

Abstract
Single immunoglobulin (Ig) interleukin-1R-related molecule (SIGIRR) is an Ig-like membrane protein critical for negative regulation of Toll-like receptor (TLR)-4-mediated signalling. We investigated SIGIRR expression and its regulation mechanism in intestinal epithelial cells (IECs) during inflammation. Endoscopic biopsy specimens were obtained from active and inactive colonic mucosa of ulcerative colitis (UC) patients, then SIGIRR expression was examined using real-time polymerase chain reaction (PCR) and immunohistochemistry (IH). Mice experimental colitis models were established by administrations of sulphonic acid (TNBS) and dextran sodium sulphate (DSS), and epithelial expression of SIGIRR was examined using real-time PCR, IH and flow cytometry. The effects of lipopolysaccharide (LPS) and tumour necrosis factor (TNF)-α on SIGIRR expression were evaluated in vitro using cultured IECs. To elucidate SIGIRR expression regulation in IECs, binding ability of the transcription factor SP1 at the responsive element of the SIGIRR promoter was examined using gel-shift and chromatin immunoprecipitation (ChIP) assays. In human colonic samples, SIGIRR was expressed mainly in IECs at levels significantly higher in inactive compared to active mucosa. In the mice, SIGIRR colonic expression decreased rapidly after colitis development and returned gradually to basal levels. Experimental colitis-mediated down-regulation of SIGIRR in IECs was also confirmed by IH and flow cytometry results. Further, inflammatory conditions induced by TLR ligands and TNF-α caused significant down-regulation of SIGIRR expression in IECs, which was dependent upon decreased SP1 binding at the responsive element of the SIGIRR promoter. We found that SIGIRR is expressed in IECs and serves as a negative regulator to maintain gut innate immunity, which is down-regulated during inflammation by inhibition of an SP1-mediated pathway.
Keywords: intestinal inflammation, negative regulator, SIGIRR, TLR
Introduction
The monolayer of intestinal epithelial cells (IECs) acts as the first line of defence against gut luminal microbial pathogens [1]. IECs can recognize conserved pathogen-associated molecular patterns (PAMPs) via several kinds of pattern recognition receptors (PRRs), including Toll-like receptors (TLRs) [2–6]. TLR signalling in IECs induces innate immune responses, which regulate the gut under physiological and pathological conditions [7,8]. Dysregulation of the intestinal innate immune system leads to various immune-mediated disorders, including human inflammatory bowel diseases (IBD) [9–12]. Studies focused upon the gut innate immune system have been crucial for understanding the pathogenesis of IBD.
TLR-mediated signalling activates a variety of nuclear factor-κB (NF-κB)-related genes, which induce inflammatory responses in various organs [13–17]. Conversely, several negative regulatory mechanisms control TLR-mediated inflammatory responses, and maintain a balance between activation and inhibition of the innate immune system [18,19]. Recently, we demonstrated that up-regulation of intracellular negative regulators, including the zinc-finger protein A20, interleukin (IL)-1-receptor-associated kinase (IRAK)-M and Toll-interacting protein (Tollip), inhibits significantly the transcription of several inflammatory genes in TLR-activated IECs [20]. These findings indicated that intracellular negative regulators of IECs are up-regulated by TLR signalling during gut inflammation to attenuate TLR response in a negative feedback loop.
Apart from intracellular negative regulators, several transmembrane negative regulators of TLR signalling have also been reported [21,22]. Single immunoglobulin (Ig) IL-1-related receptor/Toll IL-1 receptor 8 (SIGIRR/TIR8), one of the transmembrane negative regulators of TLR signalling, has a single extracellular Ig domain and an intracellular TIR domain, and is expressed in a variety of cells such as epithelial cells, dendritic cells, macrophages, fibroblasts and endothelial cells [23–25]. Overexpression of SIGIRR inhibits TLR-induced NF-κB activation and attenuates the production of inflammatory cytokines in vitro[26,27]. In SIGIRR-deficient mice, lipolysaccharide (LPS)-induced inflammatory responses have also been found to be enhanced. These observations suggest that SIGIRR functions as an important negative regulator of TLR-signalling to maintain innate immune conditions in various organs [28,29]. Although the role of SIGIRR has been demonstrated recently in mice experimental colitis models [30,31], little is known regarding the regulation mechanism of SIGIRR expression in IECs during inflammation.
In the present study, we investigated SIGIRR expression in colonic mucosa with or without inflammation, as well as its regulation mechanism in IECs. Our results indicate that SIGIRR is expressed mainly in IECs at levels significantly higher in inactive compared to active mucosa. Furthermore, in vitro results showed that inflammatory conditions induced by LPS and tumour necrosis factor (TNF)-α cause significant down-regulation of SIGIRR expression in IECs, which is dependent upon decreases in binding of the transcription factor SP1 at the responsive element of the SIGIRR promoter. These are the first known results to show that SIGIRR is expressed constitutively in IECs to maintain gut innate immunity and then down-regulated during inflammation by inhibition of an SP1-mediated pathway.
Materials and methods
Reagents and antibodies
Trinitrobenzene sulphonic acid (TNBS; Sigma, St Louis, MO, USA), dextran sodium sulphate (DSS, 5 kDa; Wako Pure Chemicals, Osaka, Japan), ultra-pure Escherichia coli LPS (0111:B4 strain; Invitrogen, Carlsbad, CA, USA), TNF-α (Sigma, St Louis, MO, USA), lipofectamine 2000 (Invitrogen) and human IL-8 enzyme immune assay (EIA) kits (Biosource, Camarillo, CA, USA) were acquired from their respective suppliers. The antibodies used were anti-mouse SIGIRR (R&D Systems, Minneapolis, MN, USA), anti-human SIGIRR (R&D Systems), anti-SIGIRR (Santa Cruz Biotechnology, Santa Cruz, CA, USA), phycoerythrin (PE)-conjugated anti-mouse E-cadherin (R&D Systems), fluorescein isothiocyanate (FITC)-conjugated anti-goat immunoglobulin (Ig)G (BD Biosciences, San Jose, CA, USA) and anti-goat IgG (Santa Cruz Biotechnology).
Patients and colonic specimens
Ten patients with ulcerative colitis (UC; five males, five females; age range: 24–66 years; mean age: 47·4 years) were studied. The diagnosis of UC was based on clinical, endoscopic and histological features of the associated diseases. To evaluate the effect of inflammation on SIGIRR expression in colonic mucosa, multiple endoscopic biopsy specimens were taken from both active and inactive mucosa of each patient. For immunohistochemical assessment of SIGIRR expression in normal colonic mucosa, only tumour-free tissues among the surgical colonic samples obtained from the patients with colon cancer were used. Those samples were snap-frozen immediately in liquid nitrogen and stored at −80°C until the assays. The study protocol was prepared according to the declaration of Helsinki and approved by the ethics committee of Shimane University Faculty of Medicine. Written informed consent was obtained from all subjects prior to beginning the study.
Animals and experimental colitis
We obtained 7-week-old male specific pathogen-free BALB/c mice from Charles Rivers (Yokohama City, Japan). The animals were cared for and handled in accordance with guidelines from the National Institutes of Health and Institute for Animal Experimentation of Shimane University. The animals were housed under constant environmental conditions with circadian light–dark cycles and given an initial adaptation period of 1 week. To evaluate the distribution of SIGIRR expression in the large intestine of normal mice, the large intestine was divided into three portions (proximal, medial and distal portions; Fig. 2a) and the intestinal tissues were used for real-time PCR to detect SIGIRR. For a DSS-induced colitis model, the mice were given 2·5% DSS dissolved in distilled water for 7 days. For a TNBS-induced colitis model, mice were anaesthetized for 90–120 min with an intraperitoneal pentobarbital injection and then given an intrarectal administration of TNBS (100 µl, 125 mg/kg) dissolved in 50% ethanol. After the colitis induction period, the mice were euthanized at each experimental time-point, and the colonic tissues were dissected for real-time PCR and immunohistochemistry assays.
Fig. 2.
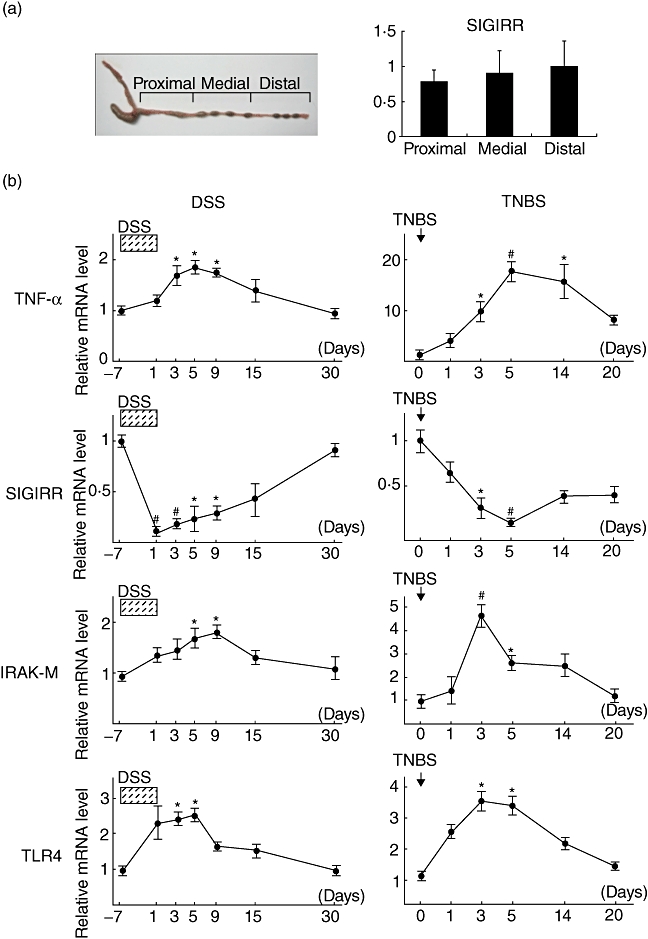
Down-regulation of single immunoglobulin (Ig) interleukin-1R-related molecule (SIGIRR) expression in colonic tissues during mice experimental colitis. (a) Distribution of SIGIRR expression in large intestines of normal mice without colitis induction. (b) Time-dependent mRNA expressions of tumour necrosis factor (TNF)-α, SIGIRR, IL-1-receptor-associated kinase (IRAK)-M and Toll-like receptor (TLR)-4 in colonic tissues from BALB/c mice with dextran sodium sulphate (DSS)- or trinitrobenzene sulphonic acid (TNBS)-induced inflammation. At each experimental time point, the colons were dissected for detection of mRNA by real-time polymerase chain reaction(PCR). Error bars indicate the standard error of mean values obtained independently from four mice. #P < 0·01 versus day 7 (DSS) or day 0 (TNBS).
Real-time PCR
Total RNA was extracted from each sample using Isogen (Nippon Gene, Tokyo, Japan), then equal amounts of RNA were reverse-transcribed into cDNA using a quantitative PCR (QPCR) cDNA kit (Stratagene, La Jolla, CA, USA). All primers (Table 1) utilized were flanked by intron–exon junctions using the NCBI blast tool and Primer3 software. Quantitative real-time PCR was performed using an ABI PRISM 7700 sequence detection system with SYBR Green PCR master mix (Applied Biosystems, Carlsbad, CA, USA), according to the manufacturer's instructions. The levels of SIGIRR, IRAK-M, TLR-4 and TNFα mRNA were normalized to that of β-actin using sequence detector software (Applied Biosystems).
Table 1.
Primer sequences.
| Gene (Accession no.) | Sequences (5′–3′) |
|---|---|
| (Human) | |
| SIGIRR (NM_001135054) | |
| Forward | TCAGTGGCTCTGAACTGCAC |
| Reverse | GTACCAGAGCAGCACGTTGA |
| IRAK-M (NM_007199) | |
| Forward | AGCTGCGGGATCTCCTTAGAG |
| Reverse | ACCGGCCTGCCAAACAG |
| IL-8 (NM_000584) | |
| Forward | TGTGTGTAAACATGACTTCCAAGCT |
| Reverse | TTAGCACTCCTTGGCAAAACTG |
| SP1 (NM_003109) | |
| Forward | CAGGTGCACCCAATTCAAG |
| Reverse | TGCCATACACTTTCCCACAG |
| GAPDH (NM_002046) | |
| Forward | CCACATCGCTCAGACACCAT |
| Reverse | TGACCAGGCGCCCAATA |
| (Mouse) | |
| TNF-α (NM_013693) | |
| Forward | AGACCCTCACACTCAGATCATCTTC |
| Reverse | TCCTCCACTTGGTGGTTTGC |
| SIGIRR (NM_023059) | |
| Forward | AGTCTCAGGTGGGTGGCAGT |
| Reverse | GTCTCGGAGTTCTGGGTGAG |
| IRAK-M (NM_028679) | |
| Forward | GCCAAAGCCATCCAATACTTG |
| Reverse | TGGGTTGGAGCTGGTCATC |
| TLR-4 (NM_021297) | |
| Forward | TCCTGGCTAGGACTCTGATCAT |
| Reverse | TCCAGCCACTGAAGTTCTGA |
| GAPDH (NM_008084) | |
| Forward | ACCCAGAAGACTGTGGATGG |
| Reverse | GGTCCTCAGTGTAGCCCAAG |
GAPDH: glyceraldehyde 3-phosphate dehydrogenase; IL: interleukin; IRAK: IL-1-receptor-associated kinase; SIGIRR: single immunoglobulin (Ig) IL-1R-related molecule; TLR: Toll-like receptor; TNF: tumour necrosis factor.
Immunohistochemistry
Six-mm-thick frozen sections of human biopsy and surgical samples and distal colons from experimental mice were fixed in cold acetone for 10 min. After washing, endogenous peroxidase activity was blocked with 3% H2O2 in water for 10 min at room temperature, followed by incubation with normal blocking serum for 30 min. Subsequently, the sections were incubated for 2 h at room temperature with anti-SIGIRR primary and isotype control antibodies at a 1:250 dilution, then the staining was processed using a commercial immunoperoxidase staining kit (Vectastain Elite ABC Kit; Vector Laboratories, Burlingame, CA, USA), according to the manufacturer's instructions. Sections were counterstained with haematoxylin.
Cell culture
A human colon cancer cell line, SW480, and a human monocytic leukaemia cell line, THP-1, were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA) and grown in RPMI-1640 media (Invitrogen), supplemented with 10% fetal bovine serum (FBS) (ICN Biomedicals, Aurora, OH, USA) and penicillin–streptomycin–amphotericin B (Invitrogen), then maintained at 37°C in 5% CO2 in a humidified incubator.
Flow cytometry
For in vitro studies, SIGIRR expression in SW480 and THP-1 cells was analysed by flow cytometry. In addition, alterations of SIGIRR expression in mice IECs during experimental colitis were also evaluated using flow cytometry. For this experiment, epithelial cells were isolated from the colonic tissues of normal and colitic mice, as described previously. The distal colonic parts dissected from the mice were opened and washed three times in Hanks' balanced salt solution (Gibco BRL, Carlsbad, CA, USA) containing 5% FBS in a sterile condition. Five-millimeter tissue segments were then incubated for 90 min at 37°C in 10 ml of RPMI-1640 containing 10 mg of Dispase (Gibco) in 50-ml centrifuge tubes with gentle rotation. Each digested sample was then passed through a nylon mesh sieve to remove mucus and undigested tissue fragments. Cell surface staining for E-cadherin, a specific maker of epithelial cells, and SIGIRR was evaluated. After staining, the cells were analysed using an EPICS XL (Beckman Coulter, Fullerton, CA, USA), in which 10 000 events (E-cadherin-positive cells) were counted for each condition and analysed using EXPO32™ software. Isotype controls were utilized for all the samples.
RNA interference, transient transfection and NF-κB luciferase assay
We investigated whether SIGIRR expressed in IECs and mononuclear cells functions as a negative regulator of TLR signalling in vitro using an RNA interference technique. SW480 (2·5 × 105 cells/well) and THP-1 (2·5 × 105 cells/well) cell lines were cultured in 24-well plates, then custom siRNAs (Qiagen, Valencia, CA, USA) targeting the human SIGIRR gene were transfected (33 nM/well), according to the manufacturer's protocol. The efficiency of target gene knock-down was assessed by real-time PCR and the results were compared to those of the negative control siRNA-transfected condition. The primers used for SIGIRR gene-specific real-time PCR are shown in Table 1. The functional effect of SIGIRR as a negative regulator of the innate immune system after its expression knock-down in the above cell lines was evaluated by measuring LPS (100 ng/ml)-mediated production of IL-8. IL-8 contents in SW480 and THP-1 cell culture supernatants treated with LPS were measured using IL-8 EIA kits, following the manufacturer's protocol.
The negative regulatory function of SIGIRR is mediated by modulating NF-κB activation upon TLR-ligand stimulation [32]. Based on that finding, we evaluated whether silenced SIGIRR expression has an influence on NF-κB status after stimulation with LPS in our in vitro cell culture system. SW480 cells were plated into 24-well plates at a density of 2 × 105 cells/well, then transfected with human SIGIRR-specific siRNA. After 24 h of transfection with SIGIRR siRNA, the medium was changed and retransfection was performed with an NF-κB reporter vector, pNF-κB-Luc (Stratagene, La Jolla, CA, USA), and an internal control vector, pCMV-renilla-luc (Promega, Madison, WI, USA) at concentrations of 100 and 10 ng/well, respectively, with lipofectamine 2000 reagent (2 µl/well) dissolved into a 50-µl volume of OPTIMEM media (Invitrogen). Following incubation for approximately 12–18 h, the cells were stimulated with LPS containing fresh medium for 12 h, then a dual luciferase assay was performed using the cellular extracts with a dual-luciferase assay kit system (Promega).
Effects of TNF-α and LPS on SIGIRR expression in SW480 and THP-1 cells
We investigated the direct effects of colitis-induced inflammatory mediators on SIGIRR expression. SW480 and THP-1 cells (5 × 104 cells/well) were stimulated with TNF-α (1 ng/ml) or LPS (100 ng/ml), after which the gene expressions of SIGIRR and IL-8 were examined at various time-points using real-time PCR.
Gel shift assay
Electrophoretic mobility shift assays were performed using nuclear extracts from SW480 cells treated with or without LPS at different time-points, as described previously [33]. Briefly, nuclear proteins were extracted in the presence of a protease inhibitor cocktail using a NucBuster protein extraction kit (Novagen, Madison, WI, USA), according to the manufacturer's protocol. An SP1 binding consensus probe, 5′-CAGCAGAGAAGGGGCGGGGCCTAGGTTGGG-3′, was produced to represent a 30-base pairs (bp)-long sequence (nt −77 to nt −48) in the human SP1 promoter region. The oligonucleotide was biotin-labelled with a 3′ end labelling kit (Thermo Fisher Scientific Inc., Rockford, IL, USA) using terminal deoxynucleotidyl transferase (TdT). For the binding reactions, 15 µg of each nuclear protein was incubated in binding buffer [100 mM Tris–HCl (pH 7·5), 500 mM KCl, 10 mM dithiothreitol (DTT), 5 mM MgCl2, 2·5% glycerol and 0·05% NP-40] with 50 ng/µl of poly(dI-dC) and 20 fmol of a biotin end-labelled probe for 30 min at room temperature in a final volume of 20 µl. DNA-protein complexes were then loaded onto 4% non-denaturing polyacrylamide gels. Finally, the chemiluminescent signals were imaged using a nucleic acid detection module (Pierce) according to the manufacturer's protocol.
Chromatin immunoprecipitation (ChIP) assay
Chromatin from SW480 cells treated with or without LPS at different time-points was fixed and immunoprecipitated using a ChIP assay kit (Upstate Biotechnology, Lake Placid, NY, USA). Sequences were identified for the human SIGIRR promoter and primers constructed, as follows: primer 1 (nt −158 to nt −139) CCCATCTTCACTTCCTGCAT and primer 2 (nt −18 to nt −36) GGTAAGGAGGAGGGAGCAG. Purified chromatin was immunoprecipitated using 10 µg of anti-SP1 (Santa Cruz Biotechnology) and eluted DNA fragments were purified to serve as templates. The input fraction corresponded to 0·1% of the chromatin solution before immunoprecipitation. The average size of the sonicated DNA fragments subjected to immunoprecipitation was 500 bp as determined by ethidium bromide gel electrophoresis. Polymerase chain reaction primers used for the SP1 ChIP assays produced a 242-bp fragment.
Statistical analysis
All data are expressed as the mean ± standard error of the mean (s.e.m.). Values were analysed using a Mann–Whitney U-test with Stat-View 4·0 software (Abacus Concepts Inc., Berkeley, CA, USA). For comparison of multiple values, analysis of variance (anova) was used. P-values less than 0·05 were considered significant.
Results
Down-regulation of SIGIRR expression in epithelial cells during colonic inflammation in UC patients
To verify the influence of inflammation on SIGIRR expression in colonic mucosa, we obtained endoscopic biopsy specimens from both active and inactive mucosa of each UC patient. The expressions of SIGIRR and IRAK-M were examined using real-time PCR. Relative levels of SIGIRR expression were significantly lower in active compared to inactive mucosa (Fig. 1a, P < 0·05 versus inactive). In contrast, IRAK-M expression was up-regulated significantly in active mucosa (Fig. 1a, P < 0·05 versus inactive). In addition, immunohistochemical analysis was performed for the detection of SIGIRR expression in histological sections. Obtained images showed clearly abundant expression of SIGIRR in both normal (Fig. 1b,i) and non-inflamed colonic tissue sections (Fig. 1b,ii), with immunoreactive signals detected mainly in epithelial cells, which were significantly higher than those in active mucosa (Fig. 1b,iii). These findings suggest that SIGIRR is expressed constitutively mainly in colonic epithelial cells and down-regulated during inflammation. Immunohistochemical findings also indicated that the mononuclear cells expressed SIGIRR (Fig. 1b,iv–vi),
Fig. 1.
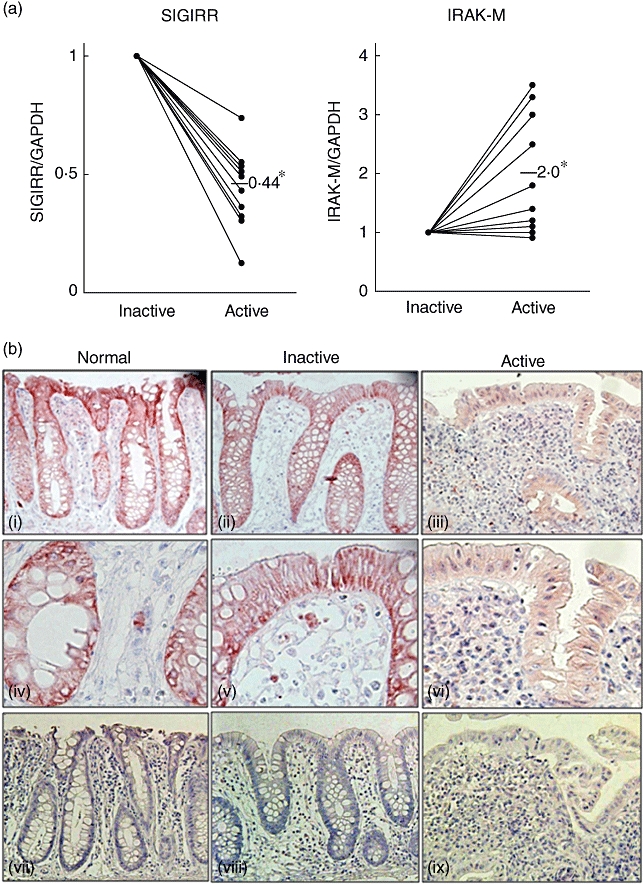
Down-regulation of single immunoglobulin (Ig) interleukin (IL)-1R-related molecule (SIGIRR) expression in human inflamed colonic mucosa. Endoscopic biopsy specimens were obtained from active and inactive mucosa of ulcerative colitis (UC) patients (n = 10). (a) The expressions of SIGIRR and IL-1-receptor-associated kinase (IRAK)-M were examined using real-time polymerase chain reaction (PCR). *P < 0·05 versus Inactive. (b) Representative immunohistochemistry results showing the expression of SIGIRR in colonic epithelial cells (vii–ix: negative controls).
Down-regulation of SIGIRR expression in colonic tissues during mice experimental colitis
To further confirm down-regulation of epithelial expression of SIGIRR during colonic inflammation, we established mice experimental colitis models and time-dependent alterations of SIGIRR expression in colonic tissues were investigated. Before performing the experiments with the colitis models, we examined the distribution of SIGIRR expression in large intestines of normal mice without colitis induction. Although SIGIRR was expressed constitutively in the large intestines, there was no difference in expression level among the various portions (Fig. 2a). Next, we used distal segments of experimental mice colons and assessed the time-course changes of colonic expressions of TNF-α, SIGIRR, IRAK-M and TLR-4 using real-time PCR (Fig. 2b). The expressions of TNF-α, IRAK-M and TLR-4 were increased significantly after DSS colitis development and gradually returned to basal levels after a period of up to 30 days. In contrast, SIGIRR expression was decreased significantly after DSS colitis development and gradually returned to basal levels after a period of up to 30 days. In addition, we observed similar time-course changes of those gene expressions in colonic tissues obtained from mice with TNBS colitis. These findings confirmed the results obtained in the experiment using human biopsy samples.
Down-regulation of SIGIRR expression in colonic epithelial cells during mice experimental colitis
Because SIGIRR expression was identified mainly in colonic epithelial cells in the human biopsy tissue sections, we confirmed further the localization of SIGIRR expression in mice colonic tissues with or without DSS colitis. Immunohistochemistry findings showed significant expression levels of SIGIRR in colonic epithelial cells of normal mice, which were clearly decreased after DSS colitis induction (day 3; Fig. 3a). In addition, we investigated SIGIRR expression in epithelial cells isolated from normal and DSS colitis mice using flow cytometry, which revealed significant down-regulation of SIGIRR expression in isolated colonic epithelial cells from mice with DSS colitis (Fig. 3b and c).
Fig. 3.
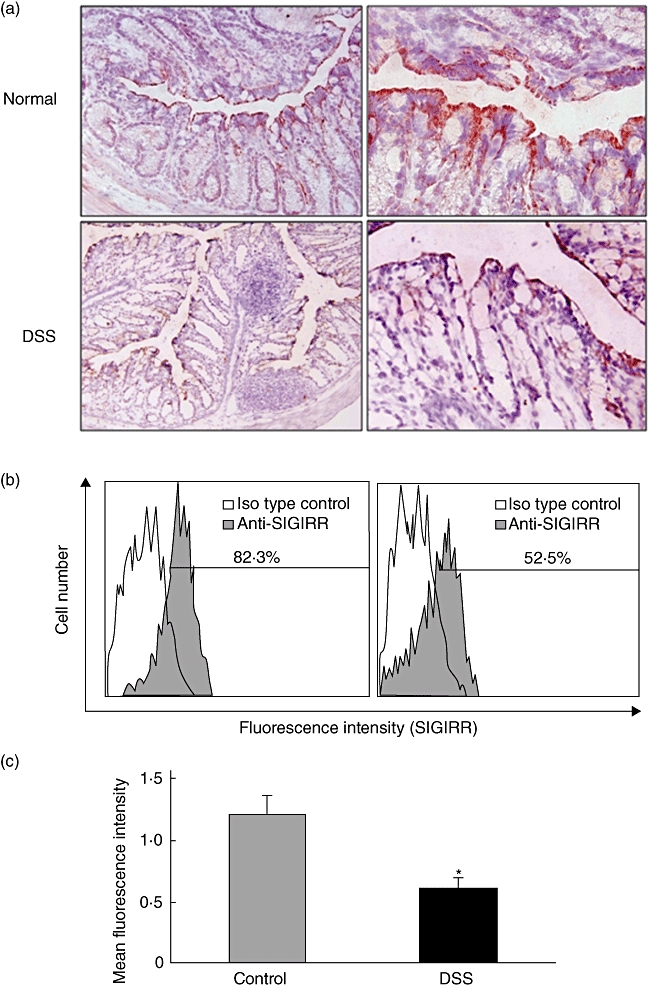
Down-regulation of single immunoglobulin (Ig) interleukin (IL)-1R-related molecule (SIGIRR) expression in colonic epithelial cells during dextran sodium sulphate (DSS)-induced colitis. (a) Representative immunohistochemistry images showing SIGIRR expression in colonic epithelial cells from normal and DSS-induced colitis mice. (b) Representative results of flow cytometry showing the expression of SIGIRR in mice colonic epithelial cells. Colonic epithelial cells were isolated from normal and DSS-induced colitis mice as described in the Materials and methods section. The fluorescence levels of cell surface SIGIRR in E-cadherin-positive cells was evaluated using flow cytometry. (c) The fluorescence levels of SIGIRR were analysed using mean fluorescence intensity (MFI). Error bars indicate the standard error of mean values obtained independently from four mice. *P < 0·05 versus Control.
SIGIRR expressed in colonic epithelial cells functions as a negative regulator in TLR-4 signalling
We also examined whether SIGIRR functions as a negative regulator in LPS-stimulated SW480 colonic epithelial cells. A monocyte-like immune-reactive cell line, THP-1, was employed to evaluate SIGIRR-mediated negative regulatory functions efficiently following LPS treatment. Constitutive high levels of expression of SIGIRR in SW480 and THP-1 cells were confirmed by flow cytometry (Fig. 4a). To clarify the role of SIGIRR in negative regulation of TLR-4 signalling in cultured colonic epithelial cells, we used a gene knock-down method with siRNA targeting SIGIRR. Cells were transfected with SIGIRR siRNA or control siRNA, then the efficiency of target gene knock-down was evaluated, which showed inhibition of SIGIRR expression by approximately 70–80% compared to the negative control siRNA (Fig. 4b). After confirming siRNA efficacy, the transfected cell lines were treated with or without LPS for 24 h, after which the IL-8 contents in culture media were examined by EIA. LPS-mediated IL-8 production in the targeted siRNA-treated cells was significantly greater than that in the control siRNA-treated cells (Fig. 4b), indicating that SIGIRR expressed in colonic epithelial cells functions as a negative regulator of TLR-4 signalling.
Fig. 4.
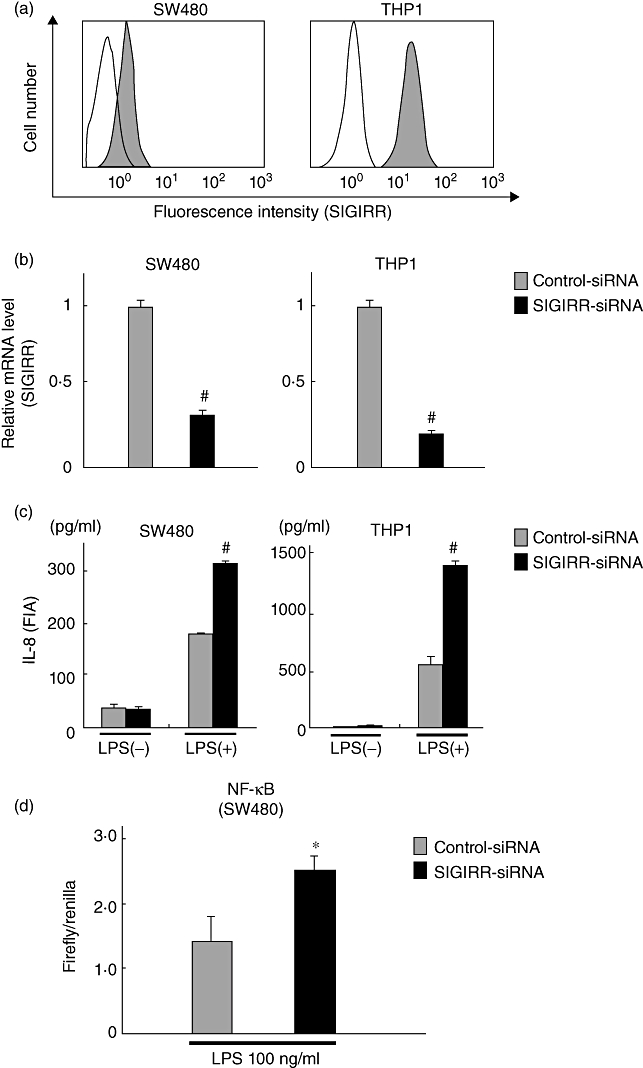
Functional role of single immunoglobulin (Ig) interleukin (IL)-1R-related molecule (SIGIRR) in Toll-like receptor (TLR)-4 signalling in colonic epithelial cells. (a) Representative results of flow cytometry showing expression of SIGIRR in SW480 and human monocytic leukaemia cell line (THP-1) cells. (b) Efficiency of SIGIRR siRNA for target gene expression knock-down in SW480 and THP-1 cells, as evaluated by real-time polymerase chain reaction (PCR). The level of SIGIRR expression in each of the samples was normalized by β-actin and compared to that of the negative control siRNA. Error bars indicate the standard error of mean values obtained from four independent experiments. #P < 0·01 versus negative control siRNA. (c) SIGIRR in colonic epithelial cells functions as a negative regulator in TLR-4 signalling. Cells were transfected with SIGIRR or control siRNAs, then cultured with or without lipopolysaccharide (LPS) (100 ng/ml) for 24 h, after which IL-8 contents in culture media were examined by enzyme immune assay (EIA). Error bars indicate the standard error of mean values obtained from four independent experiments. #P < 0·01 versus control siRNA. (d) Evaluation of nuclear factor (NF)-κB activities in SIGIRR siRNA-transfected SW480 cells after treatment with lipopolysaccharide (LPS). Firefly and renilla luciferase assays were performed with each sample, and the respective ratios were compared to that of the negative control siRNA. Error bars indicate the standard error of mean values obtained from four independent experiments. *P < 0·05 versus control siRNA.
Because NF-κB serves as one of the potent transcription factor to control IL-8 gene expression in mammalian systems, we evaluated whether silenced SIGIRR expression has an influence on the status of NF-κB in our in vitro cell culture system. To achieve this, SW480 cells were first transfected with a human SIGIRR-specific siRNA. After attaining considerable inhibition of SIGIRR gene expression, a co-transfection system was utilized, in which the same cell lines were retransfected with an NF-κB luciferase reporter construct together with an internal control renilla luciferase vector to monitor the NF-κB activity in SIGIRR siRNA transfected cells following stimulation with LPS. As shown in Fig. 4d, knock-down of SIGIRR expression by the target siRNA enhanced NF-κB activity significantly compared to that of the cells treated with the negative control siRNA when stimulated with LPS. These findings confirmed the notion that blocking of SIGIRR induces IL-8 expression in LPS-treated SW480 cells via NF-κB.
Down-regulation of SIGIRR expression in colonic epithelial cells treated with TNF-α and LPS
Results of both human and mice in vivo studies have demonstrated decreased expression of SIGIRR during colonic inflammation. Thus, we hypothesized that colitis-induced inflammatory mediators including TNF-α and LPS down-regulate SIGIRR expression directly in colonic epithelial cells, SW480. Stimulation with TNF-α and LPS clearly induced mRNA expression of the proinflammatory cytokine IL-8 in cultured SW480 cells, whereas SIGIRR expression was decreased significantly when the cells were exposed to those ligands (Fig. 5a). Similar effects of TNF-α and LPS on SIGIRR and IL-8 expression in non-epithelial THP-1 cells were also observed (Fig. 5b).
Fig. 5.
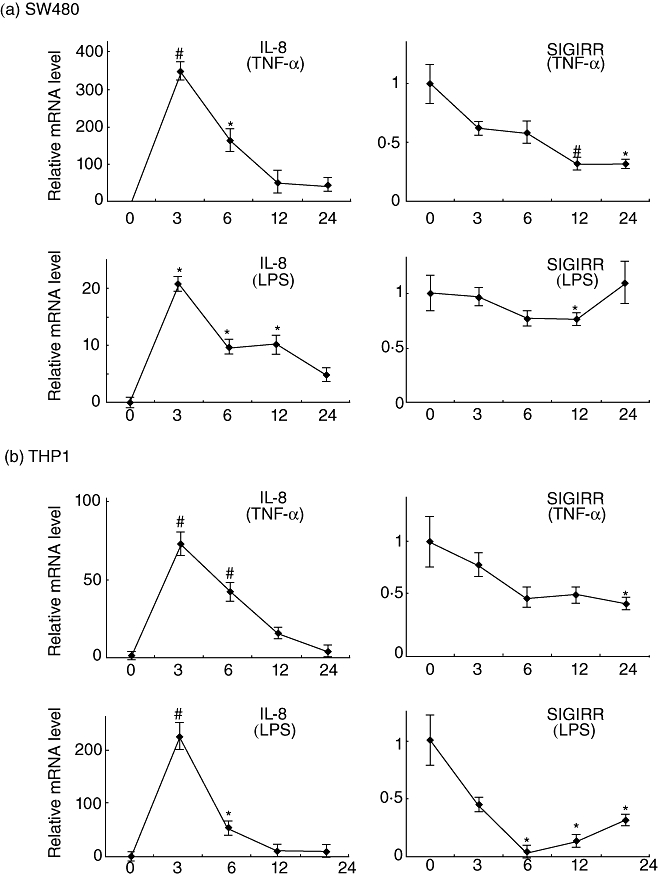
Down-regulation of single immunoglobulin (Ig) interleukin (IL)-1R-related molecule (SIGIRR) expression in colonic epithelial cells exposed to tumour necrosis factor (TNF)-α and lipopolysaccharide (LPS). SW480 (a) and human monocytic leukaemia cell line (THP-1) (b) cells (5 × 104 cells/well) were stimulated with TNF-α (1 ng/ml) or LPS (100 ng/ml), after which the gene expressions of SIGIRR and IL-8 at various time-points were examined using real-time polymerase chain reaction (PCR). Error bars indicate the standard error of mean values obtained from three independent experiments. #P < 0·01, *P < 0·05 versus non-stimulated cells.
Decreased binding of SP1 at the SIGIRR promoter in LPS-treated SW480 cells
Next, we examined the regulatory portion of the SIGIRR gene in a series of in vitro experiments. SP1, a member of a large family of zinc finger proteins, was one of the first transcription factors identified in mammalian cells [34,35]. Thereafter, the SP1 gene was shown to be expressed ubiquitously in a wide variety of mammalian cells, suggesting that mammalian cells require it as a promoter of essential genes [36]. Interestingly, we noted the presence of an SP1 binding site at the proximal end of the human SIGIRR promoter in a search conducted with transcription factor binding site software (http://www.cbrc.jp/research/db/TFSEARCH.html). To reveal whether SP1 regulates SIGIRR activity, we treated SW480 cells with various doses of the widely used SP1-specific DNA binding inhibitor mithramycin A and the expression of SIGIRR was examined using real-time PCR. As shown in Fig. 6a, SW480 cells treated with mithramycin A greatly diminished the endogenous expression of SIGIRR in a dose-dependent manner. After confirming the essential role of SP1 in SIGIRR transcription, we evaluated the status of SP1 expression in SW480 cells treated with LPS. The expression of SP1 in SW480 cells was not affected by LPS treatment (Fig. 6b), therefore we examined its binding to SIGIRR promoter consensus sites in the presence of LPS. For this experiment, SW480 cells were treated with LPS and nuclear extracts were prepared at different time-points, followed by electrophoretic mobility shift assay (EMSA). As shown in Fig. 5c, SW480 cells treated with LPS qualitatively reduced the binding of SP1 at the SIGIRR promoter in a time-dependent manner. For quantitative confirmation of SP1 binding ability to the SIGIRR promoter, chromatin immunoprecipitation was performed, which clearly showed inhibition of the binding of SP1 at the SIGIRR promoter consensus site when treated with LPS at different time-points (Fig. 6d). Taken together, these results demonstrate clearly that decreased SP1 binding to the SIGIRR promoter in LPS-treated SW480 cells may modulate the expression of SIGIRR under inflammatory conditions.
Fig. 6.
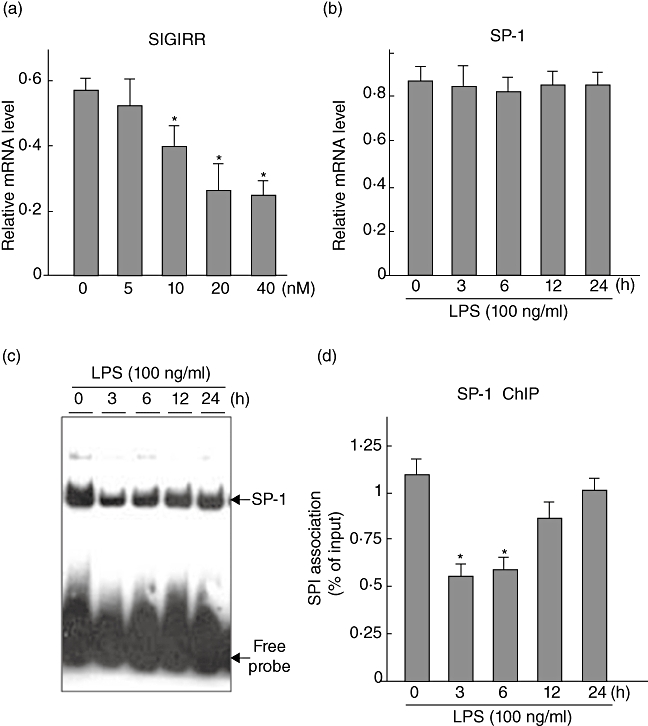
Effects of lipopolysaccharide (LPS) on SP1-mediated single immunoglobulin (Ig) interleukin (IL)-1R-related molecule (SIGIRR) expression in SW480 cells. (a) Dose-dependent effects of the SP1 inhibitor mithramycin A on endogenous SIGIRR expression in SW480 cells. SW480 cells were plated into six-well plates and stimulated with different doses of mithramycin A for 6 h, after which real-time polymerase chain reaction (PCR) assays of the inhibition of endogenous SIGIRR-expression were performed. *P < 0·05 versus mithramycin A (−). Error bars indicate the standard error of mean values obtained from three independent experiments. (b) Time-course changes of SP1 expression in LPS-treated (100 ng/ml) SW480-cells were examined using real-time PCR. Error bars indicate the standard error of mean values obtained from three independent experiments. (c) SP1 binding in the SIGIRR regulatory portion. SW480-cells were plated into 10-cm plates and then stimulated with LPS (100 ng/ml) for different time-periods, after which gel-shift-assays of nuclear extracts were performed using SP1-specific consensus probes. Representative blots obtained from three independent experiments are shown. (d) Quantitative binding of SP1 in the SIGIRR specific regulatory portion, as revealed by chromatin immunoprecipitation (ChIP) assay findings. Following stimulation with LPS (100 ng/ml), SW480 cells were subjected to ChIP assays as described in the Materials and methods section. Each ChIP assay was coupled to real-time PCR using immunoprecipitated chromatin samples. The results are expressed as a percentage of SP1 in immunoprecipitated samples compared to the respective input controls. *P < 0·05 versus LPS (−). Error bars indicate the standard error of mean values obtained from three independent experiments.
Discussion
In the present study, we identified down-regulation of SIGIRR expression in epithelial cells during intestinal inflammation. Our in vitro results demonstrated clearly that treatment with LPS or TNF-α decreased SIGIRR expression in IECs significantly, which was dependent upon the decreased binding efficiency of SP1 at the proximal end of the SIGIRR promoter. These are the first known results to show the mechanism of regulation of SIGIRR in IECs during inflammation.
In light of findings from several human and mouse studies, SIGIRR is known to be expressed prominently in a tissue-specific manner in various organs, such as the kidneys, lungs and intestines [7]. Furthermore, intestinal expression of SIGIRR and its role have been demonstrated recently in mice experimental colitis models. For example, Garlanda et al. reported that SIGIRR-deficient mice developed intestinal inflammation in response to DSS administration as well as increased susceptibility to azoxymethane-induced experimental carcinogenesis [30]. In a study that utilized conditional SIGIRR-deficient mice, Xiao et al. also noted copious amounts of SIGIRR in mouse colonic tissues, which was shown to play a crucial role in intestinal homeostasis and inflammation, as well as colitis-associated tumorigenesis by maintaining the microbial tolerance of the colonic epithelium [31]. Although those findings indicate an essential importance for SIGIRR in the gut innate immune system, alterations of SIGIRR expression and its regulation under gut inflammatory conditions have not been reported.
To investigate the influence of intestinal inflammation on SIGIRR expression, we initially used inflamed and non-inflamed colonic biopsy samples obtained from UC patients. We found that SIGIRR was expressed constitutively in colonic tissues and localized mainly in epithelial cells, and down-regulated during inflammation. Furthermore, a colonic inflammation-induced decrease in SIGIRR expression was confirmed clearly in experiments that utilized mice colitis models. In contrast to the expression of SIGIRR, that of IRAK-M, an intracellular negative regulator, was up-regulated significantly in inflamed colonic epithelial cells. Our previous study also found that expressions of intracellular negative regulators, including A20 and Tollip, were clearly induced by intestinal inflammation as well as in vitro TLR-ligation in colonic epithelial cells [20]. Together, these findings indicate that SIGIRR expression and its role may be different from those of inducible intracellular negative regulators in the gut innate immune system.
Various negative regulators control TLR-mediated cellular signalling to maintain a balance between activation and inhibition of the innate immune system [20–23]. Some of these are present constitutively to control TLR activation at a physiological level, whereas others are up-regulated by TLR signalling during inflammation to attenuate TLR response in a negative feedback loop [26,27]. Activation of TLR signalling by commensal microflora is required for homeostasis of the gut epithelium, while mice deficient in TLR signalling have dysregulated intestinal homeostasis, causing them to be more sensitive to DSS-induced injury [37–39]. Conversely, excessive commensal bacteria-induced TLR-mediated signalling in the colon is also detrimental. In this regard, TLR activation is a double-edged sword, and negative regulation of TLR signalling may be required to avoid detrimental and inappropriate inflammatory responses, suggesting that there is a fine balance between pro- and anti-inflammatory signals in the colonic epithelium, which may critically involve SIGIRR. Our findings demonstrated clearly that over-expression of TLR and proinflammatory cytokines during colonic inflammation exacerbates colitis due to an altered expression of SIGIRR. Furthermore, constitutive expression of SIGIRR in surface epithelium may regulate the gut innate immune balance under physiological conditions, whereas intracellular negative regulators may be induced after breakdown of the first-line defence system that is regulated by SIGIRR.
Next, we employed an in vitro system to evaluate SIGIRR expression and functions in colon epithelial cells treated with LPS and TNF-α. In this regard, we initially examined SIGIRR expression in the colon epithelial cell line SW480, and then confirmed the responsiveness of those cells to LPS and TNF-α in terms of elevated production of the proinflammatory cytokine IL-8. Interestingly, in concordance with our in vivo findings, we noted decreased expression of SIGIRR from the basal level when cultured epithelial cells were exposed to LPS and TNF-α. Our findings of decreased expression of SIGIRR following LPS stimulation strongly support previous reports, in which SIGIRR expression was reduced dramatically by LPS in a time-dependent manner in both in vivo and in vitro conditions [7]. Next, to delineate the role of SIGIRR in innate immune response, we knocked-down SIGIRR expression in IECs and observed that inflammation was deteriorated by an elevated production of IL-8 when the cells were treated with LPS. These findings indicate clearly that decreased expression of SIGIRR amplifies colonic inflammation by dysregulated innate immunity.
Because SIGIRR-deficient mice show enhanced sensitivity to DSS colitis, due to constitutive up-regulation of inflammatory genes and increased inflammatory responses such as interferon (IFN)-γ and IL-6 [31], a major question is whether SIGIRR expression in haematopoietic cells rather than IECs may be important. Indeed, higher expression of SIGIRR in haematopoietic cells and its effects are very important for homeostatic maintenance of innate-immune responses in various organs [27–29]. However, in the intestines, a monolayer of epithelial cells is exposed to the gut luminal environment and various microbial pathogens are present. Thus, it is reasonable to speculate that epithelial cell functions are regulated by TLR signalling and that SIGIRR expressed in IECs may contribute to restore intestinal immune balance. In particular, a distinct NF-κB-mediated immune mechanism is essential for protecting barrier integrity and eliminating invading microorganisms, thus maintaining tolerance and homeostatic balance efficiently in the intestinal mucosa. Our present in vivo findings demonstrated ubiquitous expression of SIGIRR in colonic epithelia of both normal and experimental colitic animals. Others have also demonstrated extremely elevated SIGIRR expression in intestinal and kidney epithelial cells [29]. In the present study, we also confirmed negative regulatory functions of SIGIRR in LPS-mediated SW480 cells in vitro. Based on these findings, we focused upon the effects of SIGIRR on epithelial cells during intestinal inflammation. Although we observed similar kinetics of SIGIRR expression in both IECs and THP-1 cells in our in vitro experiments, we did not elucidate fully the different comparable roles of SIGIRR that vary based upon cell type in mouse colitis models. Further studies using cell type-specific SIGIRR conditional knock-out mice may provide additional insight to compare the effects of SIGIRR on the regulation of TLR-mediated immune responses in epithelial as well as haematopoietic cells during intestinal inflammation.
Although LPS- and TNF-α-dependent decreased expression of SIGIRR has been noted in several studies, the underlying mechanism of that down-regulation has not been established. Upon recognizing pathogen products, appropriate immune responses are elicited by altering gene expression mainly through the activation of NF-κB and activator protein 1 (AP-1) [36,39]. However, in a transcription factor-binding database search [40], we did not find NF-κB and AP-1, but rather the presence of the ubiquitous transcription factor SP1 at binding sites of the proximal promoter for SIGIRR. SP1 regulates the expression of a vast number of genes that are involved in many cellular functions, including differentiation, proliferation and apoptosis [41,42]. Critical roles for SP1 in the innate immune system have also been noted in recent reports [43,44]. The SP1 transcription factor contains a zinc finger protein motif, by which it binds directly to DNA and enhances gene transcription [45]. Although our results did not demonstrate altered expression of SP1, they revealed decreased binding efficiency at the proximal end of the SIGIRR promoter. In other studies, altered binding of SP1 at its consensus site has been found with a number of physiological conditions. In addition, the DNA-binding activity of SP1 showed a dramatic age-dependent decline in vitro that was reversed by incubation with high concentrations of DTT [45]. Moreover, the binding affinity and transcriptional specificity of SP1 can be altered by interactions with other co-factors in the binding sites near the SP1 recognition motif, such as CCAAT/enhancer-binding proteins (C/EBP), NF-κB, AP-1 and AP-2 [45]. We did not detect any changes in SP1 expression following LPS stimulation in our experimental systems. However, its activity is finely modulated by a variety of stimuli through multiple post-translational modification [46] Therefore, further investigation of SP1-regulated SIGIRR transcriptional activity by focusing upon its post-translational modification via phosphorylation or sumoylation via various pathways including mitogen-activated protein (MAP) kinases may provide additional insight into its transcriptional functions. We performed the same set of experiments utilizing TNF-α as used to investigate the LPS-mediated effects of SP1 in SW480 cells (Fig. 6), although no significant effects on SP1 binding to the SIGIRR promoter with TNF-α treatment were seen in SW480 cells (data not shown). Nevertheless, the present results provide additional clues for understanding the binding of SP1 to the SIGIRR promoter, which may regulate the expression of SIGIRR under inflammatory conditions.
In summary, we investigated SIGIRR expression in colonic mucosa with or without inflammation, as well as its regulation mechanism in IECs. SIGIRR was found to be expressed mainly in IECs and down-regulated during inflammation by inhibition of an SP1-mediated pathway. Our findings provide new insights into SIGIRR-mediated negative regulation of TLR signalling, which maintains the innate immune system in the gut.
Acknowledgments
This work was supported in part by Health and Labour Sciences Research Grants for research on intractable diseases from the Ministry of Health, Labour and Welfare of Japan.
Disclosure
The authors declare that they have no conflict of interest related to the publication of this manuscript.
References
- 1.Monteleone I, Vavassori P, Biancone L, Monteleone G, Pallone F. Immunoregulation in the gut: success and failures in human disease. Gut. 2002;50:11160–4. doi: 10.1136/gut.50.suppl_3.iii60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cario E, Rosenberg IM, Brandwein SL, Beck PL, Reinecker HC, Podolsky DK. Lipopolysaccharide activates distinct signaling pathways in intestinal epithelial cell lines expressing Toll-like receptors. J Immunol. 2000;164:966–72. doi: 10.4049/jimmunol.164.2.966. [DOI] [PubMed] [Google Scholar]
- 3.Cario E. Bacterial interactions with cells of the intestinal mucosa: Toll-like receptors and NOD2. Gut. 2005;54:1182–93. doi: 10.1136/gut.2004.062794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hausmann M, Kiessling S, Mestermann S, et al. Toll-like receptors 2 and 4 are up-regulated during intestinal inflammation. Gastroenterology. 2002;122:1987–2000. doi: 10.1053/gast.2002.33662. [DOI] [PubMed] [Google Scholar]
- 5.Ortega-Cava CF, Ishihara S, Rumi MA, et al. Strategic compartmentalization of Toll-like receptor 4 in the mouse gut. J Immunol. 2003;170:3977–85. doi: 10.4049/jimmunol.170.8.3977. [DOI] [PubMed] [Google Scholar]
- 6.Ishihara S, Rumi MA, Kadowaki Y, et al. Essential role of MD-2 in TLR4-dependent signaling during Helicobacter pylori-associated gastritis. J Immunol. 2004;173:1406–16. doi: 10.4049/jimmunol.173.2.1406. [DOI] [PubMed] [Google Scholar]
- 7.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by Toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–41. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 8.Abreu MT, Fukata M, Arditi M. TLR signaling in the gut in health and disease. J Immunol. 2005;174:4453–60. doi: 10.4049/jimmunol.174.8.4453. [DOI] [PubMed] [Google Scholar]
- 9.Mizoguchi A, Mizoguchi E. Inflammatory bowel disease, past, present and future: lessons from animal models. J Gastroenterol. 2008;43:1–17. doi: 10.1007/s00535-007-2111-3. [DOI] [PubMed] [Google Scholar]
- 10.Uematsu S, Akira S. Immune responses of TLR5(+) lamina propria dendritic cells in enterobacterial infection. J Gastroenterol. 2009;44:803–11. doi: 10.1007/s00535-009-0094-y. [DOI] [PubMed] [Google Scholar]
- 11.Ishihara S, Aziz MM, Yuki T, Kazumori H, Kinoshita Y. Inflammatory bowel disease: review from the aspect of genetics. J Gastroenterol. 2009;44:1097–108. doi: 10.1007/s00535-009-0141-8. [DOI] [PubMed] [Google Scholar]
- 12.Mayer L. Evolving paradigms in the pathogenesis of IBD. J Gastroenterol. 2010;45:9–16. doi: 10.1007/s00535-009-0138-3. [DOI] [PubMed] [Google Scholar]
- 13.Ishihara S, Rumi MA, Ortega-Cava CF, et al. Therapeutic targeting of Toll-like receptors in gastrointestinal inflammation. Curr Pharm Des. 2006;12:4215–28. doi: 10.2174/138161206778743448. [DOI] [PubMed] [Google Scholar]
- 14.Horng T, Barton GM, Flavell RA, Medzhitov R. The adaptor molecule TIRAP provides signalling specificity for Toll-like receptors. Nature. 2002;420:329–33. doi: 10.1038/nature01180. [DOI] [PubMed] [Google Scholar]
- 15.O'Neill LA, Fitzgerald KA, Bowie AG. The Toll-IL-1 receptor adaptor family grows to five members. Trends Immunol. 2003;24:286–90. doi: 10.1016/s1471-4906(03)00115-7. [DOI] [PubMed] [Google Scholar]
- 16.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 17.Takeda K, Akira S. TLR signaling pathways. Semin Immunol. 2004;16:3–9. doi: 10.1016/j.smim.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 18.Liew FY, Xu D, Brint EK, O'Neill LA. Negative regulation of toll-like receptor-mediated immune responses. Nat Rev Immunol. 2005;5:446–58. doi: 10.1038/nri1630. [DOI] [PubMed] [Google Scholar]
- 19.Shibolet O, Podolsky DK. TLRs in the Gut. IV. Negative regulation of Toll-like receptors and intestinal homeostasis: addition by subtraction. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1469–73. doi: 10.1152/ajpgi.00531.2006. [DOI] [PubMed] [Google Scholar]
- 20.Oshima N, Ishihara S, Rumi MA, et al. A20 is an early responding negative regulator of Toll-like receptor 5 signalling in intestinal epithelial cells during inflammation. Clin Exp Immunol. 2010;159:185–98. doi: 10.1111/j.1365-2249.2009.04048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brint EK, Xu D, Liu H, et al. ST2 is an inhibitor of interleukin 1 receptor and Toll-like receptor 4 signaling and maintains endotoxin tolerance. Nat Immunol. 2004;5:373–9. doi: 10.1038/ni1050. [DOI] [PubMed] [Google Scholar]
- 22.Divanovic S, Trompette A, Atabani SF, et al. Negative regulation of Toll-like receptor 4 signaling by the Toll-like receptor homolog RP105. Nat Immunol. 2005;6:571–8. doi: 10.1038/ni1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thomassen E, Renshaw BR, Sims JE. Identification and characterization of SIGIRR, a molecule representing a novel subtype of the IL-1R superfamily. Cytokine. 1999;11:389–99. doi: 10.1006/cyto.1998.0452. [DOI] [PubMed] [Google Scholar]
- 24.Mantovani A, Locati M, Polentarutti N, Vecchi A, Garlanda C. Extracellular and intracellular decoys in the tuning of inflammatory cytokines and Toll-like receptors: the new entry TIR8/SIGIRR. J Leukoc Biol. 2004;75:738–42. doi: 10.1189/jlb.1003473. [DOI] [PubMed] [Google Scholar]
- 25.Polentarutti N, Rol GP, Muzio M, et al. Unique pattern of expression and inhibition of IL-1 signaling by the IL-1 receptor family member TIR8/SIGIRR. Eur Cytokine Netw. 2003;14:211–18. [PubMed] [Google Scholar]
- 26.Qin J, Qian Y, Yao J, Grace C, Li X. SIGIRR inhibits interleukin-1 receptor- and Toll-like receptor 4-mediated signaling through different mechanisms. J Biol Chem. 2005;280:25233–41. doi: 10.1074/jbc.M501363200. [DOI] [PubMed] [Google Scholar]
- 27.Li X, Qin J. Modulation of Toll-interleukin 1 receptor mediated signaling. J Mol Med. 2005;83:258–66. doi: 10.1007/s00109-004-0622-4. [DOI] [PubMed] [Google Scholar]
- 28.Boraschi D, Tagliabue A. The interleukin-1 receptor family. Vitam Horm. 2006;74:229–54. doi: 10.1016/S0083-6729(06)74009-2. [DOI] [PubMed] [Google Scholar]
- 29.Lech M, Garlanda C, Mantovani A, Kirschning CJ, Schlöndorff D, Anders HJ. Different roles of TiR8/Sigirr on toll-like receptor signaling in intrarenal antigen-presenting cells and tubular epithelial cells. Kidney Int. 2007;72:182–92. doi: 10.1038/sj.ki.5002293. [DOI] [PubMed] [Google Scholar]
- 30.Garlanda C, Riva F, Veliz T, et al. Increased susceptibility to colitis-associated cancer of mice lacking TIR8, an inhibitory member of the interleukin-1 receptor family. Cancer Res. 2007;67:6017–21. doi: 10.1158/0008-5472.CAN-07-0560. [DOI] [PubMed] [Google Scholar]
- 31.Xiao H, Gulen MF, Qin J, et al. The Toll-interleukin-1 receptor member SIGIRR regulates colonic epithelial homeostasis, inflammation, and tumorigenesis. Immunity. 2007;26:461–75. doi: 10.1016/j.immuni.2007.02.012. [DOI] [PubMed] [Google Scholar]
- 32.Khan MA, Steiner TS, Sham HP, et al. The single IgG IL-1-related receptor controls TLR responses in differentiated human intestinal epithelial cells. J Immunol. 2010;184:2305–13. doi: 10.4049/jimmunol.0900021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aziz MM, Ishihara S, Rumi MA, et al. Prolactin induces MFG-E8 production in macrophages via transcription factor C/EBPbeta-dependent pathway. Apoptosis. 2008;13:609–20. doi: 10.1007/s10495-008-0201-1. [DOI] [PubMed] [Google Scholar]
- 34.Dynan WS, Tjian R. Isolation of transcription factors that discriminate between different promoters recognized by RNA polymerase II. Cell. 1983;32:669–80. doi: 10.1016/0092-8674(83)90053-3. [DOI] [PubMed] [Google Scholar]
- 35.Suske G. The Sp-family of transcription factors. Gene. 1999;238:291–300. doi: 10.1016/s0378-1119(99)00357-1. [DOI] [PubMed] [Google Scholar]
- 36.Wald D, Qin J, Zhao Z, et al. SIGIRR, a negative regulator of Toll-like receptor-interleukin 1 receptor signaling. Nat Immunol. 2003;4:920–7. doi: 10.1038/ni968. [DOI] [PubMed] [Google Scholar]
- 37.Rakoff-Nahoum S, Medzhitov R. Role of the innate immune system and host-commensal mutualism. Curr Top Microbiol Immunol. 2006;308:1–18. doi: 10.1007/3-540-30657-9_1. [DOI] [PubMed] [Google Scholar]
- 38.Rakoff-Nahoum S, Medzhitov R. Innate immune recognition of the indigenous microbial flora. Mucosal Immunol. 2008;1:S10–14. doi: 10.1038/mi.2008.49. [DOI] [PubMed] [Google Scholar]
- 39.Latchman DS. Transcription factors: an overview. Int J Biochem Cell Biol. 1997;29:1305–12. doi: 10.1016/s1357-2725(97)00085-x. [DOI] [PubMed] [Google Scholar]
- 40.Lu S, Archer MC. Sp1 coordinately regulates de novo lipogenesis and proliferation in cancer cells. Int J Cancer. 2010;126:416–25. doi: 10.1002/ijc.24761. [DOI] [PubMed] [Google Scholar]
- 41.Deniaud E, Baguet J, Chalard R, et al. Overexpression of transcription factor Sp1 leads to gene expression perturbations and cell cycle inhibition. PLoS ONE. 2009;4:e7035. doi: 10.1371/journal.pone.0007035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tone M, Tone Y, Babik JM, Lin CY, Waldmann H. The role of Sp1 and NF-κB in regulating CD40 gene expression. J Biol Chem. 2002;277:8890–7. doi: 10.1074/jbc.M109889200. [DOI] [PubMed] [Google Scholar]
- 43.Chiang BT, Liu YW, Chen BK, Wang JM, Chang WC. Direct interaction of C/EBPdelta and Sp1 at the GC-enriched promoter region synergizes the IL-10 gene transcription in mouse macrophage. J Biomed Sci. 2006;13:621–35. doi: 10.1007/s11373-006-9101-y. [DOI] [PubMed] [Google Scholar]
- 44.Ammendola R, Mesuraca M, Russo T, Cimino F. The DNA-binding efficiency of Sp1 is affected by redox changes. Eur J Biochem. 1994;22:483–9. doi: 10.1111/j.1432-1033.1994.t01-1-00483.x. [DOI] [PubMed] [Google Scholar]
- 45.Gingras ME, Masson-Gadais B, Zaniolo K, et al. Differential binding of the transcription factors Sp1, AP-1, and NFI to the promoter of the human alpha5 integrin gene dictates its transcriptional activity. Invest Ophthalmol Vis Sci. 2009;50:57–67. doi: 10.1167/iovs.08-2059. [DOI] [PubMed] [Google Scholar]
- 46.Brightbill HD, Plevy SE, Modlin RL, Smale ST. A prominent role for Sp1 during lipopolysaccharide-mediated induction of the IL-10 promoter in macrophages. J Immunol. 2000;164:1940–51. doi: 10.4049/jimmunol.164.4.1940. [DOI] [PubMed] [Google Scholar]