

Abstract
Immunoglobulins (Igs) play important immunomodulatory effects on allergic asthma. Among these, IgG has been reported to regulate allergic inflammation in previous studies about immunotherapy and intravenous immunoglobulin therapy. In this study, to examine the immunomodulatory mechanisms of IgG and FcRs we evaluated the effects of intravenous (i.v.) rabbit IgG administration (IVIgG) on allergic airway inflammation and lung antigen-presenting cells (APCs) in a murine model of ovalbumin (OVA) sensitization and challenge. In OVA-challenged mice, IVIgG attenuated airway eosinophilia, airway hyperresponsiveness and goblet cell hyperplasia and also inhibited the local T helper type (Th) 2 cytokine levels. Additionally, IVIgG attenuated the proliferation of OVA-specific CD4+ T cells transplanted into OVA-challenged mice. Ex vivo co-culture with OVA-specific CD4+ cells and lung CD11c+ APCs from mice with IVIgG revealed the attenuated transcription level of Th2 cytokines, suggesting an inhibitory effect of IVIgG on CD11c+ APCs to induce Th2 response. Next, to analyse the effects on Fcγ receptor IIb and dendritic cells (DCs), asthmatic features in Fcγ receptor IIb-deficient mice were analysed. IVIgG failed to attenuate airway eosinophilia, airway inflammation and goblet cell hyperplasia. However, the lacking effects of IVIgG on airway eosinophilia in Fcγ receptor IIb deficiency were restored by i.v. transplantation of wild-type bone marrow-derived CD11c+ DCs. These results demonstrate that IVIgG attenuates asthmatic features and the function of lung CD11c+ DCs via Fcγ receptor IIb in allergic airway inflammation. Targeting Fc portions of IgG and Fcγ receptor IIb on CD11c+ DCs in allergic asthma is a promising therapeutic strategy.
Keywords: allergy; animal models/studies – mice/rats; asthma; dendritic cells (myeloid, plasmacytoid, monocyte-derived); Fc receptors (FcRs)
Introduction
Bronchial asthma is a disorder of the conducting airways characterized by variable airflow obstruction, but is also a chronic inflammatory disease of the airway associated with an immune response to inhaled antigens, which leads to airway infiltration of eosinophils and mast cells, goblet cell hyperplasia and airway hyperresponsiveness (AHR). These pathophysiological features are induced by T helper type (Th)2 proliferation and production of Th2 cytokines, such as interleukin (IL)-4, IL-5 and IL-13 [1]. Anti-inflammatory drugs, primarily corticosteroids, comprise the conventional treatment for chronic Th2 airway inflammation. The current anti-inflammation strategies to manage bronchial asthma have limited clinical efficacy for some patients.
Immunoglobulins (Igs) and Fc receptors (FcRs) play important roles in bronchial asthma pathogenesis. FcRs are expressed on many kinds of immune cells and control the cellular functions. Among Igs, IgE plays a crucial role in the pathogenesis of asthma by binding airborne inhalant allergen to activate various cellular inflammatory reactions of immune cells through FcεRI. Anti-IgE therapy, one of the controllers to manage bronchial asthma, reduces the free IgE available to activate effector cells [2]. In contrast, IgG reportedly has immunomodulatory effects on the immune response to common inhalant allergens. Immunotherapy by allergen vaccination is accompanied by an increase in allergen-specific IgG titres [3]. The anti-inflammatory properties of intravenous immunoglobulin (IVIG) therapy have been applied widely to attenuate disease progression in other chronic inflammatory diseases, including immune-mediated thrombocytopenia, chronic inflammatory demyelinating polyneuropathy, Kawasaki disease, Guillain–Barré syndrome and other autoimmune disorders. Intravenous (i.v.) Ig (IVIG) also provides an important adjunctive treatment to control airway inflammation, reducing oral steroid requirements in severe bronchial asthma [4–7]. The efficacy of IVIG is due largely to IgG, which is a major portion of IVIG. Several roles of IgG in IVIG therapy in autoimmunity have been proposed [8–10], and the functions of IgG in IVIG therapy in allergic diseases are also envisaged to inhibit inflammatory reaction. Although these reports suggest that i.v.-administered IgG have functions to protect against allergies and asthma, the precise target and mechanisms in allergic airway inflammation have not yet been revealed. In a murine experimental model, intranasal instillation of antigen-specific IgG reportedly reduce eosinophilic inflammation and goblet cell hyperplasia induced by antigen challenge, suggesting that topical IgG reportedly counteracts allergic pulmonary inflammation that is dependent upon Fc and interferon (IFN)-γ[11]. However, clinical use of these therapies in bronchial asthma is currently limited because of the lack of evidence. Clarifying the role of FcRs leads potentially to the development of a new strategy to manage asthmatic airway disorders.
The role of antigen-presenting cells (APCs), including dendritic cells (DCs), in the pathogenesis of asthma has been clarified. When allergens are encountered in the airways, DCs in the airway epithelium capture allergens and migrate to the draining lymph nodes, where they reside in a mature, antigen-priming mode [12]. There, antigen-specific T cells are induced to differentiate into Th effector cells or regulatory cells by these DCs. Thus, DCs are important in the initiation of T cell differentiation and activation and contribute indirectly to the development of airway inflammation. Targeting the inhibitory Fc receptor on DCs can potentially inhibit induction of the Th2 cytokine response.
We hypothesized that i.v. IgG administration (IVIgG) inhibits allergic inflammation through inhibitory FcRs on immune cells to induce a Th2 response. Among several types of FcRs, FcγRIIb is a unique inhibitory FcR which regulates immune cell function [13]. To verify the inhibitory effects of IVIgG and FcγRIIb in bronchial asthma, we pursued the mechanisms of IVIgG using murine models of allergic airway inflammation induced by ovalbumin (OVA) sensitization and aerosol challenge. As IVIgG, we analyse the effects of both mouse IgG and xenogenic (rabbit) IgG to analyse the functions on FcRs.
Methods
Allergic airway inflammation model
Pathogen-free female C57BL/6 [wild-type (WT)] FcγRIIb-deficient mice [14] and mice transgenic for the ovalbumin (OVA)323–339-specific T cell receptor (OT-II) on a C57BL/6 background were used. Body weights ranged from 20 to 23 g. All mice were housed and bred under pathogen-free conditions. All experiments were approved by the Institutional Animal Care and Use Committee and carried out according to the Kobe University Animal Experimentation Regulations.
Allergic airway inflammation was induced by intraperitoneal sensitization and airway challenge, as described previously [11]. Briefly, mice received intraperitoneal injection of 10 µg of OVA (Sigma-Aldrich, St Louis, MO, USA) and 1 mg of aluminium hydroxide (Sigma-Aldrich) in 0·5 ml of phosphate-buffered saline (PBS) on days 0, 7 and 14. Mice underwent aerosol challenge with OVA (1% in PBS) or PBS alone from days 21 to 23 daily for 30 min. Aerosolized OVA challenge using a nebulizer (NE-U07; OMRON, Kyoto, Japan) was performed in a closed aerosol chamber. For IgG administration, rabbit purified IgG (Sigma-Aldrich), F(ab′)2 (Thermo, Rockford, IL, USA), IgM (Wako, Osaka, Japan), mouse IgG (Sigma-Aldrich) or an equal volume of PBS (100 µl) alone was injected intravenously on day 20, prior to the first OVA challenge. In another experiment, OVA-sensitized mice were administered with 1 mg rabbit IgG administration after OVA challenge. The mice were challenged with OVA for 3 days before rabbit IgG administration on the third day of OVA challenge. All mice were analysed 24 h after the last OVA challenge. The experiments were repeated three times.
Assessment of airway inflammation, mucus production, AHR and OVA-specific IgE
To assess differential bronchoalveolar lavage fluid (BALF) cell counts, lungs were lavaged twice by instillation and withdrawal of 1 ml PBS through a tracheal cannula. BALF cells were counted using a haemocytometer. For differential cell counts, cytocentrifuged preparations were fixed and stained with Diff-Quick (Kokusaishiyaku, Kobe, Japan) and differentiated morphologically by counting 300 cells/slide.
For histopathological assessment, lungs were fixed and embedded in paraffin. Sections (5 µm) from all lobes were stained with haematoxylin and eosin (H&E) and periodic-acid Schiff (PAS). Airway inflammation and mucus-producing cells were graded blindly, as described previously [11]. Briefly, each tissue section was graded from 0 to 3; 0 indicated that no inflammation was detectable, 1 meant occasional cuffing with inflammatory cells, 2 indicated a thin layer of inflammatory cells surrounded most bronchi and 3 meant a thick layer of inflammatory cells surrounded most bronchi. More than five tissue sections were scored per mouse, so inflammation scores could be expressed as a mean value per animal and could be compared between groups. To estimate the presence of mucus-producing cells, we counted the number of airways per section and assigned a score of 0, 1, 2 or 3 to each airway when no, very few, <50% or >50% of the airway epithelial cells were PAS-positive. Therefore, each mouse and group was characterized by a score distribution that could be compared statistically.
AHR was assessed in conscious, unrestrained mice by whole-body plethysmography (Buxco Electronics, Sharon, CT, USA), as described previously [15]. Briefly, a mouse was placed into the main chamber of the plethysmograph. The mouse was exposed to nebulized PBS and methacholine (Sigma-Aldrich) in PBS using an ultrasonic nebulizer. As an index of in vivo airway obstruction, enhanced pause (Penh) values were measured and expressed as relative values compared to baseline Penh values following PBS exposure for each methacholine concentration (1–25 mg/ml).
Levels of plasma OVA-specific IgE (OVA-IgE) in challenged mice were measured by enzyme-linked immunosorbent assay (ELISA), as described previously [16].
Cytokine levels in BALF
Th1 and Th2 cytokine levels (IL-4, IL-5, IL-13, IFN-γ) were measured in BALF by ELISA (R&D Systems, Minneapolis, MN, USA), according to the manufacturer's instructions.
In vivo proliferation of carboxyfluorescein succinimidyl ester (CFSE)-labelled OTII cells
To estimate OVA-specific T cell proliferation in vivo, we used OTII CD4+ cells labelled with CFSE; Molecular Probes, Eugene, OR, USA). Single-cell spleen suspensions from OTII mice were depleted of dendritic cells (DCs) using CD11c microbead and automatic magnetic-activated cell sorting (autoMACS) system (Miltenyi Biotech, Auburn, CA, USA). The purity of CD4+ cells was estimated to be over 90% using a flow cytometer. Cells were incubated with 5 µM CFSE, according to the manufacturer's instructions. CFSE-labelled OTII cells (5 × 106 cells) were transferred intravenously into each IgG or PBS-administered wild-type mouse. After injection, mice were challenged with OVA for 30 min a day for 2 days. Seventy-two hours after the OTII cell transfer, mononuclear cells from the thoracic lymph nodes were stained with anti-CD4-magnetic-activated cell sorting (BD Biosciences, Franklin Lakes, NJ, USA) to analyse transferred CD4+ OTII cell proliferation using a flow cytometer. Data were analysed using Cellquest (BD Biosciences) and FlowJo software (Treestar, Ashland, OR, USA).
Isolation of lung CD11c+ antigen-presenting cells
To analyse the function of lung CD11c+ antigen-presenting cells (APCs), they were collected 24 h after the mice were administered with 1 mg of IgG or PBS, as described previously [17]. Briefly, mouse lungs were minced and then incubated in the digestion medium consisting of RPMI-1640 (Sigma-Aldrich), 5% fetal bovine serum (Sigma-Aldrich), 1 mg/ml collagenase type 4 (Roche Diagnostics, Indianapolis, IN, USA) and deoxyribonuclease I (bovine pancreas; Wako). Lung CD11c+ APCs were isolated using the CD11c microbeads and autoMACS system according to the manufacturer's instructions. The purity of CD11+ cells was estimated to be over 80% using a flow cytometer.
In vitro OTII CD4+ T cell Th2 differentiation analysis
OTII CD4+ cells were isolated from OTII mouse spleens using the MACS system. OTII CD4+ cells (2·5 × 105 cells/well) were co-cultured in a 96-well plate in complete medium with lung CD11c+ APCs (2·5 × 104 cells/well) from naive WT mice after PBS or IgG administration. Cultures were stimulated in vitro with an OVA323–339 peptide (5 µg/ml; GenWay Biotech, San Diego, CA, USA) or medium for 6 h. Cytokine mRNA levels were assessed by real-time reverse transcriptase–polymerase chain reaction (RT–PCR).
RNA isolation and real-time RT–PCR
Total RNA was extracted from cells or tissues using Isogen (Nippon Gene, Tokyo, Japan). Single-strand cDNA was synthesized using ExScript RT reagent kits (Takara, Otsu, Japan). Real-time RT–PCR was performed using an ABI PRISM 7500 Sequence Detection System (Applied Biosystems, Foster City, CA, USA), with primers described in Table 1. Amplifications were performed in duplicate with SYBR Premix Ex Taq (Takara), according to the manufacturer's instructions. Target mRNA levels were normalized against β-actin mRNA.
Table 1.
Polymerase chain reaction primers.
| IFN-γ | Forward primer | 5′-CGGCACAGTCATTGAAAGCCTA-3′ |
| Reverse primer | 5′-GTTGCTGATGGCCTGATTGTC-3′ | |
| IL-4 | Forward primer | 5′-TCTCGAATGTACCAGGAGCCATATC-3′ |
| Reverse primer | 5′-AGCACCTTGGAAGCCCTACAGA-3′ | |
| IL-5 | Forward primer | 5′-TCAGCTGTGTCTGGGCCACT-3′ |
| Reverse primer | 5′-TTATGAGTAGGGACAGGAAGCCTCA-3′ | |
| IL-13 | Forward primer | 5′-CTCTTGCTTGCCTTGGTGGTCTC-3′ |
| Reverse primer | 5′-AGGGAATCCAGGGCTACACAGAA-3′ | |
| β-actin | Forward primer | 5′-CCCTAAGGCCAACCGTGA-3′ |
| Reverse primer | 5′-GTTGAAGGTCTCAAACATGATCTG-3′ |
IFN, interferon; IL, interleukin.
Adoptive transfer of CD11c+ BMDC
Bone marrow dendritic cells (BMDC) were obtained from WT or FcγRIIb-deficient mice according to the method described previously [18]. The bone marrow cells were cultured at 1 × 106 cells/ml in the presence of 20 ng/ml murine granulocyte–macrophage colony-stimulating factor (GM-CSF). The medium was replaced with a GM-CSF-containing medium on day 4 of culture. On day 6 of culture, BMDCs were collected and CD11c+ BMDCs were purified using the autoMACS system. Sensitized FcγRIIb-deficient mice were injected i.v. with 1 × 106 CD11c+ BMDCs 24 h before i.v. administration of IgG and challenged with OVA for 3 days.
Statistical analysis
All results are expressed as mean ± standard deviation. A t-test was conducted to determine differences between two groups. As measured values were not distributed normally and the sample size was small, non-parametric analysis using a Mann–Whitney U-test confirmed that differences remained significant, even if the underlying distribution was uncertain. The P-values for significance were set at 0·05 for all tests.
Results
IVIgG prevents development of airway inflammation, hyperresponsiveness and OVA-IgE induced by OVA challenge
To estimate the effects of IVIgG on bronchial asthma, rabbit IgG was administered intravenously to the murine allergic airway inflammation model. OVA sensitization and challenge induced a substantial increase in total cells in BALF. This was due largely to increased eosinophil numbers, which is one of the characteristics of eosinophilic airway inflammation in bronchial asthma. Administration of 1 mg of rabbit IgG before airway challenge markedly decreased the number of total cells and eosinophils in BALF (Fig. 1a) in a dose-dependent manner. The treatment, such as the same amount of IgM or F(ab′)2, did not influence significantly the BALF cell counts, nor did administration of 1 mg of mouse IgG influence cell counts. In the IVIgG experiment after challenge, rabbit IgG administration after OVA challenge for 3 days also reduced the number of total cells and eosinophils significantly compared with PBS-treated mouse (Fig. 1b). Because 1 mg of rabbit IgG suppressed airway inflammation sufficiently, we used this dose to analyse the role of IVIgG before OVA challenge in our subsequent experiments.
Fig. 1.
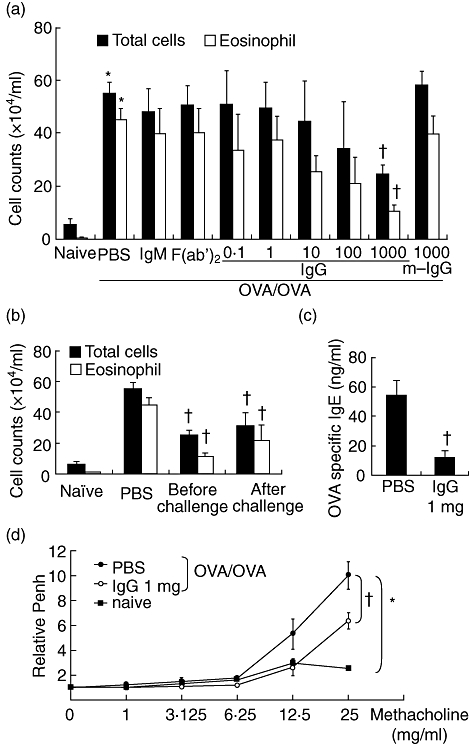
Effects of intravenous (i.v.) immunoglobulin G (IVIgG) on airway inflammation and hyperresponsiveness. (a) Ovalbumin (OVA)-sensitized mice received i.v. administration of rabbit IgG (IgG, 0·1–1000 µg), 1000 µg of IgM, F(ab′)2 or mouse IgG (m-IgG) 1 day before OVA challenge (OVA/OVA) and total cells and eosinophils in bronchoalveolar fluid (BALF) were quantified. (b) Schedules of rabbit IgG administration were compared. The number of total cells and eosinophils in BALF were measured in mice administered with rabbit IgG (1000 µg) before and after OVA challenge. (c) The level of plasma OVA-specific IgE in OVA-challenged mice was compared. (d) Airway hyperresponsiveness (AHR) was assessed using body plethysmography. At 24 h after the last challenge, increased enhanced pause (Penh) in response to inhaled methacholine were measured. The effects of IgG administration on AHR to methacholine were expressed on the Y-axis as relative values of Penh to baseline. *Significant differences (P < 0·05) versus naive mice. †Significant differences (P < 0·05), versus phosphate-buffered saline (PBS).
Plasma OVA-IgE levels were also elevated in challenged mice. This effect was suppressed by rabbit IgG administration (Fig. 1c). Next, to assess the effect of IVIgG on AHR, the relative increase of Penh in response to methacholine inhalation was evaluated. After OVA challenge, AHR was increased in OVA-sensitized mice. Administration of 1 mg of rabbit IgG inhibited the development of AHR significantly (Fig. 1d).
H&E-stained lung tissue sections showed increased numbers of inflammatory cells, including eosinophils, in the peribronchial and perivascular regions of sensitized and challenged WT mice compared to naive WT mice (Fig. 2a,b). IVIgG decreased the number of inflammatory cells (Fig. 2d–g). IVIgG also decreased airway goblet cell hyperplasia in PBS-injected mice after OVA sensitization and challenge upon analysis of PAS-stained lung tissue sections (Fig. 2c,f,h). These data suggest that IVIgG ameliorates airway inflammatory change and goblet cell hyperplasia in this murine model.
Fig. 2.
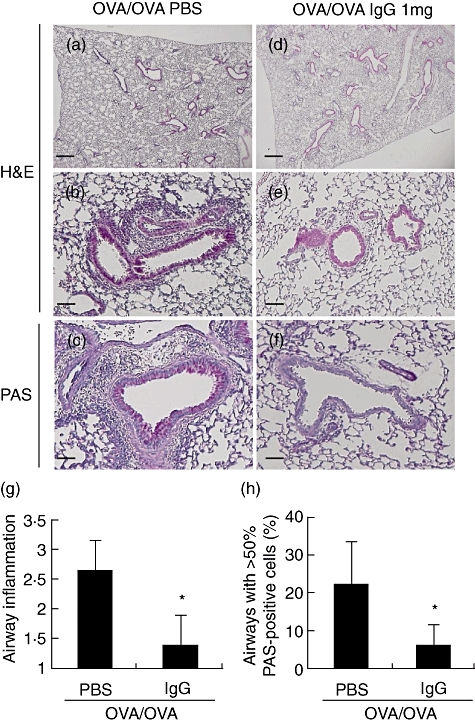
Effects of intravenous (i.v.) immunoglobulin G (IVIgG) on the development of airway inflammation and mucus production. Paraffin-embedded sections of lung of sensitized and challenged mice were prepared. Representative histological findings for lungs stained with haematoxylin and eosin (H&E) or periodic-acid Schiff (PAS) are shown. (a–c) Lung tissue from challenged mice [ovalbumin (OVA)/OVA + phosphate-buffered saline (PBS)]. (d–f) Lung tissue from mice administered with 1 mg of IgG (OVA/OVA + IgG). (a,d) Low-power field of the H&E-stained sample. (b,e) High-power field of the H&E-stained sample. (c,f) High-power fields of the PAS-stained sample, which shows mucus stained red by PAS. Scale bar equals 100 µm on low-power fields (a,d) and 50 µm on high-power fields (b,c,e,f). (g) Scores for peribronchial and perivascular inflammation are shown. (h) The number of airways free of mucus-producing cells or containing > 50% PAS-positive-cells are shown. *Significant differences (P < 0·05) versus PBS.
IVIgG decreased Th2 cytokine levels in BALF
To analyse the Th1/Th2 response in airway, cytokine levels in BALF were measured. Th2 cytokines, IL-4, IL-5 and IL-13, were increased in OVA-challenged mice (Fig. 3a–c), and the increase of IL-4 and IL-5 was attenuated significantly by IVIgG. No significant differences in IFN-γ levels were seen among challenged and administered mice (Fig. 3d). These results suggest that IVIgG modifies local Th2 response.
Fig. 3.
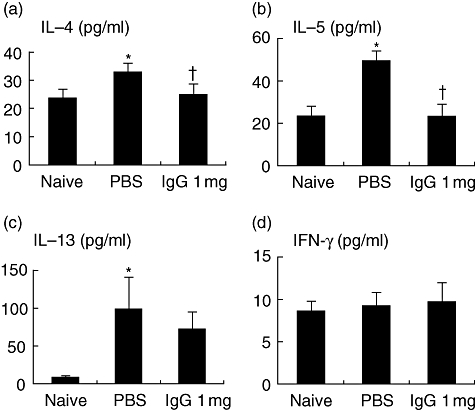
Effects of intravenous (i.v.) immunoglobulin G (IVIgG) on T helper type 2 (Th2) cytokines in bronchoalveolar fluid (BALF). Th1/Th2 cytokines in BALF were measured by enzyme-linked immunosorbent assay (ELISA). Interleukin (IL)-4 (a), IL-5 (b), IL-13 (c) and interferon (IFN)-γ (d) levels of ovalbumin (OVA)-sensitized and challenged mice are shown. *Significant differences (P < 0·05) versus naive mice. †Significant differences (P < 0·05) versus phosphate-buffered saline (PBS).
IVIgG prevents transferred OTII T cell proliferation in vivo
To assess the effect of IVIgG on allergen-specific T cells, the proliferation of transferred OTII T cells was measured. OVA challenge apparently induced CD4+ T cell proliferation, as represented by CFSE fluorescence intensity reduced by half with each cell division of transferred CFSE-labelled OTII T cells. Reduction of fluorescence was inhibited by previous administration of IVIgG compared to PBS administration (Fig. 4a). These results indicate that IVIgG inhibits the proliferation of OVA-specific CD4+ T cells.
Fig. 4.
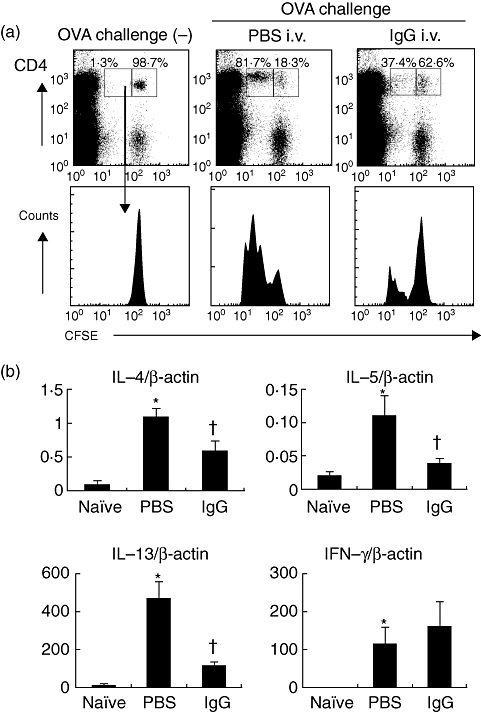
Effects of intravenous (i.v.) immunoglobulin G (IVIgG) on antigen presentation and T helper type 2 (Th2) differentiation. (a) Flow cytometric analysis of antigen presentation to ovalbumin (OVA)-specific CD4+ T cells in vivo is shown. Carboxyfluorescein succinimidyl ester (CFSE)-labelled CD4+ OVA-specific OTII lymphocytes (5 × 106 cells) were transferred into mice challenged with OVA and administered with rabbit IgG or phosphate-buffered saline (PBS). At 24 h after the last challenge, the proliferation of CD4+ T cells in thoracic lymph nodes (TLNs) was analysed. The histogram shows the reduction of CFSE intensity, which indicates cell division and proliferation of the transferred CFSE+ CD4+ cells. One representative experiment of three with similar results is presented. In the inset boxes the percentages of divided (left box) or undivided cells (right box) among CFSE+ CD4+ cells subsets in representative data are shown. (a) The mRNA levels of Th2 cytokines [interleukin (IL)-4, IL-5, IL-13] and Th1 cytokine [interferon (IFN)-γ] in co-culture of OTII CD4+ T cells and lung CD11c+ cells with OVA323–339 peptide are shown. CD4+ cells (2·5 × 105 cells) isolated from spleens of OTII mice using CD4-microbeads and magnetic-activated cell sorting (MACS) system were stimulated with the OVA peptide (5 µg/ml) and lung CD11c+ cells (2·5 × 104 cells) from IgG/PBS-administered mice. After 6 h, mRNA levels of cytokines were evaluated by real-time polymerase chain reaction (PCR) *Significant differences (P < 0·05) versus naive mice. †Significant differences (P < 0·05) versus PBS.
IVIgG inhibits Th2-induced differentiation of OTII CD4+ T cells
To examine the type of T cell proliferation and contribution of CD11c+ APCs, ex vivo antigen presentation was analysed. Co-culture of isolated lung CD11c+ APCs with OVA peptide up-regulated IL-4, IL-5 and IL-13 from OT-II CD4+ T cells. This up-regulation was decreased significantly in the co-culture with lung CD11c+ APCs from mice administered with IVIgG (Fig. 4b). IVIgG did not affect IFN-γ levels significantly (Fig. 4b). These results indicate that IVIgG inhibits the function of lung CD11c+ APCs to induce a Th2 reaction.
IVIgG did not affect airway inflammation in FcγRIIb-deficient mice
To clarify the hypothesis that the target of IVIgG in allergic airway inflammation is inhibitory FcR, the effect on airway inflammation was evaluated in OVA-challenged FcγRIIb-deficient mice. First, in our experimental model, OVA-sensitized FcγRIIb-deficient mice did not develop inflammation spontaneously in lung tissue without antigen challenge. No significant difference in BALF cell counts was seen between WT and FcγRIIb-deficient mice sensitized and challenged with OVA (Fig. 5a). Similarly, histological findings indicated that FcγRIIb-deficient mice developed airway inflammation to the same extent as WT mice (Fig. 5b). However, in FcγRIIb-deficient mice, the effects of IVIgG on the increase of total cells and eosinophils in BALF were not observed (Fig. 5a). Furthermore, histological change such as inflammatory cell infiltration into airways and goblet cell hyperplasia also failed to reveal any apparent effects of IVIgG in FcγRIIb-deficient mice (Fig. 5b).
Fig. 5.
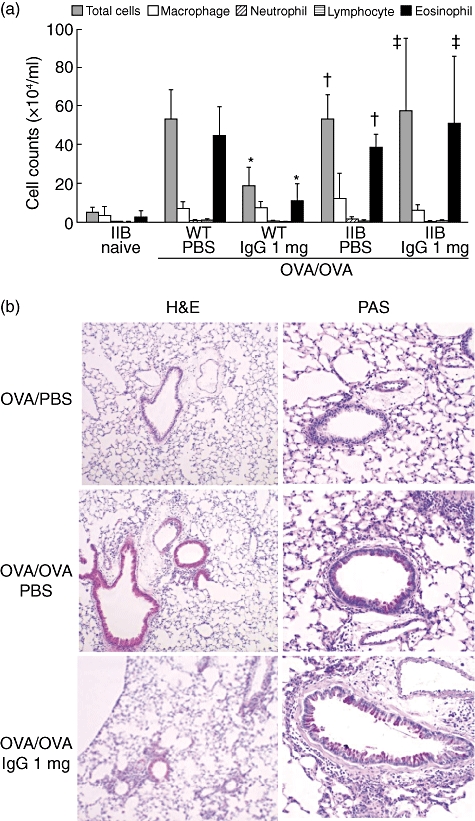
Effects of intravenous (i.v.) immunoglobulin G (IVIgG) on airway inflammation in FcγRIIb-deficient mice. (a) The cellular composition of bronchoalveolar fluid (BALF) in wild-type (WT) and FcγRIIb-deficient mice (IIB) are shown. Sensitized mice received IgG/ phosphate-buffered saline (PBS)-administration and challenged with ovalbumin (OVA). *Significant differences (P < 0·05) versus WT OVA/OVA + PBS, †versus IIB naive, ‡versus WT OVA/OVA + 1 mg IgG. (b) Representative histological findings of haematoxylin and eosin (H&E) or periodic-acid Schiff (PAS)-stained lung sections from FcγRIIb-deficient mice challenged with OVA are shown. Scale bar equals 50 µm.
IgG effects on FcγRIIb-deficient mice were restored by adoptive transfer of wild-type CD11c+ BMDCs
To evaluate the role of FcγRIIb on DCs in allergic airway inflammation, CD11c+ BMDCs were transferred into FcγRIIb-deficient mice. The effects of IVIgG on the increase of total cells and eosinophils in BALF, which were absent in FcγRIIb-deficient mice, were restored by transfer of WT CD11c+ BMDC (Fig. 6). CD11c+ BMDCs from FcγRIIb-deficient mice did not influence cell counts significantly in BALF from PBS- or IgG-administered mice. These findings suggest that the effects of IVIgG on allergic airway inflammation is largely dependent upon FcγRIIb of CD11c+ DCs.
Fig. 6.
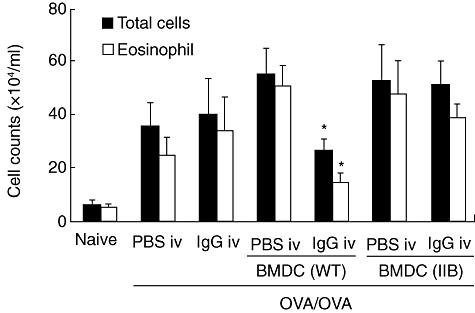
Restoration of intravenous (i.v.) immunoglobulin G (IVIgG) effects on FcγRIIb-deficient mice by adoptive transfer of bone marrow-derived CD11c+ dendritic cells (BMDCs). The total cells and eosinophils in bronchoalveolar fluid (BALF) from FcγRIIb-deficient mice are shown. Sensitized and IgG or phosphate-buffered saline (PBS)-administered mice were transferred BMDCs derived from wild-type mice (WT) or FcγRIIb-deficient mice (IIB) before OVA challenge. *Significant differences (P < 0·05) versus OVA/OVA + BMDC (IIB) + IgG.
Discussion
Here we show for the first time that IgG and its Fc portion can act on inhibitory FcR expressed by DC to attenuate the local Th2 response and following allergic airway inflammation. We have shown the effects of IVIgG to reduce local Th2 cytokine production and subsequent development of eosinophilic inflammation and AHR. These effects were clarified to be dependent upon FcγRIIb, the unique inhibitory FcR for IgG. Our data also demonstrated the inhibitory mechanism through FcRs on CD11c+ APCs in the pathogenesis of allergic airway inflammation. FcγRIIb expressed on immune cells regulates cellular behaviour, such as the proliferation of B cells, phagocytosis by macrophages and degranulation of mast cells [13,19]. In the present study, we focused upon the function of CD11c+ cells and showed that it was regulated negatively via FcγRIIb. Lung CD11c+ cells are APCs, including alveolar macrophages (AMs) and DCs. In the pathogenesis of asthma, CD11c+ DCs are especially potent APCs that have characteristics compatible with myeloid DCs and stimulate Th2 reactions, such as production of IL-4, IL-5, IL-13, resulting AHR and airway eosinophilia. Airway CD11c+ DCs reportedly induce Th2 cell stimulation during ongoing airway inflammation [20]. Lambrecht et al. stated that a Th2 reaction and eosinophilic inflammation were diminished upon CD11c+ cell depletion, showing that CD11c+ myeloid DCs are necessary for the development and continuation of airway inflammation by CD11c+ cells [21]. Meanwhile, pulmonary macrophages stimulate the naive T cell proliferation insufficiently and immunosuppress the APC function of lung DCs in situ[22]. These reports indicate that lung CD11c+ DCs play an important role in antigen presentation to induce a Th2 reaction and exacerbate allergic inflammation. Our results, using transferred BMDCs, emphasize that CD11c+ myeloid DCs play important roles among various types of cells involved in developing allergic inflammation. The effect of promoting Th2 reaction and inflammation was found to be regulated by FcγRIIb in the development of asthmatic features. Additionally, IVIgG exerts its effects on developed allergic inflammation even after OVA challenge, suggesting the therapeutic effects on airway inflammation. Our data also support the Th2 response of antigen-specific CD4+ cells as an important step in the development of asthmatic features.
Previous work in animal models indicates that the development of many human autoimmune diseases might be caused by impairment of the FcR regulatory system [13]. It has been shown that FcγR triggering determination of APC behaviour is an important step in developing a Th2 response and subsequent allergic inflammation. An asthma model using Fc receptor gamma chain (FcRγ)-deficient mice has demonstrated that expression of FcRγ on APCs is important for the development of allergic airway inflammation and AHR [23]. Deletion of FcRγ results in the deficiency of activating-type FcRs, including FcγRI, FcγRIII and FcεRI, which play important roles as activating Fc receptors, but does not affect the expression or inhibitory function of FcγRIIb. Regnault et al. reported that DCs derived from FcRγ-deficient mice failed to mature normally or promote efficient antigen presentation of peptides from exogenous IgG-complexed antigens [24]. Conversely, a recent report has shown the inhibitory mechanisms of FcγRIIb on CD11c+ APCs in allergic airway inflammation [25]. In this study, FcγRIIb on DCs reportedly controls the cellular maturation state. DCs derived from FcγRIIb-deficient mice showed proliferation of antigen-specific T cells in vitro and in vivo[26]. These reports indicate that signalling through both activating and inhibitory FcRs regulates the activity of APCs in the immune system in the pathogenesis of asthma.
In bronchial asthma, IgE and FcεRI are generally considered to be important and logical therapeutic targets. Omalizmab is available as anti-IgE therapy and binds to free IgE; this results in the reduction of FcεRI on mast cells and basophils [27]. It has been reported that cross-linking of FcεRI with FcγRIIb on mast cells and basophils inhibits the degranulation and release of potent inflammatory mediators [19]. In the alum–OVA model used in this study, development of allergic airway inflammation is not dependent upon the existence of B cells or IgE, but instead on CD4+ T cells [28,29]. These facts suggest that allergic airway inflammation with a Th2 response can be regulated by the FcγRIIb-mediated inhibitory pathway on DCs independently of IgE-FcεRI binding. However, there are a few cases of refractory asthma whose pathogenesis seems to be independent of IgE. To modify the function of lung DCs via FcγRIIb might be one of the additional therapeutic strategies in refractory asthma. For the management of bronchial asthma, it is necessary to approach the pathogenesis with sensitivity to multiple allergens. In one possible candidate for treatment of allergic airway inflammation, Sehra et al. showed that specific allergen–IgG interactions repressed inflammatory responses triggered by bystander allergen, thus suggesting that allergen-specific IgG suppress the immune response induced by other allergens [11]. Our results show that IVIgG, which is not specific for allergens, represses DC function and inflammatory responses induced by OVA through Fc portion and FcγRIIb. These results confirm the evidence that IgG, Fc portion and its receptors are potential therapeutic target candidates in the management of bronchial asthma. Manipulation of the pathway optimizes immunotherapeutic strategies by the negative regulatory effect of FcγRIIb [30].
Dharajiya et al. reported that FcγRIIb-deficient mice showed increased BALF cellularity, eosinophilia and mucin content in a mice model upon ragweed extract (RWE) intranasal instillation [25], while our results using OVA inhalation showed no difference between FcγRIIb-deficient mice and WT mice. The difference in the structure or biological properties of challenged allergen or the airway challenge methods might have influenced the consequent asthmatic features. Their experiments analysing Th2 cytokine levels from splenocytes showed that FcγRIIb deficiency did not affect DC function [25]. In our study, isolated lung CD11c+ APCs co-cultured with specific CD4+ T cells and OVA-induced Th2 responses. Moreover, our data showing restoration of IVIgG effects by transfer of WT BMDC suggests that FcγRIIb inhibits DC function to induce the following Th2 response. DCs, which have various cellular states, can influence polarization of T cells depending upon their lineage, maturation status and the local environment they are in. Together, the Th2 response in local asthmatic airway disorders is surmised to be controlled by FcγRIIb on local lung DCs.
In our results, rabbit IgG exerted its effects as IVIgG while the same dose of mouse IgG did not. In conjunction with the results that rabbit IgM or F(ab′)2 did not attenuate the inflammatory cells in BALF, an immune reaction induced by rabbit Fc portion is suggested to exerts its effects via FcγRIIb. A previous report mentioned the inhibitory mechanisms of immune complex and FcγRIIb on CD11c+ DCs [31]. From the above, our results suggest the possibility that generation of the immune complex may exert stronger effects on FcγRIIb of DCs. The dose of mouse IgG used in our experiments was 1 mg/mouse, which is approximately equivalent to 50 mg/kg body weight. In clinical application, IVIG therapy is used at much higher doses, 400–500 mg/kg or more. Our results suggest the possibility that the effects of allogeneic IgG might be exerted in larger doses while rabbit IgG modified CD11c+ cell function and asthmatic responses in other mechanisms. The mechanisms of IVIG have been reported to be involved in Fc receptors; however, formation of the immune complex and its structural and functional differences might influence the effects on immune responses. Further research into the mechanisms of receptors on DCs needs to be conducted.
Although our data represent the function of CD11c+ APCs as DCs, APCs and DCs themselves include a heterogeneous population in peripheral organs such as the lungs. This study clarified that the function of CD11c+ myeloid DCs is important in the regulation of allergic airway inflammation. Another DC subset, the plasmacytoid DCs, induces peripheral tolerance under non-inflammatory conditions in the spleen and lymph nodes [12]. Further studies on DC subsets in the lungs are necessary to distinguish the role of DCs in asthma and design more effective preventative or therapeutic strategies for asthma [12].
Both DCs and FcγR are implicated in the development of allergic airway inflammation in bronchial asthma. FcRs on APCs and DCs and their signalling also play important roles in the development and control of the pathogenesis of asthma. The present report demonstrates that manipulation of the inhibitory FcR pathway is a practical therapeutic means for controlling allergic airway inflammation. Targeting IgG-Fc and FcγRIIb on CD11c+ DC is a promising therapeutic strategy in allergic asthma.
Acknowledgments
We appreciate the advice and expertise of Drs Tetsuya Takagawa and Kentarou Minagawa. We would also like to thank Drs Kazumi Kaneshiro, Haruko Shinke, Emi Kuramoto, Yuko Kono, Akihiro Sakashita, Natsumi Hara, Nobuko Hazeki, Keiko Okuno, Suya Okamoto and Daisuke Tamura for their helpful discussions. This study was supported by KAKENHI (19790557). M. Yoshida was supported, in part, by grants for the Global Center of Excellence (COE) Program ‘Global Center of Excellence for Education and Research on Signal Transduction Medicine in the Coming Generation’ from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, The Mother and Child Health Foundation and the Long-range Research Initiative of Japan Chemical Industry Association.
Disclosure
The authors declare no conflicts of interest.
References
- 1.Wills-Karp M. Immunologic basis of antigen-induced airway hyperresponsiveness. Annu Rev Immunol. 1999;17:255–81. doi: 10.1146/annurev.immunol.17.1.255. [DOI] [PubMed] [Google Scholar]
- 2.Djukanovic R, Wilson SJ, Kraft M, et al. Effects of treatment with anti-immunoglobulin E antibody omalizumab on airway inflammation in allergic asthma. Am J Respir Crit Care Med. 2004;170:583–93. doi: 10.1164/rccm.200312-1651OC. [DOI] [PubMed] [Google Scholar]
- 3.Muller U, Akdis CA, Fricker M, et al. Successful immunotherapy with T-cell epitope peptides of bee venom phospholipase A2 induces specific T-cell anergy in patients allergic to bee venom. J Allergy Clin Immunol. 1998;101:747–54. doi: 10.1016/S0091-6749(98)70402-6. [DOI] [PubMed] [Google Scholar]
- 4.Haque S, Boyce N, Thien FC, O'Hehir RE, Douglass J. Role of intravenous immunoglobulin in severe steroid-dependent asthma. Intern Med J. 2003;33:341–4. doi: 10.1046/j.1445-5994.2003.t01-1-00419.x. [DOI] [PubMed] [Google Scholar]
- 5.Salmun LM, Barlan I, Wolf HM, et al. Effect of intravenous immunoglobulin on steroid consumption in patients with severe asthma: a double-blind, placebo-controlled, randomized trial. J Allergy Clin Immunol. 1999;103:810–15. doi: 10.1016/s0091-6749(99)70424-0. [DOI] [PubMed] [Google Scholar]
- 6.Kishiyama JL, Valacer D, Cunningham-Rundles C, et al. A multicenter, randomized, double-blind, placebo-controlled trial of high-dose intravenous immunoglobulin for oral corticosteroid-dependent asthma. Clin Immunol. 1999;91:126–33. doi: 10.1006/clim.1999.4714. [DOI] [PubMed] [Google Scholar]
- 7.Landwehr LP, Jeppson JD, Katlan MG, et al. Benefits of high-dose i.v. immunoglobulin in patients with severe steroid-dependent asthma. Chest. 1998;114:1349–56. doi: 10.1378/chest.114.5.1349. [DOI] [PubMed] [Google Scholar]
- 8.Nimmerjahn F, Ravetch JV. The antiinflammatory activity of IgG: the intravenous IgG paradox. J Exp Med. 2007;204:11–15. doi: 10.1084/jem.20061788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Samuelsson A, Towers TL, Ravetch JV. Anti-inflammatory activity of IVIG mediated through the inhibitory Fc receptor. Science. 2001;291:484–6. doi: 10.1126/science.291.5503.484. [DOI] [PubMed] [Google Scholar]
- 10.Li N, Zhao M, Hilario-Vargas J, et al. Complete FcRn dependence for intravenous Ig therapy in autoimmune skin blistering diseases. J Clin Invest. 2005;115:3440–50. doi: 10.1172/JCI24394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sehra S, Pynaert G, Tournoy K, et al. Airway IgG counteracts specific and bystander allergen-triggered pulmonary inflammation by a mechanism dependent on Fc gamma R and IFN-gamma. J Immunol. 2003;171:2080–9. doi: 10.4049/jimmunol.171.4.2080. [DOI] [PubMed] [Google Scholar]
- 12.Kuipers H, Lambrecht BN. The interplay of dendritic cells, Th2 cells and regulatory T cells in asthma. Curr Opin Immunol. 2004;16:702–8. doi: 10.1016/j.coi.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 13.Takai T. Roles of Fc receptors in autoimmunity. Nat Rev Immunol. 2002;2:580–92. doi: 10.1038/nri856. [DOI] [PubMed] [Google Scholar]
- 14.Takai T, Ono M, Hikida M, Ohmori H, Ravetch JV. Augmented humoral and anaphylactic responses in Fc gamma RII-deficient mice. Nature. 1996;379:346–9. doi: 10.1038/379346a0. [DOI] [PubMed] [Google Scholar]
- 15.Hamelmann E, Schwarze J, Takeda K, et al. Noninvasive measurement of airway responsiveness in allergic mice using barometric plethysmography. Am J Respir Crit Care Med. 1997;156:766–75. doi: 10.1164/ajrccm.156.3.9606031. [DOI] [PubMed] [Google Scholar]
- 16.Busse PJ, Zhang TF, Srivastava K, Schofield B, Li XM. Effect of ageing on pulmonary inflammation, airway hyperresponsiveness and T and B cell responses in antigen-sensitized and -challenged mice. Clin Exp Allergy. 2007;37:1392–403. doi: 10.1111/j.1365-2222.2007.02775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gonzalez-Juarrero M, Orme IM. Characterization of murine lung dendritic cells infected with Mycobacterium tuberculosis. Infect Immun. 2001;69:1127–33. doi: 10.1128/IAI.69.2.1127-1133.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inaba K, Inaba M, Romani N, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176:1693–702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wigginton SJ, Furtado PB, Armour KL, et al. An immunoglobulin E-reactive chimeric human immunoglobulin G1 anti-idiotype inhibits basophil degranulation through cross-linking of FcepsilonRI with FcgammaRIIb. Clin Exp Allergy. 2008;38:313–19. doi: 10.1111/j.1365-2222.2007.02896.x. [DOI] [PubMed] [Google Scholar]
- 20.van Rijt LS, Lambrecht BN. Dendritic cells in asthma: a function beyond sensitization. Clin Exp Allergy. 2005;35:1125–34. doi: 10.1111/j.1365-2222.2005.02321.x. [DOI] [PubMed] [Google Scholar]
- 21.van Rijt LS, Jung S, Kleinjan A, et al. In vivo depletion of lung CD11c+ dendritic cells during allergen challenge abrogates the characteristic features of asthma. J Exp Med. 2005;201:981–91. doi: 10.1084/jem.20042311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vermaelen K, Pauwels R. Accurate and simple discrimination of mouse pulmonary dendritic cell and macrophage populations by flow cytometry: methodology and new insights. Cytometry A. 2004;61:170–77. doi: 10.1002/cyto.a.20064. [DOI] [PubMed] [Google Scholar]
- 23.Kitamura K, Takeda K, Koya T, et al. Critical role of the Fc receptor gamma-chain on APCs in the development of allergen-induced airway hyperresponsiveness and inflammation. J Immunol. 2007;178:480–8. doi: 10.4049/jimmunol.178.1.480. [DOI] [PubMed] [Google Scholar]
- 24.Regnault A, Lankar D, Lacabanne V, et al. Fcgamma receptor-mediated induction of dendritic cell maturation and major histocompatibility complex class I-restricted antigen presentation after immune complex internalization. J Exp Med. 1999;189:371–80. doi: 10.1084/jem.189.2.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dharajiya N, Vaidya SV, Murai H, et al. FcgammaRIIb inhibits allergic lung inflammation in a murine model of allergic asthma. PLoS ONE. 2010;5:e9337. doi: 10.1371/journal.pone.0009337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kalergis AM, Ravetch JV. Inducing tumor immunity through the selective engagement of activating Fcgamma receptors on dendritic cells. J Exp Med. 2002;195:1653–9. doi: 10.1084/jem.20020338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Prussin C, Griffith DT, Boesel KM, Lin H, Foster B, Casale TB. Omalizumab treatment downregulates dendritic cell FcepsilonRI expression. J Allergy Clin Immunol. 2003;112:1147–54. doi: 10.1016/j.jaci.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 28.Korsgren M, Erjefalt JS, Korsgren O, Sundler F, Persson CG. Allergic eosinophil-rich inflammation develops in lungs and airways of B cell-deficient mice. J Exp Med. 1997;185:885–92. doi: 10.1084/jem.185.5.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.MacLean JA, Sauty A, Luster AD, Drazen JM, De Sanctis GT. Antigen-induced airway hyperresponsiveness, pulmonary eosinophilia, and chemokine expression in B cell-deficient mice. Am J Respir Cell Mol Biol. 1999;20:379–87. doi: 10.1165/ajrcmb.20.3.3291. [DOI] [PubMed] [Google Scholar]
- 30.Nimmerjahn F, Ravetch JV. Fcgamma receptors: old friends and new family members. Immunity. 2006;24:19–28. doi: 10.1016/j.immuni.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 31.Desai DD, Harbers SO, Flores M, et al. Fc gamma receptor IIB on dendritic cells enforces peripheral tolerance by inhibiting effector T cell responses. J Immunol. 2007;178:6217–26. doi: 10.4049/jimmunol.178.10.6217. [DOI] [PubMed] [Google Scholar]