

Abstract
Bacteriophages are the most numerous organisms in the biosphere. In spite of their biological significance and the spectrum of potential applications, little high-resolution structural detail is available on their receptor-binding fibers. Here we present the crystal structure of the receptor-binding tip of the bacteriophage T4 long tail fiber, which is highly homologous to the tip of the bacteriophage lambda side tail fibers. This structure reveals an unusual elongated six-stranded antiparallel beta-strand needle domain containing seven iron ions coordinated by histidine residues arranged colinearly along the core of the biological unit. At the end of the tip, the three chains intertwine forming a broader head domain, which contains the putative receptor interaction site. The structure reveals a previously unknown beta-structured fibrous fold, provides insights into the remarkable stability of the fiber, and suggests a framework for mutations to expand or modulate receptor-binding specificity.
Keywords: gene product 37, host cell attachment, octahedral coordination, viral fibers, X-ray crystallography
Bacteriophages are exploited in an emergent array of applications including the typing of bacteria (1), peptide display (2), and experimental phage therapy (3–5). Bacteriophages have also been extensively studied as model systems for fundamental processes such as viral infection and replication, gene transfer, protein folding, and assembly. Escherichia coli bacteriophage T4 (6), a member of the Myoviridae family of the Caudovirales order, has an exclusively lytic lifecycle. Host recognition occurs through a reversible interaction of the tip of the long tail fibers with lipopolysaccharides or with the outer membrane porin protein C (7) (Fig. 1). Upon receptor binding, a recognition signal is sent to the baseplate (8–11), causing the short tail fibers to extend and irreversibly bind to the outer core region of the lipopolysaccharides (12). This binding is followed by contraction of the outer tail sheath (13, 14), penetration of the bacterial membrane by the hollow inner tail tube (Fig. 1B), and ejection of the viral DNA into the bacterium (15). The T4 long tail fibers are an assembly of four different proteins [gene product (gp) 34, gp35, gp36, and gp37; ref. 16] and can be separated into proximal and distal half-fiber segments of approximately 70 nm (17), hinged at an angle of around 160° (Fig. 1C). Proximal half-fibers are composed of trimers of gp34, followed by a monomer of gp35 forming the hinge or “kneecap,” whereas the distal half-fibers contain a trimer of gp36 (closest to gp35) and a trimer of gp37. Gene product 34 and gp37, as well as the short tail fiber protein gp12, need the chaperone gp57 for proper trimeric assembly; gp37 also requires gp38 (18). Eleven domains (D1–11) have been observed in the distal half-fiber by electron microscopy and D3–11 were assigned to gp37. Gene product 37 comprises 1,026 amino acids per monomer and D9 is predicted to start at residue 651, D10 at residue 803, and D11 at residue 926 (assuming that the protein chain is colinear with the fiber; ref. 17). Antibodies to gp37 inactivate T4 by blocking infection (19, 20). Experiments with hybrid phages suggest the receptor-binding region encompasses residues 907–996 (21).
Fig. 1.

Bacteriophage T4 and its long tail fibers. (A and B) Schematic representations of bacteriophage T4 attached to a bacterial membrane before (Left) and after (Right) contraction of the outer tail tube. The tip domain of gp37 is boxed. (C) Schematic representation of the bacteriophage T4 long tail fiber. Domains P1–5 correspond to gp34; the kneecap domain (K-C) is formed by gp35, whereas the distal part of the fiber, consisting of gp36 and gp37, is divided into regions D1–11. The expressed protein, gp37(651–1026), corresponds to D9-11 (larger gray box), whereas the crystallized fragment, gp37(785–1026), corresponds to D10–11 (smaller gray box).
Results
Protein Expression, Purification and Crystallization.
His-tagged gp37(651–1026) was coexpressed with its chaperones gp38 and gp57 similarly to previously described for gp37(12–1026) (22) and purified by metal affinity chromatography and anion exchange chromatography. After mild heat treatment (similar to that performed for gp12; ref. 23), trypsinization yielded the stable fragment gp37(785–1026), which was purified by size exclusion chromatography. Manganese (II) chloride was included in the final step of the preparation as it was identified as a stabilizing agent, although no evidence for ordered manganese ions was found in the final structure. Average yields of purified gp37(651–1026) were around 1.7 mg per liter of bacterial culture before proteolysis and highly purified gp37(785–1026) was obtained in yields of 0.36 mg/L. A stable fragment of 24.6 kDa, gp37(785–1026), was identified by N-terminal sequence analysis and mass spectroscopic peptide fingerprinting. Clusters of small gp37(785–1026) crystals were grown by vapor diffusion from solutions containing polyethyleneglycol and sodium citrate at pH 5.0.
Data Collection and Structure Solution.
An X-ray fluorescence emission spectrum measured from a single crystal measuring 50 × 20 × 5 μm3 indicated the presence of iron, presumably Fe2+, because the crystals were not appreciably colored. Therefore, a multiwavelength anomalous diffraction dataset was collected around the iron absorption edge from the same crystal. Seven iron ion sites were located during experimental phasing, consistent with the presence of seven His-X-His motifs in the gp37(785–1026) sequence. After solvent flattening, the resulting map displayed a solvent boundary corresponding well with the expected outlines of the tip of the T4 long tail fiber. Although detail in the initial experimental map was poor, it was possible to manually position the homologous part of the collar domain of gp12 (24, 25) into the globular region. The density surrounding the iron ions was compatible with an octahedral coordination sphere involving six histidine residues per ion. This observation, combined with the realization that both the N and C termini are located in the collar domain, the geometric impossibility of the chain going all the way to the tip and back if it did not form an almost continuous extended strand, and the fact that the distance between the iron ions and the spacing of the His-X-His motifs in the sequence allowed unequivocal assignment of each iron ion to the correct His-X-His motif, made it possible to manually trace a partial model. Using this model and the peak wavelength data, a combined single anomalous diffraction/Fourier synthesis map was calculated, allowing tracing of additional residues. Subsequently, a complete simulated annealing omit map was calculated, which allowed further additions. Finally, using positive difference maps resulting from refinement, the model could be completed and refined to satisfactory geometry (Table 1). The final model contains residues 811–1026 from all three chains; residues 785–810 are not visible in the electron density maps. The final model, refined at 2.2-Å resolution, includes residues 811–1026 from all three chains and has been refined to Rwork/Rfree of 17.9/23.8%.
Table 1.
Crystallographic data and refinement statistics
| Data collection | |||
| Space group | C2 | ||
| Cell parameters (a, b, c), Å | 157.3, 54.0, 112.8 (β = 100.4°) | ||
| peak | inflection | remote | |
| Beamline (ESRF) | ID23-1 | ID23-1 | ID23-2 |
| Detector | ADSC Q315r CCD | ADSC Q315r CCD | MarMOSAIC CCD |
| Distance, mm | 179.9 | 179.9 | 210.9 |
| Wavelength, Å | 1.73945 | 1.74115 | 0.87260 |
| Resolution, Å | 22-3.0 (3.16-3.00)* | 22-2.5 (2.64-2.50) | 22-2.2 (2.32-2.20) |
| Observed reflections† | 19,041 (2,732) | 32,101 (4,563) | 47,653 (6,906) |
| Multiplicity | 3.5 (3.5) | 3.6 (3.6) | 3.8 (3.8) |
| Completeness, % | 99.2 (99.0) | 99.2 (98.5) | 99.9 (99.9) |
| Rsym,‡ % | 14.8 (44.4) | 10.4 (43.9) | 13.2 (44.0) |
| 〈I/sigma(I)〉 | 4.0 (1.6) | 5.3 (1.5) | 5.0 (1.6) |
| Phasing | |||
| Resolution range used, Å | 22-2.2 (2.32-2.20) | ||
| No. of reflections | 46,766 (6,722) | ||
| Heavy atom sites§ | 7 Fe2+ | ||
| Correlation coefficient (all/weak) | 46.15/20.72 | ||
| Patterson figure of merit | 62.59 | ||
| Correlation coefficient (E) | 0.456 | ||
| R-Cullis¶ | |||
| Isomorphous (acentric/centric) | 0.835/0.855 | 0.457/0.479 | —/— |
| Anomalous (acentric) | 0.964 | 0.986 | 0.998 |
| Phasing power¶ | |||
| Isomorphous (acentric/centric) | 0.268/0.230 | 0.873/0.654 | —/— |
| Anomalous (acentric) | 0.426 | 0.246 | 0.125 |
| Figure of merit cos(phase error) (acentric/centric)¶ | 0.178/0.195 | ||
| Solvent flattening (27 cycles with 56.0% solvent content)∥ | |||
| R factor (before/after density modification)∥ | 0.5250/0.2820 | ||
| Overall correlation on |E|2 (before/after density modification)∥ | 0.1549/0.7196 | ||
| Hand score = correlation on |E|2/contrast (original/inverted) | 0.2190/0.2178 | ||
| Refinement statistics | |||
| Resolution range used, Å | 20-2.2 (2.32-2.20) | ||
| No. of reflections used | 45,619 (6,609) | ||
| No. of reflections used for R free | 2,028 (226) | ||
| R factor** | 0.179 (0.237) | ||
| R-free | 0.238 (0.292) | ||
| No. of protein/water atoms | 4,656/665 | ||
| Ions | 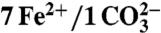 |
||
| Average B-value protein/water atoms, Å2 | 19.5/27.5 | ||
| Average B-value iron/carbonate ions, Å2 | 22.3/50.9 | ||
| Wilson B, Å2 | 22 | ||
| Ramachandran statistics, †† % | 98.4/100.0 | ||
| rmsd‡‡ (bonds, Å; angles, °) | 0.014/1.4 | ||
*Values in parentheses are for the highest resolution bin, where applicable.
†No sigma cutoff was used for inclusion of observed reflections.
‡Rsym = ΣhΣi|Ihi-〈Ih〉|/ΣhΣi|Ihi|, where Ihi is the intensity of the ith measurement of the same reflection and 〈Ih〉 is the mean observed intensity for that reflection.
§Determined with SHELXD (39).
¶Calculated with SHARP (40).
∥According to SOLOMON (41).
**R = Σ||Fobs(hkl)|-|Fcalc(hkl)||/Σ|Fobs(hkl)|.
††According to the program MOLPROBITY (46). The percentages are indicated of residues in favored and allowed regions of the Ramachandran plot, respectively.
‡‡Estimates provided by the program REFMAC (45).
Description of the Structure.
The structure (Figs. 2 and 3) reveals an elaborately interwoven trimer formed by a globular collar domain (about 45-Å wide), an elongated needle domain (around 15-Å wide), and a small head domain with a width of around 25 Å. The total length is just over 200 Å (20 nm). Each of the three chains runs from the collar domain to the end of the tip and twists around a neighboring chain before turning back, with both the N and C terminus located near the bottom of the collar domain. Amino acids 811–860 and 1016–1026 form the trimeric collar domain (Fig. 3A). Each monomer comprises a sandwich of two antiparallel beta-sheets (one containing three strands, the other two) with an alpha-helix (residues 843–850) at one end of the beta-sandwich.
Fig. 2.

Structure of gp37(785–1026). Chains A, B, and C are colored red, green, and blue, respectively; iron ions are shown in yellow. (A) Ribbon representation. The N and C termini and every 10th residue of chain A are labeled. (B and C) Surface representations of the structure of gp37(785–1026) seen from the side (B) and top (C) to illustrate the extensive intertwining of the three protein chains in the trimer; domains are indicated; i.r. is the intertwined region between the collar and needle domains.
Fig. 3.

Domain structure of gp37(785–1026). (A) Walleyed stereo representation of the collar domain viewed from the direction of the needle domain. (B) The needle domain viewed from the side. Iron ions are represented as yellow balls. The histidine doublets coordinating the iron ions are shown and labeled with letters a-g; iron ions with numbers 1–7. (C and D) Side (C) and top (D) view of the head domain. Aromatic and basic side chains are shown and labeled (R954 is hidden behind Y953 in D). (E) Comparison of gp37(811–1026) with the bacteriophage T4 baseplate proteins gp12 and gp10. Superposition of monomers of gp37(811–1026) in red, onto gp12(330–527) in blue, and gp10 in green. Iron ions belonging to the gp37(811–1026) structure are shown in yellow, whereas the zinc ion identified in gp12 is shown in gray.
Located next to the collar domain is an intricately intertwined region in which residues 861–880 encircle residues 1009–1015 of the neighboring chain. This region is followed by the needle domain, which is a 150-Å-long six-stranded antiparallel right-handed twisted circular sheet formed by residues 881–933 and 960–1008 from each of the three chains (Fig. 3B). Its diameter is roughly 15 Å, with each of the chains completing one-and-a-half turns (about 540°) around the fiber axis. In the core of the needle domain, hydrophobic and hydrophilic regions alternate, with the latter forming the metal-binding sites. The seven iron ions are coordinated octahedrally by the Nϵ atoms of two histidine residues from each chain, in a similar fashion to the zinc ion in the structure of gp12 (25). Iron ions 1, 5, and 7 are bound by the His-883/His-885, His-915/His-917, and His-929/His-931 doublets, respectively, whereas the His-966/His-968, His-980/His-982, His-989/His-991, and His-998/His-1000 pairs from the returning strand coordinate iron ions 6, 4, 3, and 2 (Fig. 3B). There are no water molecules on the threefold central axis, apart from in the collar domain, near the border with the needle domain. We also modeled a triad of water molecules around the threefold fiber axis between iron ions 6 and 7. The biological significance, if any, of these water molecules is not clear at this point, but they may indicate some flexibility in these regions.
Residues 934–959 from each of the three chains form a compact, interwoven head domain of 22 Å in diameter and 18-Å high (Fig. 3 C and D). Amino acids 934–947 loop around a neighboring chain; the chain then threads through the loop of a neighbor, before turning back into the needle domain. The head domain, located at the extreme distal end of the long tail fiber, is likely to play a primary role in receptor binding. As gp37 is known to interact with the glucosyl-alpha-1,3-glucose terminus of lipopolysaccharides (7) and protein–saccharide interactions almost always involve stacking of sugar residues onto aromatic amino acid side chains (26), aromatic surface amino acids (Tyr-932, Trp-936, Tyr-949, and Tyr-953) are attractive candidates for receptor binding. Lys-945 (near Trp-936) and Arg-954 (near Tyr-932 of a neighboring chain) may also interact with the lipopolysaccharides phosphate groups.
Stability and Folding.
Of the total surface area of each gp37(811–1026) monomer, 57% (14.5 × 103 Å2) is buried within the trimer interface, whereas the estimated energy gain upon complex formation is 270 kcal/mol. Like gp37(12–1026) (21), gp37(785–1026) is resistant to denaturation by sodium dodecylsulphate at room temperature. The complex interweaving suggests that gp37 is unlikely to exist as a stable monomer and potentially accounts for the requirement for chaperones gp57 and gp38 for correct folding. Gene product 57, also necessary for the productive folding of the short tail fiber protein gp12 and the proximal long tail fiber protein gp34, may be involved in keeping unfolded monomers apart until the collar domain trimerizes. Gene product 38 is exclusively required for gp37 folding and may have a more specialized function.
Structural Homologues.
The closest structural homologue identified by the DALI server (27) is gp12 (24, 25), which trimerizes to form the T4 short tail fiber. The five beta-strands and the alpha-helix of the gp37 collar domain can be superimposed onto five of the six beta-strands and an alpha-helix of the gp12 collar domain (Fig. 3E) with an rmsd of 2.7 Å over 115 C-alpha atoms. Similarity extends to the intertwined region adjacent to the collar domain and the first metal-binding site. His-885 and His-887 superpose well onto His-445 and His-447 of gp12, respectively, although gp12 binds a zinc ion instead of iron (25, 28). Gene product 10 (29) also exhibits significant structural homology to gp37, and 87 C-alpha atoms can be superposed with an rmsd of 4.8 Å. Three beta-strands of the collar domain of gp37 can be superimposed onto similar strands in the collars of gp10 and the closely related protein, gp11 (30). In the case of gp10, structural similarity also extends into the intertwined region next to the collar domain (Fig. 3G). The structural similarity of gp10, gp11, gp12, and gp37 makes it probable that these genes evolved from a common ancestor (29).
Discussion
Bioinformatic analysis (31) reveals sequence similarity to fibers from various phages and prophages (including several pathogens such as Shigella dysenteria, Yersinia pestis, and Salmonella enterica). When the sequences of the tip domains of bacteriophage T4 gp37, TuIa, and TuIb gp37 (all Myoviridae) and of the Siphovirus bacteriophage lambda Ur side tail fibers (32) are aligned (Fig. 4), extensive similarity is evident for residues 811–931 and 966–1026, i.e., for the whole tip domain except the putative receptor-binding head domain. The conservation pattern suggests the structural framework of the tip domain is maintained intact, whereas the head domain has diverged to acquire specific receptor-binding properties. It has been suggested (33) that T4, TuIa, and TuIb may have evolved from the T2 lineage and incorporated the C-terminal segment of the side tail fiber and the lambda tail fiber assembly protein by recombination with lambda or a close relative. This hypothesis was proposed based on experimental data showing that the C-terminal region of lambda side tail fibers can functionally substitute for gp37 in receptor binding, whereas the lambda tail fiber assembly protein ltfa can functionally substitute for gp38 in mediating the correct folding of gp37.
Fig. 4.

Alignment of the tip domains of the long tail fiber gp37 proteins of bacteriophages T4 (UniProt code P03744), TuIa (S13237), and TuIb (S13239) and of the side tail fiber of bacteriophage lambda (P03764). Of the latter two, only the sequence of the C-terminal 382 and 267 amino acids are known, respectively, although their entire gp37 proteins are expected to be similar in size to gp37 of T4. Identical residues are indicated with asterisks and similar ones with dots. Hydrophobic residues contributing to the central longitudinal core of the needle domain are boxed in gray; His-X-His motifs are also labeled with letters on top of the alignment. A deletion of 10 amino acids in the T4 needle domain after residue 909 with respect to the others is compensated for by a deletion of nine amino acids just before residue 979 in the “return” strand; these last nine amino acids contain a putative eighth metal-binding site His-Ala-His for TuIa, TuIb, and lambda.
The outer diameter of the head domain and the inner diameter of the surface cavity of the also trimeric outer membrane porin protein C (34) are both just under 25 Å. The very tip of the bacteriophage long tail fiber fits snugly into the mainly negatively charged outer cavity of its receptor outer membrane porin protein C (the gp37 head domain is uncharged apart from two small positive patches on the sides corresponding to Lys-945 and Arg-954). Automated docking experiments were performed with outer membrane porin protein C and gp37(811–1026) or the head domain plus a part of the needle domain (residues 918–973). Of the solutions obtained, many docked the side of the gp37 tip onto the hydrophobic side of the porin. Because this region would normally be covered by lipids in the membrane, these solutions were rejected. No solutions were obtained with gp37 docked on the inner membrane side of the porin. Of the remaining solutions, many aligned the threefold axes of both trimeric molecules, although some solutions with the tip oriented at an angle were observed. In both kinds of solutions, the head domain is consistently placed inside the extracellular cavity of the porin, when interactions with the hydrophobic membrane-interacting regions are excluded. In Fig. 5 the top “symmetric” solution and the solution with the largest angle are shown; in the latter, the side of the gp37 needle contacts surface loops of the porin. Interactions are either “head-on” or transversal; both are potentially relevant in recognition and infection and could represent different phage approach angles and be compatible with a conformational change of the baseplate that varies the attachment angle of the long tail fiber.
Fig. 5.

Docking of gp37 into outer membrane porin protein C. Two superimposed results of automatic docking experiments are shown with the gp37(811–1026) trimer in blue, a truncated trimeric gp37(918–973) model in red, and the three subunits of the outer membrane porin protein C trimer (PDB code 2J1N) in green, magenta, and cyan.
The present structure provides insights into the conserved molecular architecture of the T4 bacteriophage fiber tip and suggests the surface and residues that are most likely to be involved in receptor binding. Several surface-exposed aromatic and positive residues are prime candidates for mutagenesis studies to dissect the binding determinants and modulate the receptor-binding properties of this fiber. Future studies directed at the remaining components of the long tail fiber will provide valuable insights into this remarkable molecular machine.
Materials and Methods
Construction of Expression Vectors.
Sequences representing coding regions for gp38 and gp37(651–1026) were cloned into pCDF-Duet and pET30a(+) (Merck), respectively. The resulting plasmids were designated pCDF(Sm)g38 and pET(Kn)g37(651-1026). The vector pET(Ap)g57 was provided by Stefan Miller. Gene product 37(651–1026) was expressed with an additional N-terminal six-histidine tag.
Protein Expression and Purification.
Four liters of growth media (22) supplemented with ampicillin (50 mg/L), streptomycin (50 mg/L), and kanamycin (25 mg/L) were inoculated with the E. coli strain JM109(DE3) (Promega) cotransformed with pET(Kn)g37(651-1026), pET(Ap)g57, and pCDF(Sm)g38. Expression was induced at 16 °C and harvesting of bacteria performed as described (22). The cells were resuspended in 40 mL of 50 mM sodium phosphate pH 8.0, 0.3 M sodium chloride, 10 mM beta-mercaptoethanol, 10 mM imidazole, 1% glycerol, and protease inhibitors, frozen at -20 °C, and lysed by a double pass through an Avestin C5 emulsifier (Avestin, Europe, GmbH). Lysates were centrifuged at 39,000 × g and 10 °C for 40 min. Supernatant containing soluble His-tagged gp37(651–1026) was loaded onto a 5 ml nickel-nitrilotriacetic acid agarose (Qiagen) column preequilibrated with elution buffer (50 mM sodium phosphate pH 8.0, 0.3 M sodium chloride, 10 mM beta-mercaptoethanol). The recombinant protein was eluted with a step gradient of imidazole in elution buffer. The 0.25–0.4 mM imidazole fractions contained gp37(651–1026) and were combined and dialyzed overnight at 4 °C against 10 mM Tris•HCl pH 8.5. The protein was applied to a 6 mL Uno-Q column (Bio-Rad) equilibrated with the same buffer and eluted with a sodium chloride gradient. Highly purified gp37(651–1026) eluted at around 0.1 M sodium chloride. Gene product 37(651–1026) was concentrated to 10 mg/mL using 10 kDa cutoff centrifuge filters (Millipore) and buffer exchanged into 20 mM ammonium bicarbonate pH 7.8, 150 mM sodium chloride in the same step. One milliliter fractions of the concentrated protein were heat treated by incubation at 56 °C for 30 min. After cooling to 37 °C, 13.3 μg of sequencing grade modified trypsin (Promega) was added to the protein and the mixture was incubated for 80 min at 37 °C. The reaction was stopped using 1 mM phenylmethylsulfonyl fluoride. The resulting mixture was loaded onto a Hiload 16/60 sephacryl 300 column (GE Healthcare Bio-Sciences) equilibrated with 10 mM Tris•HCl pH 8.5, 150 mM sodium chloride, 1 mM manganese (II) chloride. Elution was done in the same buffer at a flow rate of 0.5 mL/ min. Peak fractions containing proteolyzed gp37(785–1026) were concentrated to 8 mg/mL and buffer exchanged into 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid-NaOH pH 7.5, 1 mM manganese (II) chloride. Manganese (II) chloride was identified as a stabilizing agent by a thermofluor assay performed according standard protocols (35).
Crystallization, Data Collection, and Structure Solution.
Crystallization was by vapor diffusion in sitting drops of 2 μL protein solution plus 2 μL of a reservoir solution containing 5% (wt/vol) polyethyleneglycol 6,000 and 0.1 M sodium citrate pH 5.0. Crystals were harvested in reservoir solution supplemented with 20% (wt/vol) glycerol, mounted into a cryoloop, and flash-frozen in liquid nitrogen for data collection. The presence of iron in the crystals was determined from X-ray fluorescence emission spectra recorded on BM30A at the European Synchrotron Radiation Facility (ESRF). A three-wavelength multiwavelength anomalous diffraction experiment was subsequently carried out on beamline ID23-1 at the ESRF. Due to radiation damage, the remote dataset was not included in the analysis; instead, a higher resolution dataset collected on ID23-2 was used as a remote and reference dataset. All data were processed and scaled using MOSFLM (36) and SCALA (37) and further analyzed using programs from the CCP4 suite (38). Reflections for calculation of Rfreewere selected in thin resolution shells. The initial sites were located using SHELXD (39) and refined within AUTOSHARP (40). Solvent flattening was with SOLOMON (41) and model building was performed with COOT (42). Combined single wavelength anomalous diffraction/Fourier synthesis and simulated annealing omit maps were calculated using PHENIX (43), in which PHASER performs the phase combination using a maximum likelihood procedure (44). Refinement was performed with REFMAC (45) and validation was carried out using MOLPROBITY (46). Loose noncrystallographic restraints were used in the final refinement step. Complex assembly parameters were estimated with PISA (47). Automated docking was performed with PATCHDOCK (48) and HEX (49), using their respective Web servers and default parameters, inputting Protein Data Bank files from which water molecules had been removed. Structure figures were prepared with PYMOL (PyMOL Molecular Graphics System, Schrödinger, LLC).
Acknowledgments.
We thank Javier Varela for N-terminal sequence analysis, Jana Alonso for mass spectroscopy, Stefan Miller for providing the gp57 expression vector and the ESRF for measurement time on beamlines BM30A, ID23-1, and ID23-2. This research was sponsored by Grant BFU2008-01588 (to M.J.v.R.), a José Castillejo fellowship (J.M.O.), and a Formacion del Profesorado Universitario Fellowship (C.G.D.) from the Spanish Ministry of Education and Science. This work was also supported by the European Commission under Contract NMP4-CT-2006-033256 and by the Xunta de Galicia via an Angeles Alvariño fellowship (J.M.O.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. J.A.K. is a guest editor invited by the Editorial Board.
Data deposition: The coordinates and structure factors (of remote, peak, and inflection point data) have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 2XGF).
References
- 1.Hagens S, Loessner MJ. Application of bacteriophages for detection and control of foodborne pathogens. Appl Microbiol Biotechnol. 2007;76:513–519. doi: 10.1007/s00253-007-1031-8. [DOI] [PubMed] [Google Scholar]
- 2.Petrenko VA, Vodyanoy VJ. Phage display for detection of biological threat agents. J Microbiol Methods. 2003;53:253–262. doi: 10.1016/s0167-7012(03)00029-0. [DOI] [PubMed] [Google Scholar]
- 3.Parisien A, Allain B, Zhang J, Mandeville R, Lan CQ. Novel alternatives to antibiotics: Bacteriophages, bacterial cell wall hydrolases, and antimicrobial peptides. J Appl Microbiol. 2008;104:1–13. doi: 10.1111/j.1365-2672.2007.03498.x. [DOI] [PubMed] [Google Scholar]
- 4.Chanishvili N, Sharp RA. Literature Review of the Practical Application of Bacteriophage Research. Tbilisi, Georgia: Eliava Inst of Bacteriophage, Microbiology and Virology; 2009. [Google Scholar]
- 5.Wright A, Hawkins CH, Anggard EE, Harper DR. A controlled clinical trial of a therapeutic bacteriophage preparation in chronic otitis due to antibiotic-resistant Pseudomonas aeruginosa; a preliminary report of efficacy. Clin Otolaryngol. 2009;34:349–357. doi: 10.1111/j.1749-4486.2009.01973.x. [DOI] [PubMed] [Google Scholar]
- 6.Karam JD. Washington, DC: Am Society for Microbiology; 1994. Molecular biology of bacteriophage T4. [Google Scholar]
- 7.Yu F, Mizushima S. Roles of lipopolysaccharide and outer membrane protein OmpC of Escherichia coli K-12 in the receptor function for bacteriophage T4. J Bacteriol. 1982;151:718–722. doi: 10.1128/jb.151.2.718-722.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crowther RA, Lenk EV, Kikuchi Y, King J. Molecular reorganization in the hexagon to star transition of the baseplate of bacteriophage T4. J Mol Biol. 1977;116:489–523. doi: 10.1016/0022-2836(77)90081-x. [DOI] [PubMed] [Google Scholar]
- 9.Kostyuchenko VA, et al. Three-dimensional structure of bacteriophage T4 baseplate. Nat Struct Biol. 2003;10:688–693. doi: 10.1038/nsb970. [DOI] [PubMed] [Google Scholar]
- 10.Leiman PG, Chipman PR, Kostyuchenko VA, Mesyanzhinov VV, Rossmann MG. Three-dimensional rearrangement of proteins in the tail of bacteriophage T4 on infection of its host. Cell. 2004;118:419–429. doi: 10.1016/j.cell.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 11.Aksyuk AA, Leiman PG, Shneider MM, Mesyanzhinov VV, Rossmann MG. The structure of gene product 6 of bacteriophage T4, the hinge-pin of the baseplate. Structure. 2009;17:800–808. doi: 10.1016/j.str.2009.04.005. [DOI] [PubMed] [Google Scholar]
- 12.Riede I. Receptor specificity of the short tail fibres (gp12) of T-even type Escherichia coli phages. Mol Gen Genet. 1987;206:110–115. doi: 10.1007/BF00326544. [DOI] [PubMed] [Google Scholar]
- 13.Kostyuchenko VA, et al. The tail structure of bacteriophage T4 and its mechanism of contraction. Nat Struct Mol Biol. 2005;(12):810–813. doi: 10.1038/nsmb975. [DOI] [PubMed] [Google Scholar]
- 14.Aksyuk AA, et al. The tail sheath structure of bacteriophage T4: A molecular machine for infecting bacteria. EMBO J. 2009;28:821–819. doi: 10.1038/emboj.2009.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rossmann MG, Mesyanzhinov VV, Arisaka F, Leiman PG. The bacteriophage T4 DNA injection machine. Curr Opin Struct Biol. 2004;14:171–180. doi: 10.1016/j.sbi.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 16.King J, Laemmli UK. Polypeptides of the tail fibres of bacteriophage T4. J Mol Biol. 1971;62:465–477. doi: 10.1016/0022-2836(71)90148-3. [DOI] [PubMed] [Google Scholar]
- 17.Cerritelli ME, Wall JS, Simon MN, Conway JF, Steven AC. Stoichiometry and domainal organization of the long tail-fiber of bacteriophage T4: A hinged viral adhesin. J Mol Biol. 1996;260:767–780. doi: 10.1006/jmbi.1996.0436. [DOI] [PubMed] [Google Scholar]
- 18.Hashemolhosseini S, Stierhof YD, Hindennach I, Henning U. Characterization of the helper proteins for the assembly of tail fibers of coliphages T4 and lambda, J Bacteriol. 1996;178:6258–6265. doi: 10.1128/jb.178.21.6258-6265.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edgar RS, Lielausis I. Serological studies with mutants of phage T4D defective in genes determining tail fiber structure. Genetics. 1965;52:1187–1200. doi: 10.1093/genetics/52.6.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.King J, Wood WB. Assembly of bacteriophage T4 tail fibers: The sequence of gene product interaction. J Mol Biol. 1969;39:583–601. doi: 10.1016/0022-2836(69)90147-8. [DOI] [PubMed] [Google Scholar]
- 21.Montag D, Hashemolhosseini S, Henning U. Receptor-recognizing proteins of T-even type bacteriophages. The receptor-recognizing area of proteins 37 of phages T4 TuIa and TuIb. J Mol Biol. 1990;216:327–334. doi: 10.1016/S0022-2836(05)80324-9. [DOI] [PubMed] [Google Scholar]
- 22.Bartual SG, Garcia-Doval C, Alonso J, Schoehn G, van Raaij MJ. Two-chaperone assisted soluble expression and purification of the bacteriophage T4 long tail fibre protein gp37. Protein Expression Purif. 2010;70:116–121. doi: 10.1016/j.pep.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 23.van Raaij MJ, et al. Identification and crystallisation of a heat- and protease-stable fragment of the bacteriophage T4 short tail fibre. Biol Chem. 2001;382:1049–1055. doi: 10.1515/BC.2001.131. [DOI] [PubMed] [Google Scholar]
- 24.van Raaij MJ, Schoehn G, Burda MR, Miller S. Crystal structure of a heat and protease-stable part of the bacteriophage T4 short tail fibre. J Mol Biol. 2001;314:1137–1146. doi: 10.1006/jmbi.2000.5204. [DOI] [PubMed] [Google Scholar]
- 25.Thomassen E, et al. The structure of the receptor-binding domain of the bacteriophage T4 short tail fibre reveals a knitted trimeric metal-binding fold. J Mol Biol. 2003;331:361–373. doi: 10.1016/s0022-2836(03)00755-1. [DOI] [PubMed] [Google Scholar]
- 26.Vyas NK. Atomic features of protein-carbohydrate interactions. Curr Opin Struct Biol. 1991;1:732–740. [Google Scholar]
- 27.Holm L, Kaariainen S, Rosenstrom P, Schenkel A. Searching protein structure databases with DaliLite v.3. Bioinformatics. 2008;24:2780–2781. doi: 10.1093/bioinformatics/btn507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zorzopulos J, Kozloff LM. Identification of T4D bacteriophage gene product 12 as the base-plate zinc metalloprotein. J Biol Chem. 1978;253:5543–5547. [PubMed] [Google Scholar]
- 29.Leiman PG, Shneider MM, Mesyanzhinov VV, Rossmann MG. Evolution of bacteriophage tails: Structure of T4 gene product 10. J Mol Biol. 2006;358:912–921. doi: 10.1016/j.jmb.2006.02.058. [DOI] [PubMed] [Google Scholar]
- 30.Leiman PG, et al. Structure of bacteriophage T4 gene product 11, the interface between the baseplate and short tail fibers. J Mol Biol. 2000;301:975–985. doi: 10.1006/jmbi.2000.3989. [DOI] [PubMed] [Google Scholar]
- 31.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 32.Hendrix RW, Duda RL. Bacteriophage lambda PaPa: Not the mother of all lambda phages. Science. 1992;258:1145–1148. doi: 10.1126/science.1439823. [DOI] [PubMed] [Google Scholar]
- 33.Montag D, Schwarz H, Henning U. A component of the side tail fiber of Escherichia coli bacteriophage lambda can functionally replace the receptor-recognizing part of a long tail fiber protein of the unrelated bacteriophage T4. J Bacteriol. 1989;171:4378–4384. doi: 10.1128/jb.171.8.4378-4384.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Basle A, Rummel G, Storici P, Rosenbusch JP, Schirmer T. Crystal structure of osmoporin OmpC from E. coli at 2.0 Å. J Mol Biol. 2006;362:933–942. doi: 10.1016/j.jmb.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 35.Ericsson UB, Hallberg BM, Detitta GT, Dekker N, Nordlund P. Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal Biochem. 2006;357:289–298. doi: 10.1016/j.ab.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 36.Leslie AG. The integration of macromolecular diffraction data. Acta Crystallogr, Sect D: Biol Crystallogr. 2006;62:48–57. doi: 10.1107/S0907444905039107. [DOI] [PubMed] [Google Scholar]
- 37.Evans P. Scaling and assessment of data quality. Acta Crystallogr, Sect D: Biol Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 38.Collaborative Computational Project Number 4. The CCP4 Suite: Programs for Protein Crystallography. Acta Crystallogr, Sect D: Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 39.Sheldrick GM. A short history of SHELX. Acta Crystallogr, Sect A: Found Crystallogr. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 40.Vonrhein C, Blanc E, Roversi P, Bricogne G. Automated structure solution with AUTOSHARP. Methods Mol Biol. 2007;364:215–230. doi: 10.1385/1-59745-266-1:215. [DOI] [PubMed] [Google Scholar]
- 41.Abrahams JP, Leslie AG. Methods used in the structure determination of bovine mitochondrial F1 ATPase. Acta Crystallogr, Sect D: Biol Crystallogr. 1996;52:30–42. doi: 10.1107/S0907444995008754. [DOI] [PubMed] [Google Scholar]
- 42.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adams PD, et al. PHENIX—a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McCoy AJ, Read RJ. Experimental phasing: Best practice and pitfalls. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:458–469. doi: 10.1107/S0907444910006335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr, Sect D: Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 46.Chen VB, et al. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Krissinel E, Hendrick K. Inference of macomolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 48.Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005;33:W363–367. doi: 10.1093/nar/gki481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Macindoe G, Mavridis L, Venkatraman V, Devignes MD, Ritchie DW. HexServer: An FFT-based protein docking server powered by graphics processors. Nucleic Acids Res. 2010;38:W445–W449. doi: 10.1093/nar/gkq311. [DOI] [PMC free article] [PubMed] [Google Scholar]