer of HO-1 in the peripheral immune system.
Fig. 4.
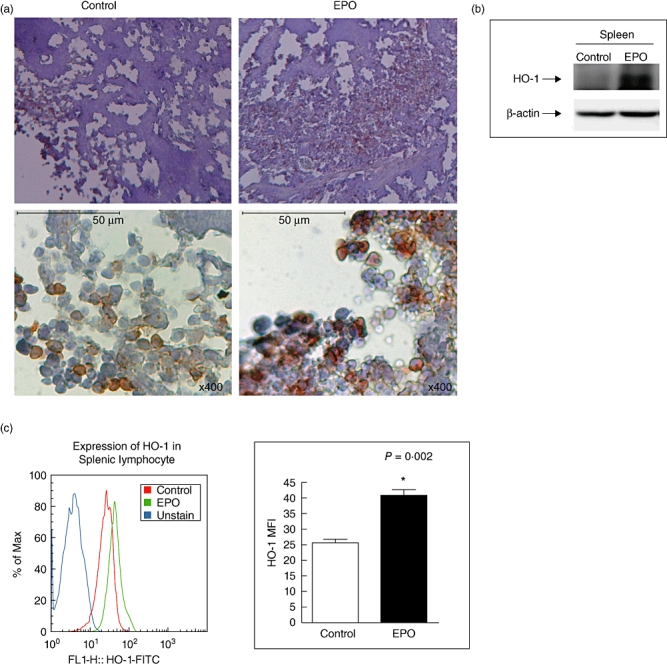
Expression of splenic haem oxygenase-1 (HO-1). (a) Immunohistochemical staining for HO-1 of spleen from erythropoietin (EPO)-treated mice (right) or controls (left) on day 14 after myelin oligodendrocyte glycoprotein (MOG) injection. Images are at either 40× (top) or 400× (bottom) magnification, and the length of the bar represents 50 µm. (b) Splenic lymphocytes either from EPO-treated mice or controls on day 14 of autoimmune encephalomyelitis (EAE) were stained with fluorescein isothiocyanate-conjugated HO-1 for flow cytometry. Mean fluorescence intensity of HO-1 staining was analysed (b, left). Representative of three experiments. *P < 0·05.
EPO enhances splenic Tregs and Th2
Recently, Yuan et al. identified a novel potential of EPO in modulation of peripheral inflammation in the murine MOG–EAE model [17]. We were interested to evaluate the effects of EPO on peripheral immunity during EAE. We thus analysed subsets of splenic immune cells and Th lineages of splenocytes by flow cytometry on day 21 after EAE induction. We demonstrated no significant difference between EPO and control groups in the numbers of CD19+, CD4+, CD8+ or CD11c+ cells (Fig. 5a) or the populations of CD44lowCD62Lhigh (naive), CD44highCD62Llow (memory) and CD69+ (active) cells within both CD4+ and CD8+ T cell populations (Fig. 5b). However, we observed a significantly lower proportion of encephalitogenic Th1 and Th17 cells in the CNS of EPO-treated MOG–EAE mice (Fig. 3), but found only a mild trend towards decreased splenic Th1 and Th17 subsets in EPO-treated MOG–EAE mice (Fig. 5c). Instead, we detected a significantly higher proportion of splenic Th2 cells (Fig. 5c) in the EPO-treated group compared with controls, and a highly significant elevation of splenic CD25+FoxP3+CD4+ Tregs was identified in EPO-treated MOG–EAE mice (Fig. 5d). These data are consistent with our previous report, and they reinforce that the mechanism of EPO-mediated peripheral immunomodulation during EAE may be through inhibition of Th1 and Th17 responses and marked augmentation of the tendency to generate Tregs and Th2 cells [17].
Fig. 5.
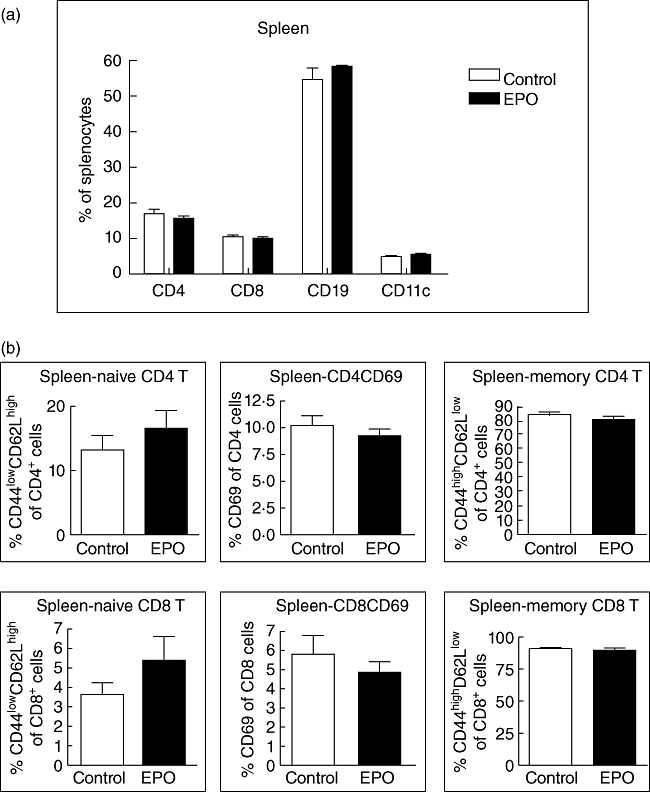
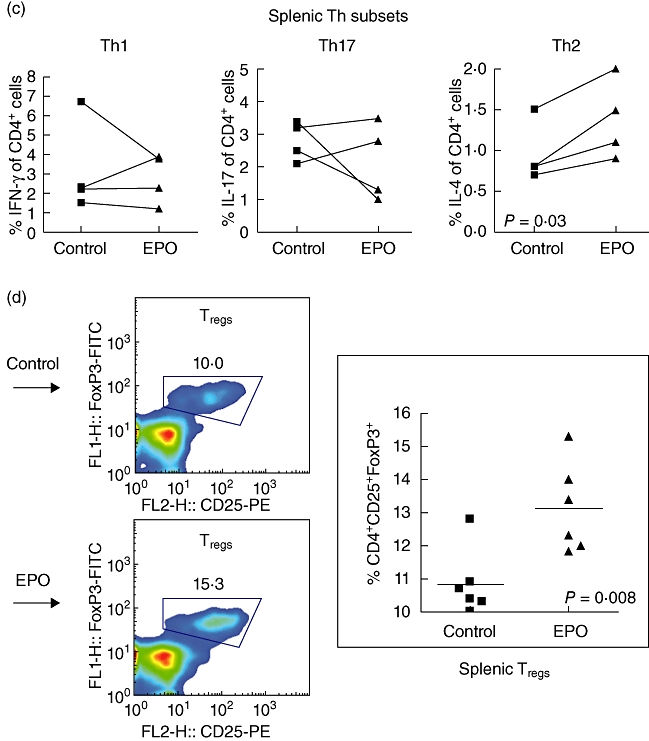
Erythropoietin (EPO) causes no difference in the distributions of splenic immune cells and T cell lineages but enhances splenic T helper type 2 (Th2) and regulatory T cells (Tregs). (a) The subsets of splenocytes from the EPO group or from controls on day 21 were analysed by flow cytometry and no difference was detected in CD4+, CD8+, CD19+ or CD11c+ cells. (b) The lineages of both CD4+ and CD8+ cells including CD44lowCD62Lhigh (naive), CD69 (active) and CD44highCD62Llow (memory) were evaluated by flow cytometry. Data represent three experiments. (c) Splenic lymphocytes either from EPO-treated mice or from controls were stained for Th1, Th2 and Th17 for flow cytometry analysis of their proportion of total CD4 T cells. Data represent four experiments. (d) These splenic lymphocytes were also stained for CD25+forkhead box P3+ CD4+ cells (Tregs) and analysed by flow cytometry. An increased ratio of splenic Tregs was detected on day 21 in EPO-treated myelin oligodendrocyte glycoprotein (MOG)–autoimmune encephalomyelitis (EAE) mice compared with controls. Data of Tregs represent six experiments.
EPO inhibits antigen-specific T cell proliferation and adoptive transfer assay
To test further the suppressive potential of EPO on T cell-mediated effector functions in EAE, we investigated the MOG-specific T cell response. We isolated splenocytes either from EPO-treated MOG–EAE mice or from control mice on day 21 post-EAE. The cultured splenocytes were treated with various concentrations of MOG as specific antigen. We observed a dose-dependent antigen-specific stimulation of T cell proliferation in both groups (Fig. 6a). A significantly lower response at each concentration of MOG was revealed in the EPO-treated group compared with controls. To elucidate further the immune modulation of T cell function in vivo by EPO, we isolated splenocytes from EPO-treated MO-EAE mice or controls on day 21 after MOG35–55/CFA induction, and transferred these cells passively into C57BL/6 recipients. One day after transfer, recipients received a half-dose of MOG35–55 (50 µg/mouse) in CFA. Recipients transferred with splenocytes from EPO-treated MOG–EAE mice had a less severe clinical score and a more rapid recovery from disease than did recipients transferred with splenocytes from controls (Fig. 6b). This indicates that immunomodulatory splenocytes from EPO-treated mice seem to preserve the ability to lessen neuroinflammation in EAE, implying that EPO may be a specific repressor of the generation of immunopathogenic T cells in EAE. Nevertheless, more evidence is needed to clarify this point and to determine the mechanisms of the interaction between EPO and HO-1 during EAE.
Fig. 6.
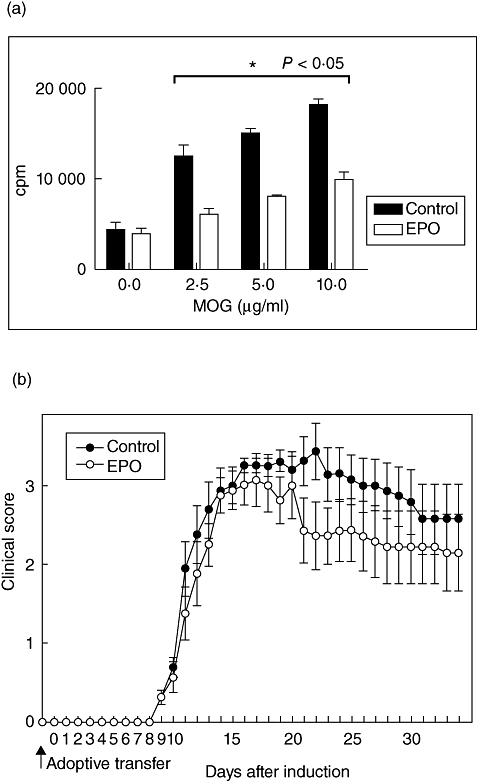
Splenocytes from erythropoietin (EPO)-treated mice inhibit T cell proliferation and ameliorate autoimmune encephalomyelitis (EAE) severity after adoptive transfer. (a) Splenocytes from either EPO-treated mice or from controls on day 14 were restimulated with various concentrations of myelin oligodendrocyte glycoprotein (MOG)35–55 for 72 h. Proliferation was measured by [3H]-thymidine incorporation and is displayed as counts per minute. Vertical bars signify standard error. *P < 0·05. Results represent triplicates. (b) Splenocytes were collected from mice treated with either EPO or controls, and each recipient was transferred adoptively with 2 × 107 splenocytes obtained from the indicated mice on day 21 after MOG35–55/CFA immunization. Each group contained eight mice. Data represent means ± standard error of the mean.
Discussion
The efficient neuroprotective ability of EPO has been demonstrated in stroke models after either intracerebral injection [5] or intranasal delivery [7], but not after intravenous (i.v.), subcutaneous (s.c.) or i.p. administration [7]. This suggests that this variation in the results obtained after different routes of administration of EPO may be a result of dilution that weakens the remote effects of i.v., s.c. or i.p. delivery of EPO. However, the progressive immunopathogenesis of MS is an extremely complicated process to be explored adequately by direct test-tube analysis. EAE has become the most popular animal model of MS in the field of central nervous system inflammation and demyelination to investigate complex pathogenic hypotheses and to test novel therapeutic agents [26]. The protective role of EPO in EAE and MS has been demonstrated in the last decade [10,17,27], and EPO has been shown to cross the blood–brain barrier (BBB) to protect astrocytes and neurones against experimental brain injury in the CNS [28]. Thus, i.p. delivery of EPO, as used in this study, elicits extensive immunomodulatory effects on both peripheral and central manifestations of EAE.
HO-1 possesses both anti-oxidative and anti-inflammatory properties, and is highly inducible by a variety of stimuli, including its substrate haem and oxidative stress. The cytoprotective effect of HO-1 induced by EPO has been reported in experimental models such as hypoxic–ischaemic heart disease and ischaemic renal injury [18,19]. Nevertheless, the role of the effect of EPO on HO-1 in autoimmune diseases such as EAE has not yet been reported. In the present study, we confirmed the neuroprotective effect of i.p. EPO in EAE and addressed for the first time the immunomodulatory effects of EPO in EAE. We demonstrate that these are mediated at least in part through up-regulation of HO-1 both centrally and peripherally as well as by counteracting the generation of Th1 and Th17 cells in situ and peripheral down-regulation of Tregs and Th2.
However, there are some limitations of this work. First, Chora demonstrated that induction of HO-1 (encoded by HMOX1) by cobalt protoporphyrin IX (CoPPIX) administration after onset of EAE reversed the inflammation. They also found augmented CNS demyelination, paralysis and mortality in Hmox1–/– C57BL/6 mice compared with Hmox1+/+ mice after EAE initiation [14]. Our study did not use HO-1 enhancers such as CoPPIX to enforce expression of HO-1 or use an HO-1 knock-out mouse model such as Hmox1–/– C57BL/6 mice to investigate the clinical score and related immunomodulation in EPO-treated mice with EAE to provide direct evidence of the efficacy of EPO on EAE with or without HO-1 induction. Secondly, Savino et al. demonstrated that prolonged administration of EPO for longer than 1 month caused anaemia in about 50% of EPO-treated mice with EAE, but they did not find a significant association between the haematocrit and the EAE score [29]. In our study, we only prescribed EPO in a preventive schedule four times in the first week after MOG induction of EAE. We did not adopt a longer therapeutic schedule of EPO in EAE, and we also did not analyse the haematocrit of EPO-treated mice with EAE to evaluate the potential erythropoietic effect. Thirdly, we used EPO as a preventive treatment but not a therapy for EAE and did not study the HO-1 activity and the related immune responses at different time-points or after therapeutic EPO introduced after the onset of EAE. Nevertheless, we did show this cross-talk between EPO, HO-1 and adaptive immunity in EAE.
HO-1 is expressed at low levels in most tissues under physiological conditions, and HO-1 can be up-regulated during cellular stress for cytoprotection [30]. In the CNS, several cell types including astrocytes, oligodendrocytes and microglia can induce expression of HO-1 in the CNS after damage or in disease conditions. Thus, HO-1 induction in cerebrovascular endothelial cells is suggested to be an important protective mechanism against neurological injury [31]. Colombrita et al. demonstrated that the expression of HO-1 mRNA in aged rats was increased in extracts from the hippocampus and cerebellum but not from the cortex [32]. In contrast, Hirose et al. showed that HO-1 protein levels assessed by IHC were increased with age in the cortex and hippocampus of autopsied human brains from patients without traumatic brain injury or neurodegenerative disease [33]. This discrepancy between studies on HO-1 mRNA expression and HO-1 protein in aged mammalian brains is a dilemma [31]. In addition, the brain is highly vascularized and contains blood vessels with particular cerebrovascular endothelial cells that form tight junctions and contribute to the BBB [31], and that may play a role in HO-1 expression during the disease process of EAE. We observed a trend to increased expression of HO-1 mRNA but a notable increase in HO-1 protein in the spinal cords of EPO-treated MOG–EAE mice compared with controls. In contrast, a significant elevation of HO-1 mRNA but only a slight increment in HO-1 protein compared with controls was observed in the brains of EPO-treated MOG–EAE mice (Fig. 1c and d). However, we detected enhanced IHC staining for HO-1 over the layer of infiltrating inflammatory cells and parenchyma of spinal cords obtained from EPO-treated MOG–EAE mice compared with controls (Fig. 1e). Thus, the complex kinetic inflammatory process and different HO-1 expressing cells in CNS may lead to this discrepancy between mRNA and protein levels of HO-1 in spinal cords and brains from EPO-treated mice.
We were interested in the immunomodulatory mechanisms that could account for this phenomenon. Currently, Th17 cells are known to play a vital role in the immunopathogenic mechanisms of EAE, and Th1 cells possessing the ability to facilitate the entry of Th17 cells to the CNS during EAE were demonstrated recently [24]. Yuan et al. showed with an elegant experimental design that short-term EPO therapy for EAE can down-regulate major histocompatibility complex (MHC) class II expression on peripheral DCs and counteract Th17 responses [17]. Our data also confirmed that EPO counteracts both Th17 and Th1 mediated inflammatory responses in situ during EAE. However, we did not evaluate the effect of EPO on the expression of MHC classes I and II on DCs. We evaluated the encephalitogenic Th subsets isolated from pooled brain and spinal cord, but we did not analyse encephalitogenic Th subsets from separate brain and spinal cord samples. Recently, Murphy et al. demonstrated an apparently comparable trend in the proportion and numbers of Th1 and Th17 cells in brain and spinal cord, respectively, on day 21 after initiation of EAE [34], implying a minor difference in the distribution of encephalitogenic Th subsets between collection and separation of brain and spinal cord obtained on day 21 after initiation of EAE. EPO reduced IL-6 markedly in the spinal cord and decreased inflammation and the clinical score of EAE, suggesting that this immunomodulation may be partly dependent upon reduction of IL-6 [27]. Tron et al. studied the elevated expression of HO-1 in a localized inflammation model after intramuscular injection of inflammatory material and showed that IL-6-specific transcripts were also detected in the injured muscle, and levels were in accordance with serum levels of IL-6. This implies that the induction of HO-1 in local inflammation may modulate anti-inflammatory effects such as the local IL-6 concentration [35]. Our data demonstrated a repression of IL-6 mRNA in CNS from EPO-treated MOG–EAE mice that may be attributed to an over-expression of endogenous HO-1 to counteract the IL-6 in CNS (Fig. 2). Batten et al. observed a de novo effect of IL-27 that suppressed IL-6-mediated T cell proliferation and the development of Th17 cells to restrict autoimmune encephalomyelitis [36]. Similarly, Veldhoen et al. demonstrated that TGF-β plays a key role in the initiation of EAE and verified that disease progression of EAE may require ongoing chronic inflammation and production of IL-23 [37]. However, the expression of IL-27EBI3 and TGF-β mRNA did not differ between the EPO-treated group and controls in either brains or spinal cords, implying a minor effect of EPO on IL-27 and TGF-β in EAE. Thus, we demonstrated that EPO preserves the capacity to inhibit inflammatory cytokines in CNS, suggesting that endogenous HO-1 may play a role in modulation of in situ inflammation and that the suppressive effect of EPO on IL-6 may not be through the effect of IL-27 in situ. Nevertheless, more evidence is needed to confirm this.
Ponomarev et al. showed that increased expression of IL-4 in glial cells was associated with reduced severity of EAE and that IL-4 production in the CNS is essential for controlling autoimmune inflammation by inducing an alternative regulation of microglial cells [25]. Lifshitz et al. showed, by over-expressing human EPO in transgenic mice, that DCs are a direct target of EPO in the initiation of the immune response in vivo, and confirmed a higher expression of Epo-R mRNA in bone marrow-derived DCs [38]. Falcon et al. showed that IL-4 is a critical modulator in EAE by using IL-4–/– mouse strains [39]. Subsequently, Zhang et al. demonstrated that mature DCs selectively down-regulate the antigen-specific response in EAE through suppression of Th1 and induction of Th2 cytokines [40]. Lee and Chau demonstrated that over-expression of HO-1 from macrophages can inhibit the proinflammatory response induced by lipopolysaccharide (LPS) stimulation, and that IL-10 and HO-1 activate a positive feedback circuit to enhance the anti-inflammatory response both in vitro and in vivo[41]. Although we did not approach the role of DCs in the EPO-treated MOG–EAE model, our data showed a higher level of IL-10 and IL-4 mRNA in CNS and a trend to increased Th2 cells in situ in EPO-treated MOG–EAE mice. Accordingly, we suggest that EPO may target resident DCs to enhance endogenous HO-1 and to polarize local Th2 bias through activation of IL-10 and IL-4 production in situ.
Ruuls et al. demonstrated a marked aggravation of the clinical signs of EAE in rats treated with NO synthase inhibitors and suggested an important role of NO as an immune modulator in the disease process during EAE [42]. Kumral et al. demonstrated that EPO exerts neuroprotection through the selective inhibitory effect of EPO on NO overproduction and that the inhibition of NO via EPO has a neuroprotective effect in neonatal hypoxic–ischaemic brain injury [43]. The signal pathway of neuroprotection by EPO in ischaemic and CNS degenerative models has been established to involve Janus-tyrosine kinase 2 (Jak2) signalling subsequent to activation of phosphatidyl inositol 3-kinase (PI3K)/protein kinase B (Akt) phosphorylation and nuclear factor (NF)-κB cascades to repress the CNS damage due to excitotoxins and consequent generation of free radicals, including NO [44]. Interestingly, PI3K/Akt-pathway-related responses to oxidative stress and apoptosis have also been demonstrated at the level of transcriptional regulation of HO-1 [45]. Liu showed that inhibition of the expression of HO-1 markedly exacerbated EAE, suggesting an important protective role of endogenous HO-1 in EAE, and that targeted induction of HO-1 over-expression may exemplify a novel therapy for the treatment of MS [46]. Panahian et al. proved in an HO-1 transgenic model that an over-expression of HO-1 protects cells and tissues of the CNS from ischaemic damage [47]. In this study, another limitation is that we did not examine the effect of EPO on inducible NO synthase (iNOS) and NO production during EPO-treated MOG–EAE or identify the signal pathways involved in the induction of endogenous HO-1, nor did we test the influence on iNOS in CNS in our EPO-treated MOG–EAE model. Thus, the cross-talk of associated oxidative stress factors such as iNOS with the effect of EPO on HO-1 and the complicated signal transduction pathways for the neuroprotective mechanisms of EPO in EAE must be clarified in the near future.
Kohm et al. observed that Tregs inhibit effectively both the proliferation of, and cytokine production by, MOG-specific Th1 cells and that adoptive transfer of Tregs confers significant protection from EAE [48]. Recently, Stephens proved that in vitro-expanded myelin-reactive Tregs can prevent disease relapse when delivered after the onset of clinical EAE [49]. Correspondingly, a high level of splenic Tregs may provide an immunosuppressive mechanism to protect EPO-treated MOG–EAE mice. Zelenay et al. demonstrated by a comparison of HO-1 deficient mice and wild-type controls under physiological conditions that the development, maintenance and function of Tregs are independent of HO-1 activity [50]. Accordingly, the enhancement of HO-1 may not contribute to the over-expression of peripheral Tregs in EPO-treated MOG–EAE mice seen in our study, and the precise mechanism by which EPO induces peripheral Tregs is still puzzling. Nevertheless, we identified a significant suppression of MOG-specific T cell proliferation and lower clinical severity after adoptive transfer of splenocytes from the EPO-treated group, which may be attributed at least in part to the up-regulation of peripheral Tregs and Th2 lineages in EPO-treated MOG–EAE mice.
In conclusion, we confirmed that giving exogenous EPO augments the induction of endogenous HO-1 centrally and peripherally, and represses Th1 and Th17 responses in situ as well as enhancing systemic Th2 and Tregs populations to ameliorate EAE. Collectively, this study suggests that the neuroprotection of EPO in EAE involves various immunomodulatory mechanisms for systemic and local inhibition of inflammation. In summary, we conclude that EPO can up-regulate endogenous HO-1 and modulate inflammatory immune responses centrally and peripherally in the MOG–EAE model, supporting the clinical therapeutic potential of EPO in autoimmune CNS disorders like MS.
Acknowledgments
We would like to thank Shu-Ying Tsai and Shin-Yi Hsieh for their technical assistance. This work was supported by grants from the TSGH-C99-22 to S.-J. Chen, National Science Council, Taiwan, Republic of China (NSC-93-2320-B-016-020 to H.-K. Sytwu) and partly by the C. Y. Foundation for Advancement of Education, Sciences and Medicine.
Disclosure
The authors declare that there are no conflicts of interest.
References
- 1.Hemmer B, Nessler S, Zhou D, Kieseier B, Hartung HP. Immunopathogenesis and immunotherapy of multiple sclerosis. Nat Clin Pract Neurol. 2006;2:201–11. doi: 10.1038/ncpneuro0154. [DOI] [PubMed] [Google Scholar]
- 2.Schreibelt G, van Horssen J, van Rossum S, Dijkstra CD, Drukarch B, de Vries HE. Therapeutic potential and biological role of endogenous antioxidant enzymes in multiple sclerosis pathology. Brain Res Rev. 2007;56:322–30. doi: 10.1016/j.brainresrev.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 3.Hisahara S, Okano H, Miura M. Caspase-mediated oligodendrocyte cell death in the pathogenesis of autoimmune demyelination. Neurosci Res. 2003;46:387–97. doi: 10.1016/s0168-0102(03)00127-5. [DOI] [PubMed] [Google Scholar]
- 4.Rabie T, Marti HH. Brain protection by erythropoietin: a manifold task. Physiology. 2008;23:263–74. doi: 10.1152/physiol.00016.2008. [DOI] [PubMed] [Google Scholar]
- 5.Bernaudin M, Marti HH, Roussel S, et al. A potential role for erythropoietin in focal permanent cerebral ischemia in mice. J Cereb Blood Flow Metab. 1999;19:643–51. doi: 10.1097/00004647-199906000-00007. [DOI] [PubMed] [Google Scholar]
- 6.Sinor AD, Greenberg DA. Erythropoietin protects cultured cortical neurons, but not astroglia, from hypoxia and AMPA toxicity. Neurosci Lett. 2000;290:213–15. doi: 10.1016/s0304-3940(00)01361-6. [DOI] [PubMed] [Google Scholar]
- 7.Fletcher L, Kohli S, Sprague SM, et al. Intranasal delivery of erythropoietin plus insulin-like growth factor-I for acute neuroprotection in stroke. J Neurosurg. 2009;111:164–70. doi: 10.3171/2009.2.JNS081199. [DOI] [PubMed] [Google Scholar]
- 8.Nadam J, Navarro F, Sanchez P, et al. Neuroprotective effects of erythropoietin in the rat hippocampus after pilocarpine-induced status epilepticus. Neurobiol Dis. 2007;25:412–26. doi: 10.1016/j.nbd.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 9.Zhang J, Li Y, Cui Y, et al. Erythropoietin treatment improves neurological functional recovery in EAE mice. Brain Res. 2005;1034:34–9. doi: 10.1016/j.brainres.2004.11.036. [DOI] [PubMed] [Google Scholar]
- 10.Ehrenreich H, Fischer B, Norra C, et al. Exploring recombinant human erythropoietin in chronic progressive multiple sclerosis. Brain. 2007;130:2577–88. doi: 10.1093/brain/awm203. [DOI] [PubMed] [Google Scholar]
- 11.Schluesener HJ, Seid K. Heme oxygenase-1 in lesions of rat experimental autoimmune encephalomyelitis and neuritis. J Neuroimmunol. 2000;110:114–20. doi: 10.1016/s0165-5728(00)00352-0. [DOI] [PubMed] [Google Scholar]
- 12.van Horssen J, Schreibelt G, Drexhage J, et al. Severe oxidative damage in multiple sclerosis lesions coincides with enhanced antioxidant enzyme expression. Free Radic Biol. 2008;45:1729–37. doi: 10.1016/j.freeradbiomed.2008.09.023. [DOI] [PubMed] [Google Scholar]
- 13.Emerson MR, LeVine SM. Heme oxygenase-1 and NADPH cytochrome P450 reductase expression in experimental allergic encephalomyelitis: an expanded view of the stress response. J Neurochem. 2000;75:2555–62. doi: 10.1046/j.1471-4159.2000.0752555.x. [DOI] [PubMed] [Google Scholar]
- 14.Chora AA, Fontoura P, Cunha A, et al. Heme oxygenase-1 and carbon monoxide suppress autoimmune neuroinflammation. J Clin Invest. 2007;117:438–47. doi: 10.1172/JCI28844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park H, Li Z, Yang XO, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–41. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tzima S, Victoratos P, Kranidioti K, Alexiou M, Kollias G. Myeloid heme oxygenase-1 regulates innate immunity and autoimmunity by modulating IFN-beta production. J Exp Med. 2009;206:1167–79. doi: 10.1084/jem.20081582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yuan R, Maeda Y, Li W, Lu W, Cook S, Dowling P. Erythropoietin: a potent inducer of peripheral immuno/inflammatory modulation in autoimmune EAE. PLoS ONE. 2008;3:e1924. doi: 10.1371/journal.pone.0001924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Katavetin P, Inagi R, Miyata T, et al. Erythropoietin induces heme oxygenase-1 expression and attenuates oxidative stress. Biochem Biophys Res Commun. 2007;359:928–34. doi: 10.1016/j.bbrc.2007.05.207. [DOI] [PubMed] [Google Scholar]
- 19.Burger D, Xiang F, Hammoud L, Lu X, Feng Q. Role of heme oxygenase-1 in the cardioprotective effects of erythropoietin during myocardial ischemia and reperfusion. Am J Physiol. 2009;296:H84–93. doi: 10.1152/ajpheart.00372.2008. [DOI] [PubMed] [Google Scholar]
- 20.Chen SJ, Wang YL, Kao JH, et al. Decoy receptor 3 ameliorates experimental autoimmune encephalomyelitis by directly counteracting local inflammation and downregulating Th17 cells. Mol Immunol. 2009;47:567–74. doi: 10.1016/j.molimm.2009.09.017. [DOI] [PubMed] [Google Scholar]
- 21.Wu CC, Lu KC, Chen JS, et al. HO-1 induction ameliorates experimental murine membranous nephropathy: anti-oxidative, anti-apoptotic and immunomodulatory effects. Nephrol Dial Transplant. 2008;23:3082–90. doi: 10.1093/ndt/gfn247. [DOI] [PubMed] [Google Scholar]
- 22.Hsieh SL, Chen NJ, Tarbell K, et al. Transgenic mice expressing surface markers for IFN-γ and IL-4 producing cells. Mol Immunol. 2000;37:281–93. doi: 10.1016/s0161-5890(00)00052-3. [DOI] [PubMed] [Google Scholar]
- 23.Bettelli E, Carrier Y, Gao W, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–8. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 24.O'Connor RA, Prendergast CT, Sabatos CA, et al. Cutting edge: Th1 cells facilitate the entry of Th17 cells to the central nervous system during experimental autoimmune encephalomyelitis. J Immunol. 2008;181:3750–4. doi: 10.4049/jimmunol.181.6.3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ponomarev ED, Maresz K, Tan Y, Dittel BN. CNS-derived interleukin-4 is essential for the regulation of autoimmune inflammation and induces a state of alternative activation in microglial cells. J Neurosci. 2007;27:10714–21. doi: 10.1523/JNEUROSCI.1922-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schreiner B, Heppner FL, Becher B. Modeling multiple sclerosis in laboratory animals. Semin Immunopathol. 2009;31:479–95. doi: 10.1007/s00281-009-0181-4. [DOI] [PubMed] [Google Scholar]
- 27.Agnello D, Bigini P, Villa P, et al. Erythropoietin exerts an anti-inflammatory effect on the CNS in a model of experimental autoimmune encephalomyelitis. Brain Res. 2002;952:128–34. doi: 10.1016/s0006-8993(02)03239-0. [DOI] [PubMed] [Google Scholar]
- 28.Brines ML, Ghezzi P, Keenan S, et al. Erythropoietin crosses the blood–brain barrier to protect against experimental brain injury. Proc Natl Acad Sci USA. 2000;97:10526–31. doi: 10.1073/pnas.97.19.10526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Savino C, Pedotti R, Baggi F, et al. Delayed administration of erythropoietin and its non-erythropoietic derivatives ameliorates chronic murine autoimmune encephalomyelitis. J Neuroimmunol. 2006;172:27–37. doi: 10.1016/j.jneuroim.2005.10.016. [DOI] [PubMed] [Google Scholar]
- 30.Ryter SW, Choi AM. Heme oxygenase-1/carbon monoxide: from metabolism to molecular therapy. Am J Respir Cell Mol Biol. 2009;41:251–60. doi: 10.1165/rcmb.2009-0170TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Syapin PJ. Regulation of haeme oxygenase-1 for treatment of neuroinflammation and brain disorders. Br J Pharmacol. 2008;155:623–40. doi: 10.1038/bjp.2008.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Colombrita C, Calabrese V, Stella AM, Mattei F, Alkon DL, Scapagnini G. Regional rat brain distribution of heme oxygenase-1 and manganese superoxide dismutase mRNA: relevance of redox homeostasis in the aging processes. Exp Biol Med. 2003;228:517–24. doi: 10.1177/15353702-0322805-16. [DOI] [PubMed] [Google Scholar]
- 33.Hirose W, Ikematsu K, Tsuda R. Age-associated increases in heme oxygenase-1 and ferritin immunoreactivity in the autopsied brain. Leg Med. 2003;5(Suppl 1):S360–6. doi: 10.1016/s1344-6223(02)00133-5. [DOI] [PubMed] [Google Scholar]
- 34.Murphy AC, Lalor SJ, Lynch MA, Mills KH. Infiltration of Th1 and Th17 cells and activation of microglia in the CNS during the course of experimental autoimmune encephalomyelitis. Brain Behav Immun. 2010;24:641–51. doi: 10.1016/j.bbi.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 35.Tron K, Novosyadlyy R, Dudas J, Samoylenko A, Kietzmann T, Ramadori G. Upregulation of heme oxygenase-1 gene by turpentine oil-induced localized inflammation: involvement of interleukin-6. Lab Invest. 2005;85:376–87. doi: 10.1038/labinvest.3700228. [DOI] [PubMed] [Google Scholar]
- 36.Batten M, Li J, Yi S, et al. Interleukin 27 limits autoimmune encephalomyelitis by suppressing the development of interleukin 17-producing T cells. Nat Immunol. 2006;7:929–36. doi: 10.1038/ni1375. [DOI] [PubMed] [Google Scholar]
- 37.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–89. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 38.Lifshitz L, Prutchi-Sagiv S, Avneon M, Gassmann M, Mittelman M, Neumann D. Non-erythroid activities of erythropoietin: functional effects on murine dendritic cells. Mol Immunol. 2009;46:713–21. doi: 10.1016/j.molimm.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 39.Falcone M, Rajan AJ, Bloom BR, Brosnan CF. A critical role for IL-4 in regulating disease severity in experimental allergic encephalomyelitis as demonstrated in IL-4-deficient C57BL/6 mice and BALB/c mice. J Immunol. 1998;160:4822–30. [PubMed] [Google Scholar]
- 40.Zhang GX, Kishi M, Xu H, Rostami A. Mature bone marrow-derived dendritic cells polarize Th2 response and suppress experimental autoimmune encephalomyelitis. Mult Scler. 2002;8:463–8. doi: 10.1191/1352458502ms857oa. [DOI] [PubMed] [Google Scholar]
- 41.Lee TS, Chau LY. Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nat Med. 2002;8:240–6. doi: 10.1038/nm0302-240. [DOI] [PubMed] [Google Scholar]
- 42.Ruuls SR, Van Der Linden S, Sontrop K, Huitinga I, Dijkstra CD. Aggravation of experimental allergic encephalomyelitis (EAE) by administration of nitric oxide (NO) synthase inhibitors. Clin Exp Immunol. 1996;103:467–74. doi: 10.1111/j.1365-2249.1996.tb08304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kumral A, Baskin H, Gokmen N, et al. Selective inhibition of nitric oxide in hypoxic–ischemic brain model in newborn rats: is it an explanation for the protective role of erythropoietin? Biol Neonate. 2004;85:51–4. doi: 10.1159/000074958. [DOI] [PubMed] [Google Scholar]
- 44.Digicaylioglu M, Lipton SA. Erythropoietin-mediated neuroprotection involves cross-talk between Jak2 and NF-kappaB signalling cascades. Nature. 2001;412:641–7. doi: 10.1038/35088074. [DOI] [PubMed] [Google Scholar]
- 45.Martin D, Rojo AI, Salinas M, et al. Regulation of heme oxygenase-1 expression through the phosphatidylinositol 3-kinase/Akt pathway and the Nrf2 transcription factor in response to the antioxidant phytochemical carnosol. J Biol Chem. 2004;279:8919–29. doi: 10.1074/jbc.M309660200. [DOI] [PubMed] [Google Scholar]
- 46.Liu Y, Zhu B, Luo L, Li P, Paty DW, Cynader MS. Heme oxygenase-1 plays an important protective role in experimental autoimmune encephalomyelitis. Neuroreport. 2001;12:1841–5. doi: 10.1097/00001756-200107030-00016. [DOI] [PubMed] [Google Scholar]
- 47.Panahian N, Yoshiura M, Maines MD. Overexpression of heme oxygenase-1 is neuroprotective in a model of permanent middle cerebral artery occlusion in transgenic mice. J Neurochem. 1999;72:1187–203. doi: 10.1111/j.1471-4159.1999.721187.x. [DOI] [PubMed] [Google Scholar]
- 48.Kohm AP, Carpentier PA, Anger HA, Miller SD. Cutting edge: CD4+CD25+ regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J Immunol. 2002;169:4712–16. doi: 10.4049/jimmunol.169.9.4712. [DOI] [PubMed] [Google Scholar]
- 49.Stephens LA, Malpass KH, Anderton SM. Curing CNS autoimmune disease with myelin-reactive Foxp3+ Treg. Eur J Immunol. 2009;39:1108–17. doi: 10.1002/eji.200839073. [DOI] [PubMed] [Google Scholar]
- 50.Zelenay S, Chora A, Soares MP, Demengeot J. Heme oxygenase-1 is not required for mouse regulatory T cell development and function. Int Immunol. 2007;19:11–18. doi: 10.1093/intimm/dxl116. [DOI] [PubMed] [Google Scholar]