

Abstract
The debate of life on Mars centers around the source of the globular, micrometer-sized mineral carbonates in the ALH84001 meteorite; consequently, the identification of Martian processes that form carbonates is critical. This paper reports a previously undescribed carbonate formation process that occurs on Earth and, likely, on Mars. We identified micrometer-sized carbonates in terrestrial aerosols that possess excess 17O (0.4–3.9‰). The unique O-isotopic composition mechanistically describes the atmospheric heterogeneous chemical reaction on aerosol surfaces. Concomitant laboratory experiments define the transfer of ozone isotopic anomaly to carbonates via hydrogen peroxide formation when O3 reacts with surface adsorbed water. This previously unidentified chemical reaction scenario provides an explanation for production of the isotopically anomalous carbonates found in the SNC (shergottites, nakhlaites, chassignites) Martian meteorites and terrestrial atmospheric carbonates. The anomalous hydrogen peroxide formed on the aerosol surfaces may transfer its O-isotopic signature to the water reservoir, thus producing mass independently fractionated secondary mineral evaporites. The formation of peroxide via heterogeneous chemistry on aerosol surfaces also reveals a previously undescribed oxidative process of utility in understanding ozone and oxygen chemistry, both on Mars and Earth.
Keywords: nanoparticles, mineral dust, heterogeneous chemical transformation, surface chemistry, mass-independent fractionation
The search for life beyond Earth is pursued based upon the requirement of liquid water both as a solvent and transport medium, and its biochemically unique role in providing support to cell structure (1). Central to the question of potential life on Mars is whether liquid water ever existed on the surface of Mars and, if so, under what climatic conditions (2). Apart from satellite observations of valleys and channels formed by aqueous activity, there are few quantitative measures (e.g., Thermal and Electrical Conductivity probe on Phoenix Lander; Compact Reconnaissance Imaging Spectrometer for Mars; High Energy Neutron Detector on board Mars Odyssey spacecraft) of significant amounts of liquid water at the surface of Mars today (3–5). Secondary minerals such as carbonates and sulfates record physical-chemical settings of the environment in which they are formed and geochemical relations between the atmosphere and the Martian surface (6). Unlike terrestrial carbonate sediments, the Martian surface lacks large amounts of carbonates despite a CO2-rich (95% vol/vol) atmosphere, though, there is evidence of Mg-Fe rich carbonates (16–34 wt %) in the Columbia Hills and < 5% CaCO3 in Martian dust and soils (3, 7–9). In contrast to most Martian meteorites, ALH84001 contained substantial amounts of secondary carbonate minerals with an average age of 3.90 ± 0.04 billion years, contemporaneous with a wet period in Martian history (10). The formation of well-defined globular structures of a definite size range and their arrangement in linear chains was argued as being relics of early biological activity on Mars (11, 12). Unambiguous identification of bacterially induced precipitation of carbonate minerals remains elusive; therefore, the identification of previously undescribed carbonate chemical formation mechanisms is important, especially if these measurements can be linked to the atmosphere in a quantitative way.
Recent observations at the Phoenix landing site indicated the presence of water as ice in residual polar caps, adsorbed water in high latitude regoliths, and 3–5 wt % carbonates associated with surficial and airborne silica-rich dust particles and moderately alkaline pH of 7.7 ± 0.5 consistent with a carbonate-buffered solution (2, 3, 13). These conditions could facilitate heterogeneous chemical transformations on soils and aerosols, thus enabling the adsorbed CO2 in the surface layers to react with oxides such as CaO, MgO, and FeO to produce coatings of carbonates on the particle surfaces (14). This process is known to occur on Earth, both in the atmosphere and on various surfaces during controlled laboratory experiments (15–18). Therefore, a detailed study of carbonate surficial mineral formation combined with C- and O-isotopic measurements can provide previously undescribed details on the history of water on the surface of Mars.
Isotope measurements δ18O* of terrestrial carbonate sediments have been extensively used to measure ocean temperature, salinity, atmospheric CO2 concentration, and sea water pH (19, 20). On a triple isotope plot (δ17O vs δ18O) terrestrial carbonate sediments are expected to obey a mass-dependent fractionation relation (δ17O ∼ 0.52δ18O) as a result of kinetic and equilibrium processes involved in the precipitation of carbonates in aqueous solutions that depend on the relative mass difference of oxygen isotopes (21, 22). Extraterrestrial carbonates from SNC (shergottites, nakhlaites, chassignites) Martian meteorites, however, possess excess 17O† (0.7–1.1‰), and this anomaly was attributed to photochemical fractionation processes on Mars (23, 24) that are similar to those known to occur in the Earth’s upper atmosphere (25). Mechanistically understanding the origin of the O-isotopic anomaly of these carbonates will lead to a better understanding of the interaction between the Martian atmosphere, lithosphere, and hydrosphere. Studies of mass independent fractionation processes of O isotopes have been used to understand the reaction mechanism between CO2 and O3 via excited oxygen atoms (26–29) and have been used to infer biological productivity on decadal to millennium time scales (30, 31). The O3 molecule is unique as it possesses unusually high isotopic enrichments (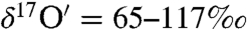 and
and 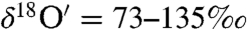 ) compared to any other atmospheric oxygen-carrying species and exhibits an oxygen isotope anomaly (17O of stratospheric O3 = 25–39‰ and Δ17O of tropospheric O3 = 18–39‰) (32, 33) that can be transferred to products of ozone oxidation, thus making it a sensitive tracer of atmospheric chemical reaction pathways. Measurements have revealed that the positive Δ17O of ozone is transferred to other oxygen-carrying species such as NO3, SO4, H2O2, and stratospheric CO2 (34, 35).
) compared to any other atmospheric oxygen-carrying species and exhibits an oxygen isotope anomaly (17O of stratospheric O3 = 25–39‰ and Δ17O of tropospheric O3 = 18–39‰) (32, 33) that can be transferred to products of ozone oxidation, thus making it a sensitive tracer of atmospheric chemical reaction pathways. Measurements have revealed that the positive Δ17O of ozone is transferred to other oxygen-carrying species such as NO3, SO4, H2O2, and stratospheric CO2 (34, 35).
From a combination of natural samples (aerosols and soils) and laboratory experiments, we show that the triple oxygen isotopic composition of atmospheric carbonates can be used to resolve chemical processes occurring at interfaces (solid–liquid–gas) in the presence of myriads of organic and inorganic species. Aerosol samples (coarse > 1 μm and fine < 1 μm) were collected weekly in La Jolla, California, and reacted with 100% H3PO4 to release CO2 (36). To obtain independent measurement of O isotopes, purified CO2 gas was fluorinated to produce oxygen. Controlled laboratory experiments indicated that CO2 produced during this reaction is strictly from inorganic carbonates. The presence of an O-isotopic anomaly (Δ17O) in terrestrial atmospheric carbonates has been observed, and its potential to uniquely resolve the processes occurring on mineral aerosol surfaces has been investigated. Our laboratory experiments furnish a previously undescribed level of understanding of Δ17O (0.7–1.1‰) observed in Martian carbonates and the activity of surface adsorbed water.
Results
Atmospheric Carbonates.
The carbonate fractions of aerosol particles have excess 17O ranging from 0.4 to 3.9‰ (Fig. 1). The coarse fraction of atmospheric carbonates possesses higher oxygen isotope anomalies of 3.9‰ (Δ17O) at Scripps Institute of Oceanography (SIO); 3.5‰ at Mt. Soledad (MSD); 2.9‰ at the Biochemistry building (BCH), University of California, San Diego; and 0.8‰ at the White Mountain Research Station (WMRS)‡ compared to the corresponding fine aerosol fraction [1‰ (SIO), 2‰ (MSD), 2.9‰ (BCH) to 0.5‰ (WMRS)]. The carbonate concentration in the coarse aerosol fraction was observed to be higher and varied from 1.83 to 6.47 μg m-3 (SI Appendix, Table S1) compared to the fine fraction (1.83–4.38 μg m-3).
Fig. 1.

Oxygen isotope anomaly (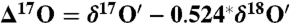 ) in the coarse (solid blue square) and fine (open blue square) fraction of atmospheric carbonates collected in La Jolla, California. For comparison isotopically normal carbonate standards (red triangle) and soil carbonates (green star) are also shown. Here
) in the coarse (solid blue square) and fine (open blue square) fraction of atmospheric carbonates collected in La Jolla, California. For comparison isotopically normal carbonate standards (red triangle) and soil carbonates (green star) are also shown. Here 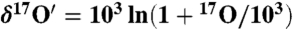 and
and 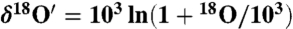 .
.
The oxygen isotopic composition of carbonates in soil samples was measured to distinguish atmospheric carbonate chemistry from other equilibrium and kinetic isotope processes associated with pedogenic and sedimentary carbonate formation. The carbonate content of measured soils varied from 0.03% in Grand Canyon Red soil to a maximum of 4.21% in Black Rock Desert dust, Nevada, and 4.42% in soil sample obtained from YaDan GanSu, China (SI Appendix, Table S2). The triple oxygen isotopic composition of soil and commercial cement sample (carbonate content 3%) were mass dependently fractionated (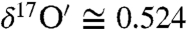
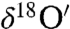 with Δ17O = -0.1 to -0.2‰) and enriched in both 17O and 18O (
with Δ17O = -0.1 to -0.2‰) and enriched in both 17O and 18O (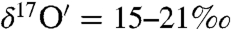 and
and 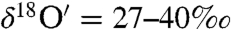 ) compared to the atmospheric carbonates with
) compared to the atmospheric carbonates with 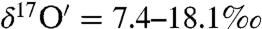 and
and 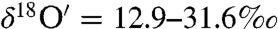 .
.
Experimental Resolution of Production of Oxygen Isotope Anomaly in Surficial Carbonates.
To determine the origin of the observed oxygen isotopic anomaly in atmospheric carbonates, pure calcite powder and ozone were used to study the kinetics and mechanism of isotope exchange reactions at aerosol surfaces. Two experiments were carried out at ambient room temperature (25 ± 2 °C). In one set of experiments, predried (100 °C for 72 h) calcite powder (3.2 ± 0.02 mg) of known isotopic composition (Δ17O = 0) was placed at the bottom of a glass reaction tube and degassed at 10-5 mT for 24 h. Following the admission of 27 ± 2 μmoles of ozone to the reaction vessel, the mixture was allowed to react for 30 and 60 min. CO2 was released from calcite by acid digestion and collected for oxygen isotopic analysis. No isotope exchange was observed in the product CO2 during these experiments. In a second set of experiments, ozone (27 ± 2 μmoles) was allowed to react with CaCO3 in the presence of water (55.5 ± 0.2 μmole) for 30, 60, and 1,320 min. The isotopic composition of CO2 released by acid digestion was measured at each time step to estimate the rate of isotopic exchange. The results clearly indicated that the isotopic anomaly from ozone was transferred to calcite (Δ17O = 1.6, 2.4, 2.8‰) through surface adsorbed water (SI Appendix, Fig. S1a). The isotopic enrichment increased at a rate of 0.07‰ and 0.06‰ min-1 for 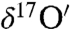 and
and 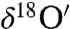 during the initial 1 h and subsequently attained a plateau (
during the initial 1 h and subsequently attained a plateau (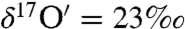 and
and 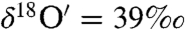 ). The steady-state value is presumably controlled by the competition among rates of adsorption, heterogeneous reaction and desorption of ozone, and its decomposition product. Experiments with varying O3 concentrations (0, 25 ± 2, 50 ± 2 μmoles) at fixed amounts of CaCO3 = 3.2 ± 0.02 mg; H2O = 55.5 ± 0.2 μmole and constant time (30 min) were conducted. The isotopic enrichment in calcite increased by 1‰ with a 10-μmole increase in ozone concentration, resulting in a concomitant increase in the oxygen isotope anomaly from 1.2‰ and 1.98‰ at 25 and 50 μmoles of ozone (SI Appendix, Fig. S1b; Table S3). In a third set of experiments, ozone and water were reacted for 11 and 72 h. After complete removal of oxygen gas from water frozen at -60 °C by vacuum pumping, the water sample was thawed and treated with KMnO4 at pH 1 (37). Oxygen produced from this reaction is derived exclusively from hydrogen peroxide (H2O2) produced during ozone–water interaction. As depicted in Fig. 2, oxygen gas produced from the decomposition of H2O2 is anomalous (Δ17O = 2.5 and 11‰ after 11 and 72 h) and enriched compared to the initial Millipore water (
). The steady-state value is presumably controlled by the competition among rates of adsorption, heterogeneous reaction and desorption of ozone, and its decomposition product. Experiments with varying O3 concentrations (0, 25 ± 2, 50 ± 2 μmoles) at fixed amounts of CaCO3 = 3.2 ± 0.02 mg; H2O = 55.5 ± 0.2 μmole and constant time (30 min) were conducted. The isotopic enrichment in calcite increased by 1‰ with a 10-μmole increase in ozone concentration, resulting in a concomitant increase in the oxygen isotope anomaly from 1.2‰ and 1.98‰ at 25 and 50 μmoles of ozone (SI Appendix, Fig. S1b; Table S3). In a third set of experiments, ozone and water were reacted for 11 and 72 h. After complete removal of oxygen gas from water frozen at -60 °C by vacuum pumping, the water sample was thawed and treated with KMnO4 at pH 1 (37). Oxygen produced from this reaction is derived exclusively from hydrogen peroxide (H2O2) produced during ozone–water interaction. As depicted in Fig. 2, oxygen gas produced from the decomposition of H2O2 is anomalous (Δ17O = 2.5 and 11‰ after 11 and 72 h) and enriched compared to the initial Millipore water (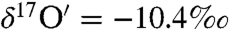 ,
, 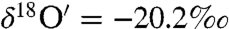 ). The isotopic exchange between water and ozone proceeded much faster on the sea sand (S25-3, Fisher Scientific) as the enrichment observed in the peroxide (
). The isotopic exchange between water and ozone proceeded much faster on the sea sand (S25-3, Fisher Scientific) as the enrichment observed in the peroxide (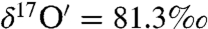 and
and 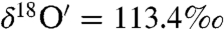 , Δ17O = 22.3‰) is highest after 28 h. The low enrichment in the oxygen derived from H2O2 (
, Δ17O = 22.3‰) is highest after 28 h. The low enrichment in the oxygen derived from H2O2 (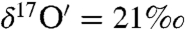 and
and 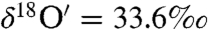 , Δ17O = 3.5‰) after 47 h of interaction between surface adsorbed water on CaO and ozone may be due to additional isotopic exchange reaction of O atoms with the CaO molecules or possibly the result of incomplete recovery of the adsorbed water (< 80%) and peroxide from the CaO aerosol surface.
, Δ17O = 3.5‰) after 47 h of interaction between surface adsorbed water on CaO and ozone may be due to additional isotopic exchange reaction of O atoms with the CaO molecules or possibly the result of incomplete recovery of the adsorbed water (< 80%) and peroxide from the CaO aerosol surface.
Fig. 2.

Oxygen isotope anomaly in peroxide (open square) formed during ozone (300 ± 10 μ mole) and water (83 ± 3 μ mole) interaction (A) Δ17O = 2.5% after 11 h, (B) interaction of ozone with adsorbed water on aerosol surface (CaO) for 47 h produced peroxide with Δ17O = 3.5%, (C) Δ17O = 11.06% after 72 h of reaction with ozone and water, and (D) ozone and water isotope exchange in the presence of sea sand with Δ17O = 22.3% after 28 h. The isotopic composition of the initial ozone (solid square) and Millipore water are shown with a closed symbol (triangle).
To investigate the mechanism of in situ carbonate formation on aerosol surfaces, a slurry of CaO was coated on a Teflon tube and ambient air was pumped at a flow rate of 2 sccm for ∼16 h. The coating was removed, dried at 100 °C overnight, and acid digested to study the oxygen isotopic composition of in situ formed carbonates (net reaction = CaO + H2CO3 → CaCO3 + H2O). The measurements revealed the oxygen isotopic composition of in situ formed carbonates was mass dependent with 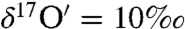 and
and 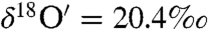 . Static experiments with ozone (300 ± 2 μmole), water (8.3 ± 0.02 mmole), CO2 (243 ± 0.2 μmole), and CaO (121 mg) revealed the O-isotopic composition of in situ formed carbonates was enriched (
. Static experiments with ozone (300 ± 2 μmole), water (8.3 ± 0.02 mmole), CO2 (243 ± 0.2 μmole), and CaO (121 mg) revealed the O-isotopic composition of in situ formed carbonates was enriched (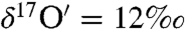 and
and 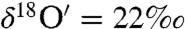 ), and anomalous (Δ17O = 0.6‰).
), and anomalous (Δ17O = 0.6‰).
Discussion
Atmospheric carbonates are anomalously enriched in 17O (0.4–3.9‰) compared to soil carbonates (-0.1 to -0.2‰) and the oxygen isotopic anomaly is aerosol size-dependent (Fig. 1) being higher in the coarse (≥1 μm) compared to the fine fraction of aerosols (≤ 1 μm). The carbonate concentration is higher (∼twice) in the coarse fractions of aerosols except SIO250507 (SI Appendix, Table S1). The higher carbonate concentrations in the coarse fraction of aerosols is expected as mineral dust particles from continental sources and sea spray are typically observed in the 1- to 10-μm range (38, 39). Mineral oxides, which serve as precursors for in situ carbonate formation, are also concentrated in the coarse aerosol fraction. Both primary and secondary mineral carbonates provide reactive surfaces for interactions with ozone in the presence of chemisorbed water, leading to the formation of isotopically anomalous hydrogen peroxide. This may explain the higher oxygen isotopic anomaly found in the coarse aerosol fraction. Future work on the size dependency of O-isotopic anomaly of atmospheric carbonates will be important in this regard.
The laboratory experiments suggest that the incorporation of the oxygen isotopic anomaly from ozone to the carbonates can occur via two different pathways (Fig. 3). Mechanism A involves the isotopic exchange reactions on preexisting carbonates, whereas mechanism B involves in situ formation of carbonates on mineral particles. In both cases, the interaction of ozone with surface adsorbed water is required to produce the carbonate O-isotopic anomaly. Atmospheric relative humidity levels between 20–90% (∼5–20 Torr water vapor pressure in the atmosphere) promote the formation of thin water films on particle surfaces (40). In the case of metal oxides (CaO, MgO, and Fe2O3) and carbonates (CaCO3 and MgCO3), dissociation of water molecules at the interfaces is energetically favorable because hydroxylated surfaces are more stable than metal-terminated surfaces under ambient conditions (15, 41, 42). Recent experiments and molecular dynamic simulations have shown the free energy of adsorption of water on calcite surfaces (43, 44) to be as low as -10.6 kcal mol-1.
Fig. 3.

The molecular mechanisms proposed to explain the origin of the oxygen isotopic anomaly in atmospheric carbonates. (A). Ozone isotope exchange on existing carbonate aerosols with dissociative adsorption of water and peroxide formation. (B). In situ formation of carbonates and interaction with ozone on particle surfaces. The red circles indicate ozone isotope exchange reaction and probable hydrogen peroxide formation sites. The abbreviations on the side bar are S, solid (MO and MCO3) such as CaO, MgO, and Fe2O3, CaCO3, MgCO3; l, liquid or adsorbed-water film; G, gas phase.
Mechanism (A) initiates with the dissociative adsorption reaction of water on mineral surfaces (1–4) and the formation of a surface complex (M(OH)(HCO3))sc. The presence of carbonic acid has been confirmed in numerous other studies on the CaCO3 surface in the presence of H2O vapor (41, 45, 46). When an O3 molecule collides with the aerosol surface, it may scatter back into the gas phase or adsorb across the hydration layer depending on the hydration shell thickness (Fig. 3). The uptake of ozone at the hydrated calcite surface is a multistep process consisting of (i) gas phase diffusion of the O3 molecules to the liquid interface, (ii) transfer across the interface, (iii) diffusion and reaction with water in the condensed phase, and (iv) diffusion out and desorption of the residual reaction products. Isotopically anomalous hydrogen peroxide formation via dissociation of O3 (1a–1c) transfers the isotopic signature in the hydration layer, which in turn transfers the isotopic anomaly to the carbonates (2–4). In the reaction scheme below, M = Ca, Mg, K, Fe, etc., and Q = 17O and 18O.
| [1a] |
| [1b] |
| [1c] |
| [2] |
| [3] |
| [4] |
Net Reaction:
The uptake of ozone into the liquid film is enhanced by more than an order of magnitude in the presence of ions in the solution compared to pure water (47), and the residence time of O3 is estimated to be ∼50 ps in aqueous salt aerosol surfaces (48). The initial interaction of ozone with adsorbed-water molecules is ascribed to the polarity of the ozone molecule (0.53D), which results from its four resonance states (49). The enrichment in peroxide (Fig. 2) formed during the reaction increased with 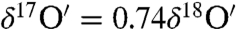 and a concomitant rise in the oxygen isotopic anomaly (2.5 to 22‰). Though the formation of a peroxide intermediate has been suggested during the decomposition of ozone on the MnO catalyst (50) based on the low activation energy (6.2 kJ·mole-1), it has been difficult to directly demonstrate its existence. We have identified peroxide on the aerosol surface and confirmed the role of ozone–water O-isotopic exchange. The formation of peroxide depends on the ozone concentration as increased ozone concentration (25 to 50 μmoles) produced higher enrichment in the carbonates (product CO2) and a higher oxygen isotopic anomaly (SI Appendix, Fig. S1b). Hydrogen peroxide formation may also depend on the number of adsorbed-water molecules; however, further experiments are required to verify the dependence of peroxide concentration on the chemisorbed water on aerosol surfaces. The enrichment in peroxide depends on the chemical composition of aerosol surfaces as the maximum enrichment and anomaly was observed on quartz rich sand.
and a concomitant rise in the oxygen isotopic anomaly (2.5 to 22‰). Though the formation of a peroxide intermediate has been suggested during the decomposition of ozone on the MnO catalyst (50) based on the low activation energy (6.2 kJ·mole-1), it has been difficult to directly demonstrate its existence. We have identified peroxide on the aerosol surface and confirmed the role of ozone–water O-isotopic exchange. The formation of peroxide depends on the ozone concentration as increased ozone concentration (25 to 50 μmoles) produced higher enrichment in the carbonates (product CO2) and a higher oxygen isotopic anomaly (SI Appendix, Fig. S1b). Hydrogen peroxide formation may also depend on the number of adsorbed-water molecules; however, further experiments are required to verify the dependence of peroxide concentration on the chemisorbed water on aerosol surfaces. The enrichment in peroxide depends on the chemical composition of aerosol surfaces as the maximum enrichment and anomaly was observed on quartz rich sand.
Mechanism B (Fig. 3B) involves in situ formation of carbonates in the presence of CO2 and water. The oxygen-bearing mineral aerosols (CaO, MgO, Fe2O3, ZnO, SiO2, and Cu2O) are naturally derived from dust, sea spray, and anthropogenic sources and may serve as nucleating sites for in situ carbonate formation. It is known that alkaline metals in solution enhance the uptake of CO2. Similarly, the presence of basic oxide and hydroxide in the aerosols facilitates the interaction between chemisorbed water and CO2 and promotes the formation of bicarbonates on aerosol surfaces. The surface layer upon drying becomes supersaturated, leading to secondary carbonate formation on mineral surfaces via heterogeneous chemistry. Secondary carbonate formation on mineral surfaces is associated with pH changes in the microenvironment near the surface. The dissociative adsorption of water on metals surfaces produces OH species, and consequently the metal surface carries a partial negative charge. Analogous to OH- ions in solution, OHγ- species adsorbed on metal surfaces act as Brłnsted bases (51, 52) and may facilitate the formation and precipitation of carbonates. The in situ formation of CO2 on iron oxide surfaces in the presence of adsorbed water and CO2 has been demonstrated using 18O labeled water (15) and labeled C16O2, C18O2 on zeolite surfaces (53). Carbonate mineral growth on concrete structures has been observed (54) with δ18O values in the range of 8.5–16.6‰. These findings suggest that small quantities of atmospheric CO2 are adsorbed in water films at particle surfaces and subsequent dissolution and evaporation leads to formation of mineral carbonates in localized environments, similar to rock weathering.
Our static experiments revealed that isotopic exchange between calcite and ozone required a hydration layer for the formation of a peroxide intermediate; however, the maximum anomaly produced in these experiments (SI Appendix, Fig S1a) was 2.67‰ compared to the anomaly observed in atmospheric carbonates (3.97‰). Future experiments with varying relative humidity in a flow tube with suspended particles are needed to closely simulate the atmospheric conditions of relevant heterogeneous chemistry. The present work using triple isotopes of oxygen demonstrates that the catalytic decomposition of O3 on surfaces leads to the formation of isotopically anomalous H2O2, which in turn transfers mass independent oxygen from ozone to carbonates. This previously unidentified channel for the formation of H2O2 may account for the regeneration of the hydroxyl radical upon photolysis during the daytime, independent of NOx concentration in the heavily polluted city Guangzhou at Pearl River delta, China (55). These findings may also explain the observations of reduced (10–20%) ozone concentrations in polluted source regions in the presence of mineral aerosols (56, 57).
The previously undescribed findings provide another pathway to understanding the origin of the O-isotopic anomaly reported in Martian carbonates (23, 24). Our laboratory results show that adsorbed surface water is required for the solid–gas interaction and isotopically anomalous carbonate formation. The authors (23, 24) suggested that anomalous CO2 (produced by the interaction of gas phase O3 and CO2 via excited O atoms at λ > 254 nm) as the most probable source of O-isotopic anomaly in Martian carbonates. This mechanism may be operative, but it cannot explain the heterogeneity of carbonates found in Martian meteorites. We suggest that the oxygen isotopic anomaly in carbonates extracted from SNC meteorites (Δ17O = 0.7–1.1‰) arises from the interaction of O3, CO2 and adsorbed water on particle surfaces in the Martian regoliths and potentially on dust aerosols that are pervasive in the Martian atmosphere (58). Secondary carbonate formation via hydrolysis of mineral particle surfaces and evaporative enrichment of ions in the microenvironment on aerosol surfaces may account for the presence of 2–5% carbonates in the Martian soils (3). A plot of Δ17O versus 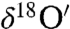 reveals a wide range of oxygen isotopic composition for the atmospheric carbonates similar to the carbonates minerals extracted from Martian meteorites (Fig. 4). These variations could be due to (i) the extent of exchange reaction between ozone and carbonates as enrichment in both
reveals a wide range of oxygen isotopic composition for the atmospheric carbonates similar to the carbonates minerals extracted from Martian meteorites (Fig. 4). These variations could be due to (i) the extent of exchange reaction between ozone and carbonates as enrichment in both 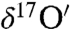 ,
, 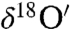 and the O-isotopic anomaly (Δ17O) increases with time; (ii) the amount of ozone present at the time of reaction because the enrichment in the oxygen isotopes of carbonates increases with ozone concentration; or, (iii) interaction of different sources of water of varying isotopic composition and evaporative enrichment. Heterogeneous carbonate formation on mineral aerosols depends upon the molecular collision of CO2 with aqueous layers on particle surfaces leading to kinetic isotope fractionation and is likely to be affected by the pCO2, thickness of water film, temperature, and chemical nature and surface area of the particles and photochemistry of the environment. The production of anomalous carbonates on Mars likely occurs via multiple pathways: (i) MIF (mass independently fractionated) CO2 production in the atmosphere and its interaction with aqueous solutions to form mineral evaporites and (ii) surfical peroxide-mediated process involving interaction of O3 and CO2 with adsorbed water similar to the mechanisms suggested for terrestrial atmospheric carbonate formation. The combination of ground observations from the Mars Phoenix Lander and orbital data from the CRISM (Compact Reconnaissance Imaging Spectrometer for Mars) and High Resolution Imaging Science Experiment indicated mixture of soil particles (∼100 μm) covered with CO2 ice and water (4, 59), an appropriate mixture for in situ carbonate formation. It is possible that heterogeneous chemical transformations may occur on aerosol particles during Martian dust events in the presence of water and ozone, as we observe in this work, providing an alternate route to carbonate formation. In this case, the carbonates thought to be life-forms, may in fact be in situ produced carbonates on aerosol surfaces. The analysis of returned Martian dust samples will provide greater detail of Mar’s geology and climate. Simultaneous O-isotopic measurements of secondary mineral such as carbonates, sulfate, and water from the same SNC meteorite can provide valuable information about the formation mechanism and evolution of Martian atmosphere through time. The heterogeneity of the O-isotopic composition of the carbonates and water in SNC meteorites was attributed to the spatial and temporal variability of the fluid form in which different minerals were precipitated (23). The possibility that carbonates were produced in the Martian regolith might be further resolved by measuring the O-isotope fractionation factors in controlled laboratory studies with various mineral aerosols and water. An additional implication of the previously undescribed carbonate formation mechanism is that the operation of this mechanism in the Martian atmosphere would generate peroxide on dust particles, which may restrict the synthesis of organics on the soil particles, thus further pointing to the need for expanded isotopic measurements of SNC meteorites.
and the O-isotopic anomaly (Δ17O) increases with time; (ii) the amount of ozone present at the time of reaction because the enrichment in the oxygen isotopes of carbonates increases with ozone concentration; or, (iii) interaction of different sources of water of varying isotopic composition and evaporative enrichment. Heterogeneous carbonate formation on mineral aerosols depends upon the molecular collision of CO2 with aqueous layers on particle surfaces leading to kinetic isotope fractionation and is likely to be affected by the pCO2, thickness of water film, temperature, and chemical nature and surface area of the particles and photochemistry of the environment. The production of anomalous carbonates on Mars likely occurs via multiple pathways: (i) MIF (mass independently fractionated) CO2 production in the atmosphere and its interaction with aqueous solutions to form mineral evaporites and (ii) surfical peroxide-mediated process involving interaction of O3 and CO2 with adsorbed water similar to the mechanisms suggested for terrestrial atmospheric carbonate formation. The combination of ground observations from the Mars Phoenix Lander and orbital data from the CRISM (Compact Reconnaissance Imaging Spectrometer for Mars) and High Resolution Imaging Science Experiment indicated mixture of soil particles (∼100 μm) covered with CO2 ice and water (4, 59), an appropriate mixture for in situ carbonate formation. It is possible that heterogeneous chemical transformations may occur on aerosol particles during Martian dust events in the presence of water and ozone, as we observe in this work, providing an alternate route to carbonate formation. In this case, the carbonates thought to be life-forms, may in fact be in situ produced carbonates on aerosol surfaces. The analysis of returned Martian dust samples will provide greater detail of Mar’s geology and climate. Simultaneous O-isotopic measurements of secondary mineral such as carbonates, sulfate, and water from the same SNC meteorite can provide valuable information about the formation mechanism and evolution of Martian atmosphere through time. The heterogeneity of the O-isotopic composition of the carbonates and water in SNC meteorites was attributed to the spatial and temporal variability of the fluid form in which different minerals were precipitated (23). The possibility that carbonates were produced in the Martian regolith might be further resolved by measuring the O-isotope fractionation factors in controlled laboratory studies with various mineral aerosols and water. An additional implication of the previously undescribed carbonate formation mechanism is that the operation of this mechanism in the Martian atmosphere would generate peroxide on dust particles, which may restrict the synthesis of organics on the soil particles, thus further pointing to the need for expanded isotopic measurements of SNC meteorites.
Fig. 4.

Oxygen isotope anomaly in Martian (solid red triangle) and Earth atmospheric (open triangle) carbonates. For comparison excess 17O of hydrogen peroxide (green asterisk) in rain water and both tropospheric (solid blue circle) and stratospheric (open blue circle) ozone are shown.
Conclusions
Analogous to terrestrial processes, the presence of carbonates in meteorites is interpreted as evidence for secondary aqueous processing of minerals on large planetary bodies with excessive water. In this article, the first observation of micrometer-sized terrestrial atmospheric carbonates possessing an O-isotopic anomaly (0.4–3.9‰) is reported. Adsorbed water on particle surfaces facilitates the interaction of the gaseous CO2 and O3 with formation of anomalous hydrogen peroxide and carbonates. The excess 17O in the terrestrial atmospheric carbonates provides direct proof of the chemical transformations and mechanisms of O-isotopic exchange on aerosol surfaces and is a process that may mediate ozone processes. This isotopically tagged carbonate records ozone interactions, and it is possible that in an appropriate environment, it could be used as a paleo-ozone recorder. The previously undescribed mechanism of carbonate formation via heterogeneous chemistry in the atmosphere may also explain the presence of carbonates in interplanetary dust particles in the presence of ice water (60). Further laboratory and modeling efforts will be instrumental to understand the nonequilibrium kinetic isotopic fractionation processes during in situ carbonate formation using different aerosol surfaces at various temperatures. These previously undescribed data and future measurements of in situ carbonate formation will allow comprehensive understanding of gas and particles interaction, and the role of water and hydrogen peroxide chemistry on the surface of Mars.
Materials and Methods
Aerosol samples were collected for a week (flow rate of 1.2 ± 0.1 m-3 min-1) on glass–fiber filter papers using a high volume cascade impactor (Thermo Anderson) with three stages (cutoff equivalent aerodynamic diameter of 7.0, 3.0, and 1.0 μm) and backup filter paper (≤ 1.0 μm). Prior to loading, glass–fiber filter paper (Staplex TFAG810) was heated for 48 h in an oven at 100 ± 2 °C to remove impurities. The aerosol samples were divided into two sets: (i) coarse fraction (particles > 1 μm; filter 1, 2, and 3) and fine fraction (particles < 1 μm contained on backup filter paper). Filter papers were evacuated overnight in a glass reaction tube containing 100% H3PO4 in the side arm. The acid was added to the filter papers at room temperature and allowed to react for 8 h at 25 ± 2 °C. CO2 and other gases were collected at liquid nitrogen temperature and purified chromatographically (Varian 3600, analytical instruments). To obtain independent 17O measurements, CO2 gas was fluorinated with BrF5 at 800 ± 5 °C for 45 h to yield O2 and CF4. Oxygen gas was purified chromatographically (DB100/120 column, Alltech) from other reaction products. The isotopic composition of pure oxygen gas thus obtained was measured using Isotope Ratio Mass spectrometer (Finnigan Mat 253) against a laboratory reference gas calibrated with respect to SMOW.
Filter papers without aerosol were acid digested and processed in a similar way, and blank correction was applied to all the samples (CO2 obtained was less than 0.2 μmoles). Two filter papers containing the coarse and fine fractions of aerosol samples collected at SIO in May (SIO180507) were used as controls. In these experiments, 5 mL of 2N HCl was added to the filter papers and allowed to react over night. This treatment ensured removal of inorganic carbonates from the sample (61). Water vapor and other gaseous substances were removed by pumping, and the remaining aerosol sample was processed as usual with 100% H3PO4. No CO2 was obtained after acid digestion, thereby ruling out the contribution of CO2 from organic sources. Soil samples from various sites (SI Appendix, Table S2) were collected during field campaigns. These samples were dried and crushed to a fine powder. The soil samples were acid digested, and CO2 gas was processed as outlined above.
To investigate the mechanism of isotope exchange on calcite surfaces, two sets of controlled laboratory experiments were conducted. Ozone (27 ± 2 μmole) was prepared using an electric discharge in a static system at liquid nitrogen temperature (-196 °C) with mean 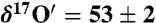 and
and 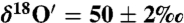 ). In the first set, ozone was allowed to react directly with calcium carbonate (32 ± 0.2 mg) in the reaction vessel (∼120 cm3) for 30 and 60 min. In the second set of experiments, 55.5 ± 0.2 μmole of Millipore water was added to CaCO3 in the reaction vessel to simulate hydration layers at the calcite surface. The reactants were condensed at -60 °C and air pumped away. The Freeze–thaw cycle was repeated to remove residual atmospheric gases prior to reaction. Ozone was added to the reaction vessel by freezing at liquid nitrogen temperature (-196 °C). Kinetics of the isotope exchange reaction was followed after 30, 60, and 1,320 min. In order to quantify the effect of ozone on the transfer of O-isotope anomaly to the carbonates, similar experiments were conducted with 25 ± 2, 50 ± 2 μmole of ozone while allowing the reaction to proceed for only 30 min. The oxygen isotopic composition of hydrogen peroxide was analyzed using KMnO4 at acidic pH (37). To study the ozone interaction during in situ carbonate formation, 121 mg of dried CaO powder was added to the reaction tube, and 8.3 ± 0.02 mmole water was added. Air, CO2, and other trace gases were removed by pumping while the mixture was frozen at -60 °C. To ensure complete removal of trace gases the freeze–thaw cycle was repeated twice. Ozone (300 ± 2 μmole) and CO2 (243 ± 0.2 μmole) were added to the reaction mixture at -196 °C and allowed to react for 48 h. Oxygen gas was pumped away, and carbonates were digested with 100% H3PO4 to produce CO2. O isotopes were measured as outlined above.
). In the first set, ozone was allowed to react directly with calcium carbonate (32 ± 0.2 mg) in the reaction vessel (∼120 cm3) for 30 and 60 min. In the second set of experiments, 55.5 ± 0.2 μmole of Millipore water was added to CaCO3 in the reaction vessel to simulate hydration layers at the calcite surface. The reactants were condensed at -60 °C and air pumped away. The Freeze–thaw cycle was repeated to remove residual atmospheric gases prior to reaction. Ozone was added to the reaction vessel by freezing at liquid nitrogen temperature (-196 °C). Kinetics of the isotope exchange reaction was followed after 30, 60, and 1,320 min. In order to quantify the effect of ozone on the transfer of O-isotope anomaly to the carbonates, similar experiments were conducted with 25 ± 2, 50 ± 2 μmole of ozone while allowing the reaction to proceed for only 30 min. The oxygen isotopic composition of hydrogen peroxide was analyzed using KMnO4 at acidic pH (37). To study the ozone interaction during in situ carbonate formation, 121 mg of dried CaO powder was added to the reaction tube, and 8.3 ± 0.02 mmole water was added. Air, CO2, and other trace gases were removed by pumping while the mixture was frozen at -60 °C. To ensure complete removal of trace gases the freeze–thaw cycle was repeated twice. Ozone (300 ± 2 μmole) and CO2 (243 ± 0.2 μmole) were added to the reaction mixture at -196 °C and allowed to react for 48 h. Oxygen gas was pumped away, and carbonates were digested with 100% H3PO4 to produce CO2. O isotopes were measured as outlined above.
Supplementary Material
Acknowledgments.
Many people helped to make this study possible. We thank current and former colleagues at Stable Isotope Laboratory at University of California at San Diego (UCSD) for useful discussions. We are grateful to reviewers Drs. J. Farquhar, D. Rumble, and G. Benedix for their valuable comments that helped to improve the manuscript. We appreciate the assistance of the staff at White Mountain Research Station and Air Pollution Control District, San Diego to collect aerosol samples. Graduate students from Prof. Kubiak’s labs at UCSD are highly acknowledged for their support to identify organic species in the samples using the IR spectrophotometer. This research was partially funded through grants from National Aeronautics and Space Administration (NNX09AG93G) and National Science Foundation (AGS0960594).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1014399107/-/DCSupplemental.
*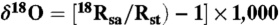 . Here delta (δ) values denote the relative deviation of the isotope ratios (18R = 18O/16O) in a sample (Rs) with respect to a standard material (Rst), which is SMOW (Standard Mean Ocean Water) in the case of oxygen.
. Here delta (δ) values denote the relative deviation of the isotope ratios (18R = 18O/16O) in a sample (Rs) with respect to a standard material (Rst), which is SMOW (Standard Mean Ocean Water) in the case of oxygen.
†The depletion or abundance of an isotope is measured with respect to a standard reference value. In the case of atmospheric carbonate, the relationship between 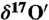 and
and 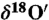 was determined using five CaCO3 samples and this value was used to define excess 17O (
was determined using five CaCO3 samples and this value was used to define excess 17O (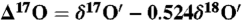 ) in carbonates. Here
) in carbonates. Here 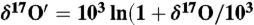 ) and
) and 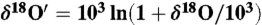 .
.
‡Mt. Soledad (250 m, −117.29°W, 32.78°N) La Jolla, California; Pier of Scripps Institute of Oceanography (−117.25°W, 32.86°N) and Urey Hall (Biochemistry building), University of California–San Diego, La Jolla, California (−117.25°W, 32.86°N); White Mountain Research Station (3,800 m, −118.326°W, 37.3605°N), Bishop, California.
References
- 1.Grady MM, Wright I. The carbon cycle on early Earth—and on Mars. Philos Trans R Soc B. 2006;361:1703–1713. doi: 10.1098/rstb.2006.1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith PH, et al. H2O at the Phoenix landing site. Science. 2009;325:58–61. doi: 10.1126/science.1172339. [DOI] [PubMed] [Google Scholar]
- 3.Boynton WV, et al. Evidence for calcium carbonate at the Mars Phoenix landing site. Science. 2009;325:61–64. doi: 10.1126/science.1172768. [DOI] [PubMed] [Google Scholar]
- 4.Cull S, et al. Seasonal H2O and CO2 ice cycles at the Mars Phoenix landing site: 1. Prelanding CRISM and HiRISE observations. J Geophys Res-Planet. 2010;115:E00D16. 10.1029/2009JE003340. [Google Scholar]
- 5.Pommerol A, Schmitt B, Beck P, Brissaud O. Water sorption on Martian regolith analogs: Thermodynamics and near-infrared reflectance spectroscopy. Icarus. 2009;204:114–136. [Google Scholar]
- 6.Bandfield JL. Global mineral distributions on Mars. J Geophys Res-Planet. 2002;107:5042. 10.1029/2001JE001510. [Google Scholar]
- 7.Morris RV, et al. Identification of carbonate-rich outcrops on Mars by the Spirit Rover. Science. 2010;329:421–424. doi: 10.1126/science.1189667. [DOI] [PubMed] [Google Scholar]
- 8.Orofino V, Blanco A, Blecka MI, Fonti S, Jurewicz A. Carbonates and coated particles on Mars. Planet Space Sci. 2000;48:1341–1347. [Google Scholar]
- 9.Tamppari LK, Smith MD, Bass DS, Hale AS. Water-ice clouds and dust in the north polar region of Mars using MGS TES data. Planet Space Sci. 2008;56:227–245. [Google Scholar]
- 10.Borg LE, et al. The age of the carbonates in Martian meteorite ALH84001. Science. 1999;286:90–94. doi: 10.1126/science.286.5437.90. [DOI] [PubMed] [Google Scholar]
- 11.McKay CP. Did Mars once have Martians? Astronomy. 1993;21:27–33. [PubMed] [Google Scholar]
- 12.McKay DS, et al. Search for past life on Mars: Possible relic biogenic activity in Martian meteorite ALH84001. Science. 1996;273:924–930. doi: 10.1126/science.273.5277.924. [DOI] [PubMed] [Google Scholar]
- 13.Hecht MH, et al. Detection of perchlorate and the soluble chemistry of Martian soil at the Phoenix Lander Site. Science. 2009;325:64–67. doi: 10.1126/science.1172466. [DOI] [PubMed] [Google Scholar]
- 14.Gardin E, Allemand P, Quantin C, Thollot P. (Defrosting, dark flow features, and dune activity on Mars: Example in Russell crater. J Geophys Res-Planet. 2010;115:E06016. 10.1029/2009JE003515. [Google Scholar]
- 15.Baltrusaitis J, Grassian VH. Surface reactions of carbon dioxide at the adsorbed water-iron oxide interface. J Phys Chem B. 2005;109:12227–12230. doi: 10.1021/jp051868k. [DOI] [PubMed] [Google Scholar]
- 16.Krischok S, Hofft O, Kempter V. The chemisorption of H2O and CO2 on TiO2 surfaces: Studies with MIES and UPS (HeI/II) Surf Sc. 2002;507:69–73. [Google Scholar]
- 17.Ochs D, Braun B, Maus-Friedrichs W, Kempter V. CO2 chemisorption at Ca and CaO surfaces: A study with MIES, UPS(HeI) and XPS. Surf Sci. 1998;417:406–414. [Google Scholar]
- 18.Ochs D, Brause M, Braun B, Maus-Friedrichs W, Kempter V. CO2 chemisorption at Mg and MgO surfaces: A study with MIES and UPS (He I) Surf Sci. 1998;397:101–107. [Google Scholar]
- 19.Prokoph A, Shields GA, Veizer J. Compilation and time-series analysis of a marine carbonate delta O-18, delta C-13, Sr-87/Sr-86 and delta S-34 database through Earth history. Earth Sci Rev. 2008;87:113–133. [Google Scholar]
- 20.Zeebe RE. An explanation of the effect of seawater carbonate concentration on foraminiferal oxygen isotopes. Geochim Cosmochim Acta. 1999;63:2001–2007. [Google Scholar]
- 21.Miller MF. Isotopic fractionation and the quantification of O-17 anomalies in the oxygen three-isotope system: An appraisal and geochemical significance. Geochim Cosmochim Acta. 2002;66:1881–1889. [Google Scholar]
- 22.Young ED, Galy A, Nagahara H. Kinetic and equilibrium mass-dependent isotope fractionation laws in nature and their geochemical and cosmochemical significance. Geochim Cosmochim Acta. 2002;66:1095–1104. [Google Scholar]
- 23.Farquhar J, Thiemens MH. Oxygen cycle of the Martian atmosphere-regolith system: Delta O-17 of secondary phases in Nakhla and Lafayette. J Geophys Res-Planet. 2000;105:11991–11997. [Google Scholar]
- 24.Farquhar J, Thiemens MH, Jackson T. Atmosphere-surface interactions on Mars: Delta O-17 measurements of carbonate from ALH 84001. Science. 1998;280:1580–1582. doi: 10.1126/science.280.5369.1580. [DOI] [PubMed] [Google Scholar]
- 25.Thiemens MH. Mass-independent isotope effects and their use in understanding natural processes. Isr J Chem. 2002;42:43–54. [Google Scholar]
- 26.Chakraborty S, Bhattacharya SK. Experimental investigation of oxygen isotope exchange between CO2 and O(1D) and its relevance to the stratosphere. J Geophys Res. 2003;108:ACH5-1-15. 10.1029/2002JD002915. [Google Scholar]
- 27.Perri MJ, Van Wyngarden AL, Boering KA, Lin JJ, Lee YT. Dynamics of the O(D - 1) + CO2 oxygen isotope exchange reaction. J Chem Phys. 2003;119:8213–8216. [Google Scholar]
- 28.Shaheen R, Janssen C, Rockmann T. Investigations of the photochemical isotope equilibrium between O-2, CO2 and O-3. Atmos Chem Phys. 2007;7:495–509. [Google Scholar]
- 29.Wen J, Thiemens MH. Multi-isotope study of the O((1)D) + CO2 exchange and stratospheric consequences. J Geophys Res-Atmos. 1993;98:12801–12808. [Google Scholar]
- 30.Hoag KJ, Still CJ, Fung IY, Boering KA. Triple oxygen isotope composition of tropospheric carbon dioxide as a tracer of terrestrial gross carbon fluxes. Geophys Res Lett. 2005;32:5. 10.1029/2004GL021011. [Google Scholar]
- 31.Luz B, Barkan E, Bender ML, Thiemens MH, Boering KA. Triple-isotope composition of atmospheric oxygen as a tracer of biosphere productivity. Nature. 1999;400:547–550. [Google Scholar]
- 32.Johnston JC, Thiemens MH. The isotopic composition of tropospheric ozone in three environments. J Geophys Res-Atmos. 1997;102:25395–25404. [Google Scholar]
- 33.Krankowsky D, et al. Stratospheric ozone isotope fractionations derived from collected samples. J Geophys Res-Atmos. 2007;112 10.1029/2006JD007855. [Google Scholar]
- 34.Brenninkmeijer CAM, et al. Isotope effects in the chemistry of atmospheric trace compounds. Chem Rev. 2003;103:5125–5161. doi: 10.1021/cr020644k. [DOI] [PubMed] [Google Scholar]
- 35.Thiemens MH. History and applications of mass-independent isotope effects. Annu Rev Earth Planet Sci. 2006;34:217–262. [Google Scholar]
- 36.Swart PK, Burns SJ, Leder JJ. Fractionation of the stable isotopes of oxygen and carbon in carbon-dioxide during the reaction of calcite with phosphoric-acid as a function of temperature and technique. Chem Geol. 1991;86:89–96. [Google Scholar]
- 37.Savarino J, Thiemens MH. Analytical procedure to determine both delta O-18 and delta O-17 of H2O2 in natural water and first measurements. Atmos Environ. 1999;33:3683–3690. [Google Scholar]
- 38.Goudie AS, Middleton N. Desert Dust in the Global System. Heidelberg, Germany: Springer; 2006. [Google Scholar]
- 39.Meszaros E, et al. Size distributions of inorganic and organic species in the atmospheric aerosol in Hungary. J Aerosol Sci. 1997;28:1163–1175. [Google Scholar]
- 40.Al-Abadleh HA, Al-Hosney HA, Grassian VH. Oxide and carbonate surfaces as environmental interfaces: The importance of water in surface composition and surface reactivity. J Mol Catal A-Chem. 2005;228:47–54. [Google Scholar]
- 41.Karagulian F, Santschi C, Rossi MJ. The heterogeneous chemical kinetics of N2O5 on CaCO3 and other atmospheric mineral dust surrogates. Atmos Chem Phys. 2006;6:1373–1388. doi: 10.1039/b506750m. [DOI] [PubMed] [Google Scholar]
- 42.Stipp SLS. Understanding interface processes and their role in the mobility of contaminants in the geosphere—The use of surface sensitive techniques. Eclogae Geol Helv. 1994;87:335–355. [Google Scholar]
- 43.Magdans U, Gies H, Torrelles X, Rius J. Investigation of the {104} surface of calcite under dry and humid atmospheric conditions with grazing incidence X-ray diffraction (GIXRD) Eur J Mineral. 2006;18:83–91. [Google Scholar]
- 44.Rahaman A, Grassian VH, Margulis CJ. Dynamics of water adsorption onto a calcite surface as a function of relative humidity. J Phys Chem C. 2008;112:2109–2115. [Google Scholar]
- 45.Al-Hosney HA, Carlos-Cuellar S, Baltrusaitis J, Grassian VH. Heterogeneous uptake and reactivity of formic acid on calcium carbonate particles: A Knudsen cell reactor, FTIR and SEM study. Phys Chem Chem Phys. 2005;7:3587–3595. doi: 10.1039/b510112c. [DOI] [PubMed] [Google Scholar]
- 46.Al-Hosney HA, Grassian VH. Water, sulfur dioxide and nitric acid adsorption on calcium carbonate: A transmission and ATR-FTIR study. Phys Chem Chem Phys. 2005;7:1266–1276. doi: 10.1039/b417872f. [DOI] [PubMed] [Google Scholar]
- 47.Schutze M, Herrmann H. Determination of phase transfer parameters for the uptake of HNO3, N2O5 and O-3 on single aqueous drops. Phys Chem Chem Phys. 2002;4:60–67. [Google Scholar]
- 48.Roeselova M, Jungwirth P, Tobias DJ, Gerber RB. Impact, trapping, and accommodation of hydroxyl radical and ozone at aqueous salt aerosol surfaces. A molecular dynamics study. J Phys Chem B. 107:12690–12699. [Google Scholar]
- 49.Beltran FJ. Ozone Reaction Kinetics for Water and Wasterwater Systems. New York: Lewis Publishers/CRC Press; 2004. [Google Scholar]
- 50.Li W, Oyama ST. Mechanism of ozone decomposition on a manganese oxide catalyst. 2. Steady-state and transient kinetic studies. J Am Chem Soc. 1998;120:9047–9052. [Google Scholar]
- 51.Barnette AL, Asay DB, Kim SH. Average molecular orientations in the adsorbed water layers on silicon oxide in ambient conditions. Phys Chem Chem Phys. 2008;10:4981–4986. doi: 10.1039/b810309g. [DOI] [PubMed] [Google Scholar]
- 52.Yamamoto S, et al. In situ X-ray photoelectron spectroscopy studies of water on metals and oxides at ambient conditions. J Phys Condens Matt. 2008:184025–184014. [Google Scholar]
- 53.Galhotra P, Navea JG, Larsen SC, Grassian VH. Carbon dioxide ((CO2)-O-16 and (CO2)-O-18) adsorption in zeolite Y materials: Effect of cation, adsorbed water and particle size. Energ Environ Sci. 2009;2:401–409. [Google Scholar]
- 54.Macleod G, Hall AJ, Fallick AE. An applied mineralogical investigation of concrete degradation in a major concrete road bridge. Mineral Mag. 1990;54:637–644. [Google Scholar]
- 55.Hofzumahaus A, et al. Amplified trace gas removal in the troposphere. Science. 2009;324:1702–1704. doi: 10.1126/science.1164566. [DOI] [PubMed] [Google Scholar]
- 56.Bian HS, Zender CS. Mineral dust and global tropospheric chemistry: Relative roles of photolysis and heterogeneous uptake. J Geophys Res-Atmos. 2003;108:4672. 10.1029/2002JD003143. [Google Scholar]
- 57.Dentener F, et al. The global atmospheric environment for the next generation. Environ Sci Technol. 2006;40:3586–3594. doi: 10.1021/es0523845. [DOI] [PubMed] [Google Scholar]
- 58.Smith MD. THEMIS observations of Mars aerosol optical depth from 2002-2008. Icarus. 2009;202:444–452. [Google Scholar]
- 59.Cull S, et al. Seasonal ice cycle at the Mars Phoenix landing site: 2. Postlanding CRISM and ground observations. J Geophys Res-Planet. 2010;115:E00E19. 0.1029/2009JE003410. [Google Scholar]
- 60.Kemper F, et al. Detection of carbonates in dust shells around evolved stars. Nature. 2002;415:295–297. doi: 10.1038/415295a. [DOI] [PubMed] [Google Scholar]
- 61.Wang Y, McDonald E, Amundson R, McFadden L, Chadwick O. An isotopic study of soils in chronological sequences of alluvial deposits, Providence Mountains, California. Geol Soc Am Bull. 1996;108:379–391. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.