

Abstract
In this study, we investigated the roles of serum amyloid A (SAA) in T helper 17 (Th17)-related cytokine induction in rheumatoid arthritis (RA) synoviocytes. Synoviocytes isolated from rheumatoid arthritis (RA) patients were stimulated with recombinant SAA and IL-23 expression was investigated using reverse transcriptase-polymerase chain reaction and Western blot. The involvement of mitogen-activated protein kineases (MAPKs) and nuclear factor (NF)-κB in SAA-induced interleukin (IL)-23 p19 expression was investigated using pharmacological inhibitors. In RA synoviocytes, SAA induced the expression of IL-23 p19 and p40 mRNA expression. The SAA-stimulated expression of p19 was rapid (< 3 h), and insensitive to polymyxin B treatment. This SAA-stimulated expression of IL-23 p19 was inhibited completely by inhibitors of NF-κB, p38MAPK and dexamethasone. Interestingly, the SAA-induced IL-23, p19 and p40 production was accompanied by enhanced expression of IL-1β, but not transforming growth factor-β. These results indicate that SAA is a significant inducer of IL-23 and IL-1β in RA synoviocytes and potentially activates the IL-23/IL-17 pathway in the RA synovium. Our data present a novel interaction between inflammation and autoimmunity by an acute-phase protein.
Keywords: IL-23, IL-1β, rheumatoid arthritis, serum amyloid A, synoviocytes, Th17 cells
Introduction
A large number of cytokines are active in the joints of rheumatoid arthritis (RA) patients [1]. A new subset of interleukin (IL)-17-producing T helper type 17 (Th17) cells has been thought to play a critical role in chronic inflammatory diseases [2]. Increased expression of IL-17 has been reported in RA patients, suggesting a role of Th17 cells in the development of RA [3,4]. Recent studies have identified an important role for IL-23 in the differentiation of Th17 cells [5]. IL-23 overexpression has also been demonstrated in rheumatoid synovium from RA patients [6,7]. However, the mechanisms related to this aberrant expression of IL-23 in the rheumatoid synovium need to be investigated.
The acute phase protein, serum amyloid A (SAA), is regulated by inflammatory cytokines and abundant levels of SAA have been demonstrated in RA sera and synovium [8,9]. In view of these inflammatory properties of SAA, the mechanism by which SAA exacerbates the inflammatory responses under autoimmune conditions remains elusive. Although IL-23, like IL-12, is thought to play an important role in autoimmune disease [10,11], the mechanism of control of IL-23 has not been understood fully in RA. IL-1β, tumour necrosis factor (TNF-α), IL-17, prostaglandin E2 and lipopolysaccharide (LPS) are known to induce the production of IL-23 [12–15]. Several recent studies have shown that SAA possesses cytokine-like activity and can stimulate the production of matrix metalloproteinase and cytokines, such TNF-α, IL-1β and IL-6 [16–20]. These findings have led us to investigate whether SAA can induce Th17-related cytokines.
In this study, we examined the effects of SAA on the induction of Th17-related cytokines in RA synoviocytes. Our results demonstrate that expression of IL-23 p19 was induced in RA synoviocytes by SAA stimulation. This suggests a novel role of SAA in promoting synovial inflammation or autoimmunity via the IL-23/IL-17 pathway.
Materials and methods
Reagents
Recombinant human SAA was purchased from Peprotech (Rocky Hills, NJ, USA). Polymyxin B was purchased from Sigma (St Louis, MO, USA). PD98059, SB203580, SP600125 and BAY11-7082 were obtained from Calbiochem (San Diego, CA, USA).
Patients
All RA patients fulfilled the American College of Rheumatology criteria for RA. Synovial tissue samples were obtained from five patients with RA during synovectomy. Synovial fibroblasts were isolated from the synovial tissues by enzymatic digestion. The isolated synovial fibroblasts were used at the third or fourth passages for subsequent experiments. All experiments were performed with the protocol approved by the ethics committee of Nagasaki Medical Center.
Measurement of cytokine secretion
RA synoviocytes (5 × 104) were seeded in 24-well plates containing RPMI plus 10% fetal calf serum (FCS) for 24 h. Following 24 h of incubation in serum-free RPMI, the cells were stimulated with SAA (0–5 µg/ml) for 24 h. Cell-free supernatants were collected by centrifugation at 400 g for 5 min and assayed for IL-23 and IL-12, p40 with enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems, Minneapolis, MN, USA), according to the manufacturer's instructions.
Reverse transcription–polymerase chain reaction (RT–PCR)
Total cellular RNA was extracted with Trizol (Invitrogen, Carlsbad, CA, USA), according to the manufacturer's protocol. First-strand cDNA was synthesized from 1 µg of total cellular RNA using an RNA PCR kit (Takara Bio Inc., Otsu, Japan) with random primers. Thereafter, cDNA was amplified using 28 cycles for IL-23 p19 and β-actin, respectively. The specific primers used were as follows – IL-23 p19: forward primer 5′-GCA GAT TCC AAG CCT CAG TC-3′, reverse primer 5′-TTC AAC ATA TGC AGG TCC CA-3′; β-actin: forward primer 5′-GTGGGGCGCCCCAGGCACCA-3′, reverse primer 5′-CTCCTTAATGTCACGCACGATTTC-3′.
The product sizes were 276 base pairs (bp) for IL-23 p19 and 234 bp for β-actin. The thermocycling conditions for the targets were as follows: 94°C for 30 s and 60°C for 60 s and 72°C for 30 s. The PCR products were electrophoresed on 2% agarose gels and visualized by ethidium bromide staining.
Amplification of the IL-23 p19, p40, IL-12 p35, IL-1β and transforming growth factor (TGF)-β transcripts was also accomplished on a Light Cycler (Roche Diagnostics, Mannheim, Germany) using specific primers. The housekeeping gene fragment of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used for verification of equal loading.
Western blot analysis
For measurement of IL-23 p19 protein expression by Western blot analysis, serum-starved RA–fibroblast-like synoviocytes (RA-FLS) seeded in six-well plates were stimulated with SAA for 24 h, and the cells were washed by ice-cold phosphate-buffered saline (PBS) and lysed with a lysis buffer [1% Nonidet P 40, 50 mM Tris, pH 7·5, 100 mM NaCl, 50 mM NaF, 5 mM ethylenediamine tetraacetic acid (EDTA), 20 mM β-glycerophosphate, 1·0 m, sodium orthovanadate, 10 µg/ml aprotinin and 10 µg/ml leupeptin] for 20 min at 4°C. Insoluble material was removed by centrifugation at 15 000 g for 15 min at 4°C. The supernatant was saved and the protein concentration was determined using the Bio-Rad protein assay kit (Bio-Rad, Hercules, CA, USA). An identical amount of protein (50 µg) for each lysate was subjected to 12% sodium dodecyl sulphate-polyacrylamide gel electrophoresis, and then transferred to a nitrocellulose membrane. Western blot analysis using the primary monoclonal antibodies against IL-23 p19 (BioLegend, San Diego, CA, USA) and β-actin (Sigma) was performed with an ECL Western blotting kit (Amersham, Little Chalfont, UK).
Statistical analysis
Differences between groups were examined for statistical significance using Wilcoxon's signed-rank test.
Results
SAA stimulates IL-23 p19 mRNA expression
We first evaluated the mRNA expression of IL-23, which is involved in Th17 immune responses, in SAA-stimulated RA synoviocytes. As shown in Fig. 1, a marked and significant increase in IL-23 p19 transcript was observed in SAA-stimulated synoviocytes compared with unstimulated synoviocytes. We next evaluated the mRNA expression quantitatively using real-time PCR. SAA stimulated IL-23 p19 mRNA expression in synoviocytes and polymyxin B did not alter SAA-induced IL-23 p19 mRNA expression (Fig. 2a). Although SAA induced IL-23 p40 mRNA expression, SAA stimulation did not elicit any significant increase in the mRNA levels of IL-12 p35 in the same treated synoviocytes (Fig. 2b,c). IL-23 p19 and p40 mRNA expression in synoviocytes was increased at 3 h post-stimulation with SAA. SAA stimulation of RA synoviocytes induced maximal expression of p19 and p40 at 6 and 9 h post-stimulation, respectively (Fig. 2d).
Fig. 1.
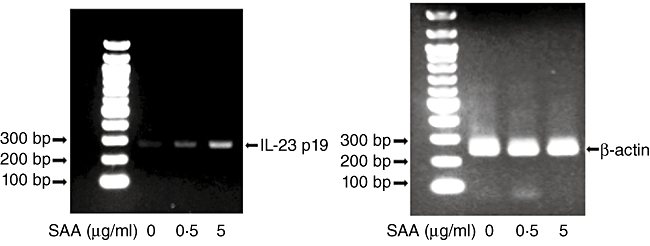
Semi-quantitative analysis of interleukin (IL)-23 p19 mRNA expression in rheumatoid arthritis (RA) synoviocytes. RA synoviocytes were stimulated by serum amyloid A (SAA) (5 µg/ml) for 6 h and total RNA was analysed by reverse transcription–polymerase chain reaction (RT–PCR) using primers specific for IL-23 p19 and β-actin, which was used as an internal control. Three experiments were performed using different RA synoviocytes and a representative finding is shown.
Fig. 2.
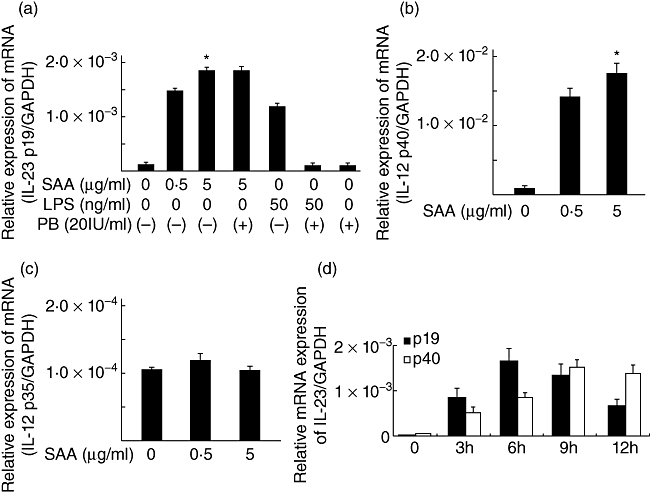
Serum amyloid A (SAA) stimulates interleukin (IL)-23 p19 and p40 mRNA expressions in rheumatoid arthritis (RA) synoviocytes. (a) RA synoviocytes were unstimulated or stimulated with SAA (0·5, 5 µg/ml) and lipopolysaccharide (LPS; 100 ng/ml) for 6 h in the presence or absence of polymyxin B (10 µg/ml). IL-23 p19 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA expression was determined by real-time polymerase chain reaction (PCR). (b) RA synoviocytes were stimulated with SAA (0·5, 5 µg/ml) for 6 h. IL-23 p40 and GAPDH mRNA expression was determined by real-time PCR. (c) RA synoviocytes were stimulated with SAA (0·5, 5 µg/ml) for 6 h. IL-12 p35 and GAPDH mRNA expression was determined by real-time PCR. (d) RA synoviocytes were stimulated with SAA (5 µg/ml) for 0, 3, 6, 9 and 12 h. IL-23 p19 and p40 mRNA expressions were determined by real-time PCR. Data are means of three different RA–fibroblast-like synoviocytes run in triplicate ± standard deviation. *P < 0·0001 compared to untreated RA synoviocytes.
Activation of nuclear factor (NF)-κB and p38 mitogen-activated protein kinease (MAPK) are required for SAA-induced expression of p19
We have demonstrated previously that the SAA-induced expression of cytokines was accompanied by activation of the NF-κB and MAPK signalling pathways and that dexamethasone inhibited the expression of these cytokines, probably by transrepressing NF-κB in RA synoviocytes [19]. We examined the effects of dexamethasone on SAA-stimulated induction of these cytokines (IL-23, p19, p40) in RA synoviocytes. Treatment of cells with dexamethasone for 2 h prior to stimulation with SAA inhibited significantly the expression of IL-23 p19 and p40 (Fig. 3). Dexamethasone exerts its biological effects by interacting with transcription factors, such as NF-κB or activator protein-1 (AP-1). To examine the involvement of these pathways in the SAA-induced IL-23 p19 expression, we used specific inhibitors for NK-κB and MAPK. Bay11-0073 and SB203580 inhibited almost completely the SAA-induced expression of IL-23 p19 transcript, indicating that activation of NF-κB and p38 MAPK is required for the SAA-induced expression of p19 (Fig. 4). Meanwhile, pretreatment with the specific inhibitors, PD98059 [extracellular-regulated 1/2 kinase (ERK1/2)] and SP600125 [c-Jun N-terminal kinase (JNK)], did not affect the SAA-induced expression of p19.
Fig. 3.
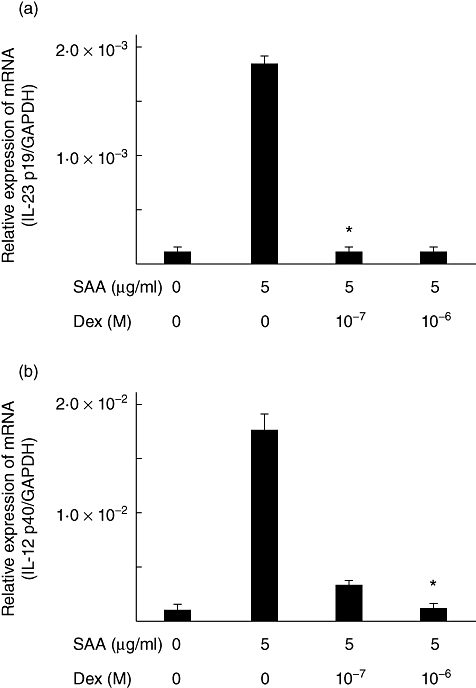
Dexamethasone suppressed the serum amyloid A (SAA)-induced p19 and p40 mRNA expressions. Rheumatoid arthritis (RA) synoviocytes were incubated with the indicated concentrations of dexamethasone for 1 h. Cells were then stimulated with 5 µg/ml of SAA for 6 h and IL-23 p19 (a), p40 (b) and glyceraldehyde-3-phosphate dehydrogenase mRNA expression was determined by real-time polymerase chain reaction (PCR). Data are the means of two different RA synoviocytes run in triplicate ± standard deviation. *P < 0·0001 compared to SAA-treated RA synoviocytes.
Fig. 4.
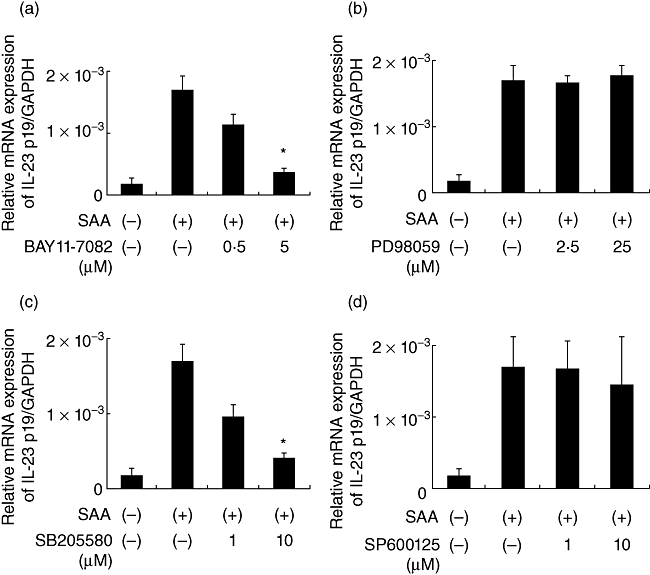
Nuclear factor (NF)-κB and p38 mitogen-activated protein kinease (MAPK) inhibition suppressed interleukin (IL)-23 p19 mRNA expression. Rheumatoid arthritis (RA) synoviocytes were incubated with vehicle (dimethylsulphoxide, media), BAY11-7082 (a, nuclear factor-κB inhibitor), PD98059 (b, extracellular regulated kinase pathway inhibitor), SB203580 (c, p38 inhibitor) and SP600125 (d, c-Jun N-terminal kinase inhibitor) for 1 h. Cells were then stimulated with 5 µg/ml of serum amyloid A (SAA) for 6 h and interleukin (IL)-23 p19 and glyceraldehyde-3-phosphate dehydrogenase mRNA expression was determined by real-time polymerase chain reaction. Data are the means of two different RA synoviocytes run in triplicate ± standard error of the mean. *P < 0·0001 compared to SAA-treated RA synoviocytes.
SAA stimulates IL-23 p19 protein expression
We next determined whether this induced expression of p19 and p40 mRNA resulted in secretion of IL-23. We could not detect IL-23 in the supernatants of SAA-stimulated synoviocytes using an IL-23-specific ELISA system. However, in the cellular lysates of SAA-stimulated RA synoviocytes, induction of p19 was confirmed at the protein level by Western blot analysis (Fig. 5). Conversely, the IL-23 subunit, p40, was induced in SAA-stimulated synoviocytes-conditioned medium, as demonstrated using a p40-specific ELISA system (Fig. 6).
Fig. 5.

Serum amyloid A (SAA) stimulates interleukin (IL)-23 p19 protein expression in rheumatoid arthritis (RA) synoviocytes. RA synoviocytes were stimulated with SAA for 24 h. IL-23 p19 protein expression was determined by Western blotting using specific antibodies against p19. Three experiments were performed using different RA synoviocytes and a representative result is shown.
Fig. 6.
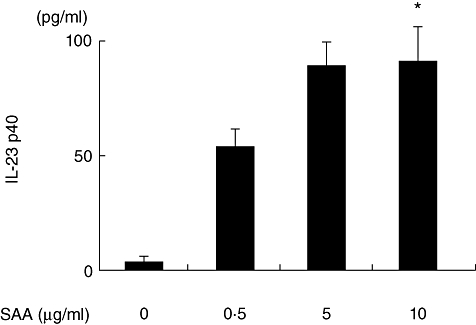
Serum amyloid A (SAA) stimulates interleukin (IL)-23 p40 protein synthesis in rheumatoid arthritis (RA) synoviocytes. RA synoviocytes were stimulated with various concentrations of recombinant SAA as indicated for 24 h. IL-23 p40 protein in the conditioned media was determined by enzyme-linked immunosorbent assay. Data represent the means of three independent experiments run in triplicate ± standard deviation. *P < 0·0001 compared to untreated RA synoviocytes.
SAA preferentially stimulated expression of IL-1β but not TGF-β mRNA
To address the question whether SAA could induce other Th17-related cytokines, we examined the effects of SAA on expression of IL-1β and TGF-β mRNA in rheumatoid synoviocytes. IL-1β mRNA was strongly up-regulated following stimulation with SAA. However, SAA stimulation did not affect TGF-β mRNA expression in rheumatoid synoviocytes (Fig. 7).
Fig. 7.
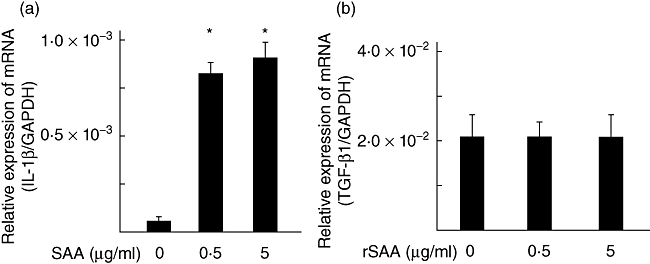
Serum amyloid A (SAA) stimulates interleukin (IL)-1β mRNA expression in rheumatoid arthritis (RA) synoviocytes. RA synoviocytes were stimulated with SAA (0·5, 5 µg/ml) for 6 h. IL-1β and transforming growth factor-β1 mRNA expression was determined by real-time polymerase chain reaction. Data are means of three different RA–fibroblast-like synoviocytes run in triplicate ± standard deviation. P < 0·0001 compared to untreated RA synoviocytes.
Discussion
RA is a common autoimmune condition, characterized by synovitis and subsequent destruction of the joint [21]. The IL-23/IL-17 axis has been shown to be a major proinflammatory pathway in chronic inflammatory disease, including RA [22]. IL-23, which is comprised of the IL-12 p40 subunit and a unique IL-23 p19 subunit, plays a critical role in the maintenance and expansion of Th17 cells [23]. The importance of IL-23 has been highlighted in a murine model of arthritis in which specific absence of the IL-23 gene conferred complete resistance to the development of collagen-induced arthritis (CIA) in mice [10]. In patients with RA, serum and synovial fluid concentrations of IL-23 have been reported to be elevated [24]. Furthermore, IL-23 p19 expression has been demonstrated in rheumatoid synovium [6,7,12].
In this study we demonstrated that SAA, which is overproduced in sera and synovial fluids from RA patients, up-regulated significantly the expression of IL-23 p19 in rheumatoid synoviocytes. Our data are consistent with previous reports demonstrating the induction of IL-23 p19 and p40 in SAA-stimulated monocytes [25]. IL-23 is composed of a p19 and p40 heterodimer. Despite the two-log fold induction of expression of p19 and p40 mRNA and the detection of their proteins, we did not detect IL-23 in the supernatant of SAA-stimulated synoviocytes by ELISA. Our findings are consistent with the abundant expression of p19 protein in the rheumatoid synovium, despite the fact that heterodimeric IL-23 was barely detectable [7]. The possibility exists that abnormalities in the dimerization of p19 and p40 may be implicated in this impaired production of IL-23.
The signalling mechanisms responsible for SAA-induced cytokine expressions have been studied. A number of proteins have been shown to interact with SAA. Among these proteins, formyl peptide receptor-like 1 (FPRL1) has been shown to be a cell surface receptor for SAA [26] and is expressed in rheumatoid synoviocytes [17]. FPRL1 is a G protein-coupled receptor that can mediate the activation of MAPKs and NF-κB [26]. Our results demonstrated that IL-23 p19 expression was reduced dramatically in response to impaired activation of NF-κB and p38 MAP kinase signalling by pharmacological inhibition. While regulation of IL-23 p19 at the molecular level is not understood fully, recent studies suggest that NF-κB and AP-1 are important for p19 gene expression [27–29]. IL-23 p19 expression induced by IL-17 can be blocked by inhibitors of NF-κB and p38 MAPK in RA synoviocytes [6,12]. Together, our results suggest the involvement of p38 MAPK and the NF-κB signalling pathway in the regulation of IL-23 p19 expression in rheumatoid synovium.
As reported previously, IL-23 overexpression may not be associated with a fully developed Th17 response [30]. In this regard, IL-23 alone is a relatively ineffective inducer of Th17 cells and requires the combination of IL-6 and IL-1β to induce differentiation of Th17 cells from naive human T cells [31]. Conversely, we have reported previously that SAA is a strong inducer of IL-6 in rheumatoid synoviocytes [19]. In this study, we showed that SAA is a potent inducer of IL-1β. Taken together with these previous findings, SAA could be a potent inducer of rheumatoid synoviocytes in promoting autoimmunity and chronic inflammation via the Th17 immune response. The results of this study contribute towards understanding the immunoregulatory properties of SAA in rheumatoid inflammation by providing the novel observation that SAA promotes Th17-related cytokines in rheumatoid synovium. Maintenance of Th17 cells requires the presence of IL-23, IL-1β and IL-6 [30], all of which are induced by SAA. In this study, we demonstrated that SAA stimulated IL-1β induction, but did not affect TGF-β expression in RA synoviocytes. Although TGF-β and IL-23 are the major cytokines that contribute towards Th17 cell differentiation in mice [32], the role of TGF-β in human Th17 cells is an area of discussion. Recent investigations have indicated that human T cells could be induced to differentiate into Th17 cells in response to either IL-1β or IL-23, but not TGF-β[33]. Therefore, SAA-mediated IL-23 and IL-1β induction may contribute to the Th17 immune response in rheumatoid synovium.
Disclosure
The authors declare that they have no competing interests.
References
- 1.Brennan FM, McInnes IB. Evidence that cytokines play a role in rheumatoid arthritis. J Clin Invest. 2008;118:3537–45. doi: 10.1172/JCI36389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fouser LA, Wright JF, Dunussi-Joannopoulos K, Collins M. Th17 cytokines and their emerging roles in inflammation and autoimmunity. Immunol Rev. 2008;226:87–102. doi: 10.1111/j.1600-065X.2008.00712.x. [DOI] [PubMed] [Google Scholar]
- 3.Kotake S, Udagawa N, Takahashi N, et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest. 1999;103:1345–52. doi: 10.1172/JCI5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chabaud M, Durand JM, Buchs N, et al. Human interleukin-17: a T cell-derived proinflammatory cytokine produced by the rheumatoid synovium. Arthritis Rheum. 1999;42:963–70. doi: 10.1002/1529-0131(199905)42:5<963::AID-ANR15>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 5.Annunziato F, Cosmi L, Liotta F, Maggi E, Romagnani S. Type 17 T helper cells-origins, features and possible roles in rheumatic disease. Nat Rev Rheumatol. 2009;5:325–31. doi: 10.1038/nrrheum.2009.80. [DOI] [PubMed] [Google Scholar]
- 6.Liu FL, Chen CH, Chu SJ, et al. Interleukin (IL)-23 p19 expression induced by IL-1beta in human fibroblast-like synoviocytes with rheumatoid arthritis via active nuclear factor-kappaB and AP-1 dependent pathway. Rheumatology. 2007;46:1266–73. doi: 10.1093/rheumatology/kem055. [DOI] [PubMed] [Google Scholar]
- 7.Brentano F, Ospelt C, Stanczyk J, Gay RE, Gay S, Kyburz D. Abundant expression of the interleukin (IL)23 subunit p19, but low levels of bioactive IL23 in the rheumatoid synovium: differential expression and Toll-like receptor-(TLR) dependent regulation of the IL23 subunits, p19 and p40, in rheumatoid arthritis. Ann Rheum Dis. 2009;68:143–50. doi: 10.1136/ard.2007.082081. [DOI] [PubMed] [Google Scholar]
- 8.Chambers RE, MacFarlane DG, Whicher JT, Dieppe PA. Serum amyloid-A protein concentration in rheumatoid arthritis and its role in monitoring disease activity. Ann Rheum Dis. 1983;42:665–7. doi: 10.1136/ard.42.6.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kumon Y, Suehiro T, Hashimoto K, Nakatani K, Sipe JD. Local expression of acute phase serum amyloid A mRNA in rheumatoid arthritis synovial tissue and cells. J Rheumatol. 1999;26:785–90. [PubMed] [Google Scholar]
- 10.Murphy CA, Langrish CL, Chen Y, et al. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. 2003;198:1951–7. doi: 10.1084/jem.20030896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oppmann B, Lesley R, Blom B, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–25. doi: 10.1016/s1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- 12.Kim HR, Cho ML, Kim KW, et al. Up-regulation of IL-23p19 expression in rheumatoid arthritis synovial fibroblasts by IL-17 through PI3-kinase-, NF-kappaB- and p38 MAPK-dependent signalling pathways. Rheumatology. 2007;46:57–64. doi: 10.1093/rheumatology/kel159. [DOI] [PubMed] [Google Scholar]
- 13.Zhan Z, Andoh A, Yasui H, et al. Interleukin-1beta and tumor necrosis factor-alpha upregulate interleukin-23 subunit p19 gene expression in human colonic subepithelial myofibroblasts. Int J Mol Med. 2005;15:79–83. [PubMed] [Google Scholar]
- 14.Sheibanie AF, Khayrullina T, Safadi FF, Ganea D. Prostaglandin E2 exacerbates collagen-induced arthritis in mice through the inflammatory interleukin-23/interleukin-17 axis. Arthritis Rheum. 2007;56:2608–19. doi: 10.1002/art.22794. [DOI] [PubMed] [Google Scholar]
- 15.Utsugi M, Dobashi K, Ishizuka T, et al. Rac1 negatively regulates lipopolysaccharide-induced IL-23 p19 expression in human macrophages and dendritic cells and NF-kappaB p65 trans activation plays a novel role. J Immunol. 2006;177:4550–7. doi: 10.4049/jimmunol.177.7.4550. [DOI] [PubMed] [Google Scholar]
- 16.Migita K, Kawabe Y, Tominaga M, Origuchi T, Aoyagi T, Eguchi K. Serum amyloid A protein induces production of matrix metalloproteinases by human synovial fibroblasts. Lab Invest. 1998;78:535–9. [PubMed] [Google Scholar]
- 17.Vallon R, Freuler F, Desta-Tsedu N, et al. Serum amyloid A (apoSAA) expression is up-regulated in rheumatoid arthritis and induces transcription of matrix metalloproteinases. J Immunol. 2001;166:2801–7. doi: 10.4049/jimmunol.166.4.2801. [DOI] [PubMed] [Google Scholar]
- 18.He R, Sang H, Ye RD. Serum amyloid A induces IL-8 secretion through a G protein-coupled receptor, FPRL1/LXA4R. Blood. 2003;101:1572–81. doi: 10.1182/blood-2002-05-1431. [DOI] [PubMed] [Google Scholar]
- 19.Koga T, Torigoshi T, Motokawa S, et al. Serum amyloid A-induced IL-6 production by rheumatoid synoviocytes. FEBS Lett. 2008;582:579–85. doi: 10.1016/j.febslet.2008.01.022. [DOI] [PubMed] [Google Scholar]
- 20.Patel H, Fellowes R, Coade S, Woo P. Human serum amyloid A has cytokine-like properties. Scand J Immunol. 1998;48:410–18. doi: 10.1046/j.1365-3083.1998.00394.x. [DOI] [PubMed] [Google Scholar]
- 21.Lee DM, Weinblatt ME. Rheumatoid arthritis. Lancet. 2001;358:903–11. doi: 10.1016/S0140-6736(01)06075-5. [DOI] [PubMed] [Google Scholar]
- 22.Iwakura Y, Ishigame H. The IL-23/IL-17 axis in inflammation. J Clin Invest. 2006;116:1218–22. doi: 10.1172/JCI28508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McGeachy MJ, Cua DJ. Th17 cell differentiation: the long and winding road. Immunity. 2008;28:445–53. doi: 10.1016/j.immuni.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 24.Melis L, Vandooren B, Kruithof E, et al. Systemic levels of IL-23 are strongly associated with disease activity in rheumatoid arthritis but not spondyloarthritis. Ann Rheum Dis. 2010;69:618–23. doi: 10.1136/ard.2009.107649. [DOI] [PubMed] [Google Scholar]
- 25.He R, Shepard LW, Chen J, Pan ZK, Ye RD. Serum amyloid A is an endogenous ligand that differentially induces IL-12 and IL-23. J Immunol. 2006;177:4072–9. doi: 10.4049/jimmunol.177.6.4072. [DOI] [PubMed] [Google Scholar]
- 26.Su SB, Gong W, Gao JL, et al. A seven-transmembrane, G protein-coupled receptor, FPRL1, mediates the chemotactic activity of serum amyloid A for human phagocytic cells. J Exp Med. 1999;189:395–402. doi: 10.1084/jem.189.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu W, Ouyang X, Yang J, et al. AP-1 activated by Toll-like receptors regulates expression of IL-23 p19. J Biol Chem. 2009;284:24006–16. doi: 10.1074/jbc.M109.025528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mise-Omata S, Kuroda E, Niikura J, Yamashita U, Obata Y, Doi TS. A proximal kappaB site in the IL-23 p19 promoter is responsible for RelA- and c-Rel-dependent transcription. J Immunol. 2007;179:6596–603. doi: 10.4049/jimmunol.179.10.6596. [DOI] [PubMed] [Google Scholar]
- 29.Carmody RJ, Ruan Q, Liou HC, Chen YH. Essential roles of c-Rel in TLR-induced IL-23 p19 gene expression in dendritic cells. J Immunol. 2007;178:186–91. doi: 10.4049/jimmunol.178.1.186. [DOI] [PubMed] [Google Scholar]
- 30.Boniface K, Blom B, Liu YJ, de Waal Malefyt R. From interleukin-23 to T-helper 17 cells: human T-helper cell differentiation revisited. Immunol Rev. 2008;226:132–46. doi: 10.1111/j.1600-065X.2008.00714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 32.Volpe E, Servant N, Zollinger R, et al. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat Immunol. 2008;9:650–7. doi: 10.1038/ni.1613. [DOI] [PubMed] [Google Scholar]
- 33.Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8:942–9. doi: 10.1038/ni1496. [DOI] [PubMed] [Google Scholar]