

Summary
Precise contact between epithelial cells and their underlying basement membrane is crucial to the maintenance of tissue architecture and function. To understand the role that the laminin receptor dystroglycan (DG) plays in these processes, we assayed cell responses to laminin-111 following conditional ablation of DG gene (Dag1) expression in cultured mammary epithelial cells. Strikingly, DG loss disrupted laminin-111-induced polarity and β-casein production, and abolished laminin assembly at the step of laminin binding to the cell surface. Dystroglycan re-expression restored these deficiencies. Investigations of the mechanism revealed that DG cytoplasmic sequences were not necessary for laminin assembly and signaling, and only when the entire mucin domain of extracellular DG was deleted did laminin assembly not occur. These results demonstrate that DG is essential as a laminin-111 co-receptor in mammary epithelial cells that functions by mediating laminin anchoring to the cell surface, a process that allows laminin polymerization, tissue polarity and β-casein induction. The observed loss of laminin-111 assembly and signaling in Dag1−/− mammary epithelial cells provides insights into the signaling changes occurring in breast carcinomas and other cancers, where the binding function of DG to laminin is frequently defective.
Keywords: Dystroglycan, Laminin, Polarity, Mammary, Epithelial, Integrin
Introduction
Laminins are important structural and signaling components of basement membranes (BMs), serving as crucial modulators of BM assembly, cellular architecture, and tissue morphogenesis and function (Miner and Yurchenco, 2004). Interaction of laminins with epithelial cells influences cellular responses, such as adhesion, polarity and survival (Li et al., 2003). Genetic defects in laminin subunits result in muscular dystrophies and skin blistering (Miner and Yurchenco, 2004) and disregulated cell-laminin interactions have been implicated in the progression of cancers (Patarroyo et al., 2002). We are using the mammary gland as a model system to understand cellular interactions with laminins that regulate signals for epithelial architecture and function. Laminin-111, previously named laminin-1 (Aumailley et al., 2005), is a key player in these processes in mammary epithelial cells (MECs), inducing polarization (Gudjonsson et al., 2002; Slade et al., 1999) and β-casein production (Streuli et al., 1995). The identities of the multiple laminin receptors that elicit these effects are not completely understood, neither are the cooperative relationships among these receptors. Thus far, the integrins have been implicated (Muschler et al., 1999; Naylor et al., 2005; Slade et al., 1999; Streuli et al., 1991; Weaver et al., 1997) and, based on indirect evidence, we have postulated an important role for dystroglycan (DG) (Weir and Muschler, 2003).
DG is a heterodimeric glycoprotein encoded by a single gene (DAG1) and is located on cell surfaces in most adult tissues (Michele and Campbell, 2003). It consists of a transmembrane β-subunit of 43 kDa and a non-covalently associated, extracellular α-subunit of 120–200 kDa (Fig. 1A). The cytoplasmic domain possess known signaling motifs and links to the actin cytoskeleton, whereas the extracellular domain is capable of interacting with extracellular matrix (ECM) proteins, such as laminins, agrin and perlecan (Michele and Campbell, 2003). Binding of DG to laminin-111 occurs at the C-terminal laminin G-like (LG) globular domains LG4 and LG5 of the laminin α subunit (Ervasti and Campbell, 1993; Gee et al., 1993) (Fig. 1B). In skeletal muscle, DG serves as a transmembrane link between laminin-2 in the ECM and the intracellular actin cytoskeleton, possibly stabilizing the muscle-cell membrane (Ervasti and Campbell, 1993). In such cells, DG forms part of the dystrophinglycoprotein complex and certain defects in these components result in distinct muscular dystrophies (Durbeej and Campbell, 2002).
Fig. 1.

Generation of DG+/+ and partial-DG−/− MEC populations by adenoviral infection of immortalized mouse MECs. (A) Diagram of DG, including the extracellular α-DG subunit, with central mucin domain, and the transmembrane β-DG subunit. (B) Diagram of laminin-111 (LN), including the three subunits (α, β, γ), and the five C-terminal LG domains, with respective receptor binding sites. (C) Western blot of cell extracts (5 μg protein) prepared on different days after infection of the immortalized, floxed DG mouse MEpG cell line with control or Cre-recombinase-expressing adenovirus to generate DG+/+ and partial-DG−/− cell populations, respectively. The first lane (far left) represents uninfected cells at time 0. Blots were incubated with antibodies specific for α-DG, C-terminal β-DG or E-cadherin (loading control), followed by HRP-conjugated secondary antibodies. Sizes of molecular mass markers are shown in kDa. (D) Vertically paired immunofluorescent images of DG+/+ and partial-DG−/− MEpG cell populations using primary antibodies specific for α-DG or C-terminal β-DG, followed by FITC-labeled secondary antibodies (upper panel). Nuclei were stained with propidium iodide (bottom panel). Bar, 60 μm.
In some tissues, DG has been shown to play a role in BM formation. Knockout of Dag1 in mice is embryonic lethal, resulting in a lack of laminin recruitment and formational defects in Reichert’s membrane, an extra-embryonic BM (Williamson et al., 1997). In embryoid bodies of Dag1−/− mice (hereafter called DG−/− mice), disruption of the BM was seen with an almost total loss in laminin cell-surface binding (Henry and Campbell, 1998). In a skeletal muscle cell line, laminin-111 and laminin-211 polymerized while interacting with DG and integrins on the cell surface, suggesting a model for receptor-facilitated self assembly of laminins (Colognato et al., 1999).
Several studies have implicated DG in BM-induced epithelial functions, consistent with its location on the basolateral surface of epithelial cells contacting the BM, including those in the mammary gland (Durbeej et al., 1998). Based on antibody perturbation studies, DG plays a role in epithelial morphogenesis in kidney, lung, and salivary gland (Durbeej et al., 1995; Durbeej et al., 2001). Genetic disruption of DG expression revealed functions in survival of Drosophila epithelial cells (Deng et al., 2003) and epiblasts of embryoid bodies (Li et al., 2002). DG has also been implicated in epithelial polarity by a study in Drosophila (Deng et al., 2003) and by overexpression in a tumorigenic human MEC line (Muschler et al., 2002).
Since DG knockout in mice is embryonic lethal (Williamson et al., 1997), DG functions have not been assessed by genetic deletion in adult mammalian epithelial cells. Here, we have used a genetic approach in cultured cells to investigate the contribution of DG to laminin-111-induced epithelial architecture and function. We examined the effect of a DG gene deletion on laminin assembly and laminin-111-induced responses in adult mouse MEC lines. Results presented here demonstrate for the first time that DG serves as a crucial MEC co-receptor mediating cell responses to the BM that include epithelial polarization and β-casein induction. We also dissect the crucial receptor domains and present evidence that DG enacts these signals solely by anchoring laminin-111 to the cell surface, thereby facilitating laminin-111 polymerization and subsequent signaling.
Results
Establishment of DG+/+ and partial-DG−/− mouse MEC populations
To assess DG function in adult mouse MECs, a culture system was developed in which DG gene expression could be conditionally abrogated using Cre-lox recombination. We established two spontaneously immortalized MEC lines, MEpG and MEpL (mammary epithelial clones G and L), from mammary glands of floxed DG transgenic mice (see Materials and Methods) (Moore et al., 2002). Infection of these cells with Cre-recombinase-expressing adenovirus resulted in recombination between loxP sites flanking exon 2 of the DG gene, subsequent DG gene inactivation and creation of DG−/− MECs.
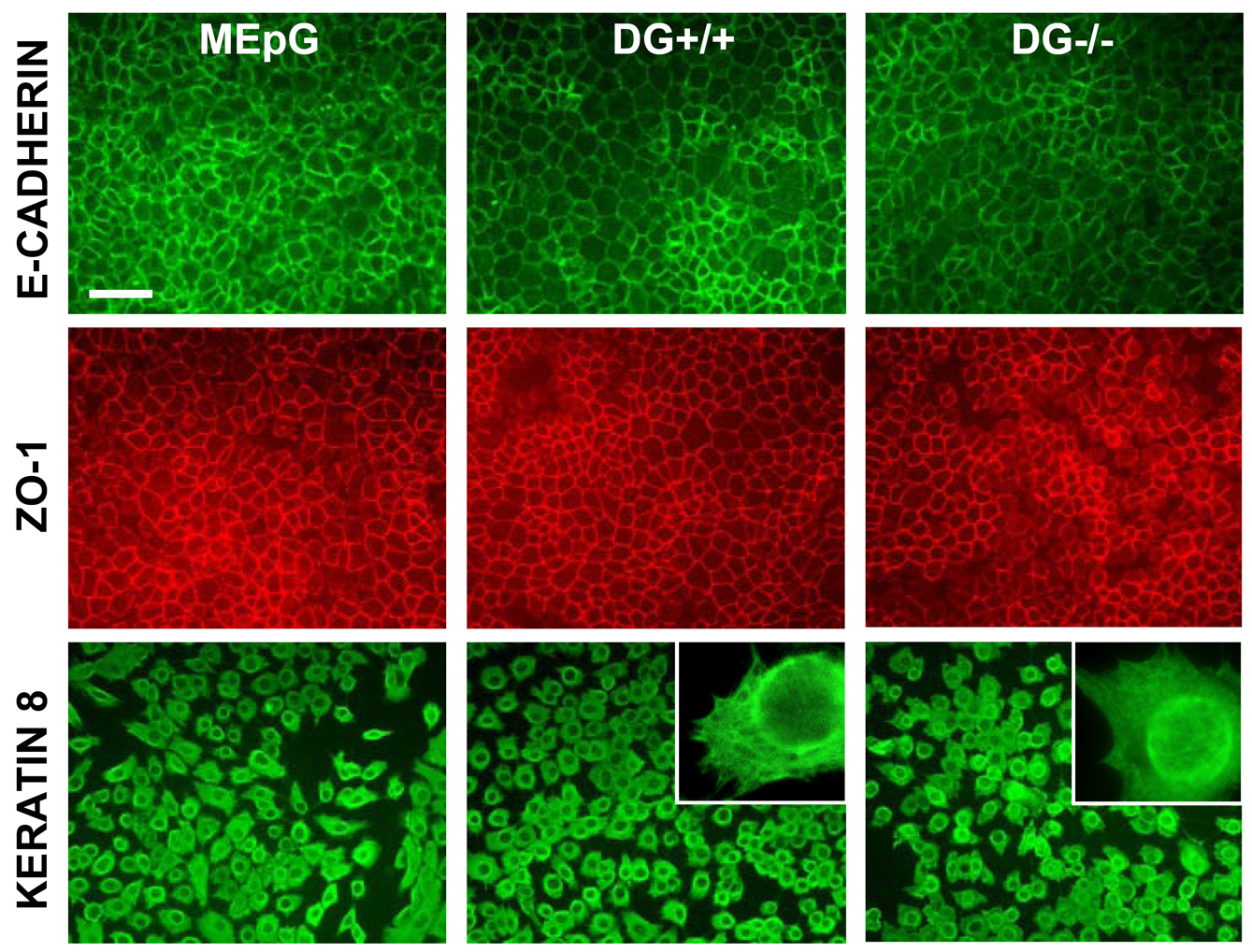
Both MEpG and MEpL cell lines were epithelial in nature, as judged by tightly packed, cobblestone-like morphologies and expression of typical MEC markers; immunodetection revealed expression of epithelial ZO-1, E-cadherin, and keratin 8 (supplementary material Fig. S1, left panel), but not myoepithelial smooth muscle α-actin or vimentin (data not shown). The normal complement of adhesion molecules, including DG, α6 and β1 integrins was also confirmed by immunodetection (below and data not shown). The MEpG cell line was used for laminin assembly and polarity assays; these cells did not express β-casein. The MEpL cell line was used for laminin assembly and β-casein assays, but not for polarity analyses. Many MEpL colonies produced pseudopod-like extensions when grown in 3D matrices, making assessment of polarization difficult.
Infection of the MEpG cell line with control adenovirus produced a control DG+/+ cell population which retained expression of DG protein over time, as shown by western blotting (Fig. 1C) and immunostaining (Fig. 1D) for α-DG and β-DG. Parallel infection of the MEpG cell line with Cre-recombinase-expressing adenovirus, to produce a DG−/− cell population, resulted in a near complete loss of DG protein expression, as demonstrated by western blotting for α-DG and β-DG (Fig. 1C). Immunostaining revealed that about 90% of the Cre-infected MECs lacked α-DG and β-DG expression (Fig. 1D). Similar results were obtained upon adenoviral infection of the MEpL cell line (supplementary material Fig. S2). DG+/+ and partial-DG−/− cell populations retained the epithelial marker expression profile seen in MEpG and MEpL parent cell lines prior to adenoviral exposure, showing that neither viral infection nor DG loss altered the epithelial phenotype (supplementary material Fig. S1 and data not shown).
DG loss and MEC polarity
To investigate the role of DG in laminin-111-induced MEC polarization, DG+/+ and partial-DG−/− cell populations were grown in 3D matrices containing collagen-1 with or without laminin-111, established culture models that can mimic the in vivo MEC response to the BM microenvironment. Polarity was assessed by examining the distribution of ZO-1, α6 integrin, nuclei and cytoskeletal actin.
Immunofluorescent staining of DG+/+ and DG−/− colonies grown in collagen I revealed a random distribution of nuclei, ZO-1 and α6 integrin (Fig. 2A, upper panel). Actin and DG (the latter in DG+/+ cells only) showed apolar patterns similar to α6 integrin (Fig. 2B, upper panel). Quantification of polarization using ZO-1 staining revealed few polar DG+/+ or DG−/− colonies in collagen I (Fig. 2C).
Fig. 2.
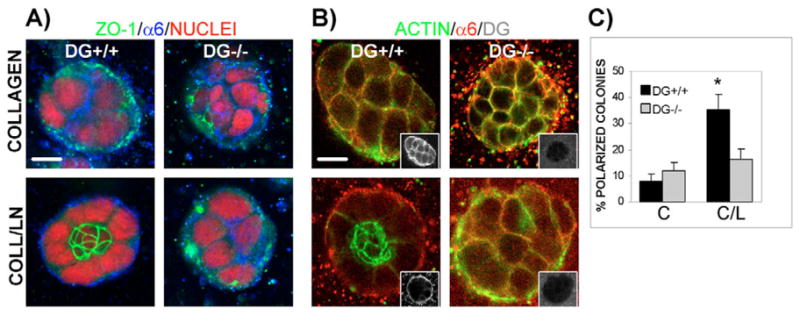
Loss of polarity in DG−/− colonies grown in a 3D matrix of collagen I–laminin-111. DG+/+ and DG−/− MEpG cells were grown in a 3D matrix of collagen I or collagen I–laminin-111 and co-immunostained. Confocal immunofluorescent images were taken at colony centers. Bars, 10 μm. (A) Staining using anti-ZO-1 and anti-α6 integrin antibodies, visualized with FITC- (green) and Cy5- (blue) labeled secondary antibodies, respectively, and propidium iodide to stain nuclei (red). (B) Staining using antibodies against α6 integrin and C-terminal β-DG (insets), detected with Rhodamine- (red) and Cy5- (blue changed to white for easier visualization) labeled secondary antibodies, respectively. Actin was seen using Alexa Fluor-488–phalloidin (green). Overlap between actin and α6 integrin staining appeared yellow. (C) Quantification of polarity in DG+/+ and DG−/− colonies grown in collagen I (C) or collagen I–laminin-111 (C/L) using ZO-1 as a polarity marker. Results are shown as the mean ± s.e.m. of four to six independent experiments, each with triplicate or quadruplicate counts. *P<0.01, for all paired combinations.
When laminin-111, a known inducer of polarization of mammary gland acini, was added to the collagen I matrix, DG+/+ cells polarized, displaying dramatic changes in the distribution of polarity markers and the cytoskeleton (bottom left images in Fig. 2A,B). ZO-1 and actin were found at the center of colonies, consistent with apical formation of tight junctions and an underlying cytoplasmic actin belt. DG and α6 integrin were localized basolaterally on cell surfaces and nuclei shifted to the colony periphery. Quantification using ZO-1 staining revealed a significant increase in polarized colonies in collagen-I–laminin-111 compared with collagen I alone (35.3% vs 8.0%, P<0.01; Fig. 2C).
Unlike DG+/+ cells, DG−/− cells did not significantly polarize in collagen-I–laminin-111 (bottom right images in Fig. 2A,B), exhibiting polarization levels similar to those seen in collagen I (Fig. 2C). Increasing laminin-111 from 35 μg to 75 μg in the collagen matrix did not elevate polarization of DG+/+ or DG−/− cells further (data not shown). The inability of DG−/− cells to polarize in response to laminin-111 was not due to a problem in tight-junction formation because ZO-1 still localized at cell-cell contacts in confluent monolayers of DG−/− cells grown on plastic (supplementary material Fig. S1, right panel, middle).
DG links laminin assembly and MEC polarity
DG has been implicated in laminin assembly in a few cell types (Colognato et al., 1999; Henry and Campbell, 1998; Williamson et al., 1997), but such a role in differentiated epithelial cells has not been investigated. To test the hypothesis that DG−/− MECs failed to polarize in response to laminin-111 because of laminin-assembly defects, DG+/+ and DG−/− cells in 3D polarity assays were immunostained using a polyclonal antibody raised against EHS (Engelbreth-Holm-Swarm) laminin subunits.
Apolar DG+/+ cells in collagen I showed punctate patterns of endogenously produced laminin on the outer surfaces of colonies (Fig. 3A, left of top panel) that co-localized with DG in many regions (Fig. 3B, top panel). Apolar DG−/− cells in collagen I lacked laminin surface staining (Fig. 3A, right of top panel). Importantly, in collagen-I–laminin-111 gels, polarized DG+/+ cells had an extensive laminin network on the outer surfaces of colonies (Fig. 3A, left of third panel) that colocalized with DG as a more continuous array than seen in collagen I alone (Fig. 3B, bottom panel). By contrast, apolar DG−/− cells in collagen-I–laminin-111 were deficient in laminin surface staining (Fig. 3A, right of third panel).
Fig. 3.

Loss of laminin binding and DG colocalization on the surface of DG−/− cells grown in a 3D matrix of collagen I or collagen-I–laminin-111. (A) Vertically paired confocal immunofluorescent images of DG+/+ and DG−/− MEpG cells grown in collagen I or collagen-I–laminin-111. Samples were co-immunostained with laminin, α6 integrin and C-terminal β-DG (insets) antibodies, followed by Rhodamine- (red), FITC- (green), and Cy5- (blue changed to white for easier visualization) labeled secondary antibodies, respectively. Images were taken at colony centers. (B) Confocal immunofluorescent images taken at the cell surface of DG+/+ colonies shown in A to reveal co-staining for laminin and β-DG, and their extent of co-localization. Arrows point to arrays of laminin. Bars, 10 μm.
To determine whether the observed lack of laminin staining on DG−/− cells was unique to the 3D ECM environment, laminin assembly was examined further using cell monolayers. Staining of DG+/+ cells for endogenous laminins revealed a diffuse, intracellular component and a punctate, extracellular pattern (Fig. 4A, left images above line). By contrast, cells lacking DG in the partial-DG−/− cell population exhibited intracellular, but not extracellular, laminin staining (Fig. 4A, right images above line). Laminin locations were confirmed by using unpermeabilized cells where only extracellular laminin staining was visible due to lack of intracellular access by the anti-laminin antibody (Fig. 4A, images below line).
Fig. 4.
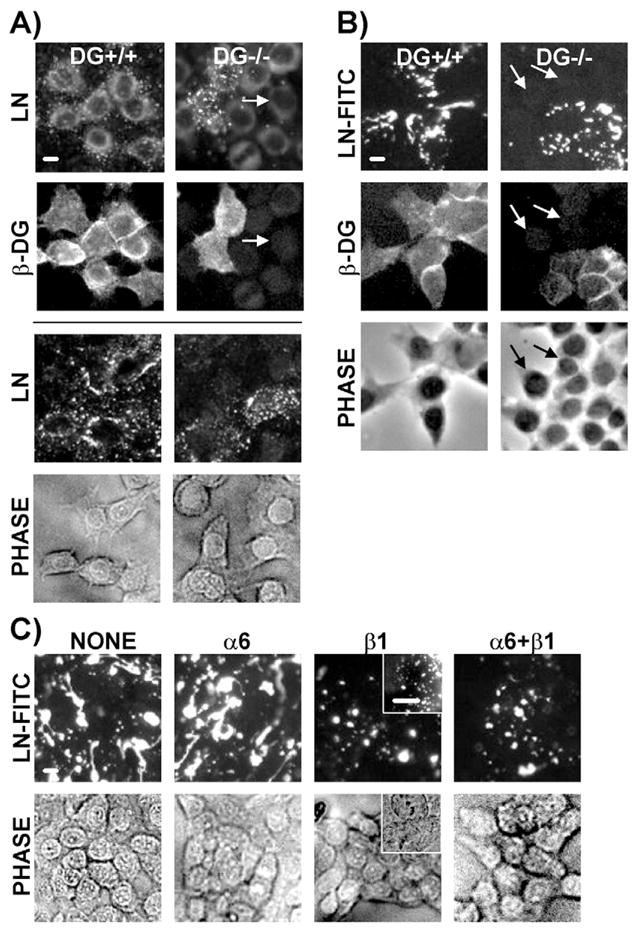
DG−/− cell monolayers failed to bind endogenous laminin or exogenous laminin-111-FITC. (A) Vertically paired immunofluorescent images of DG+/+ and partial-DG−/− MEpG cell populations co-stained using laminin and C-terminal β-DG antibodies, followed by Rhodamine- and FITC-labelled secondary antibodies, respectively, all in the presence of Tween-20 (images above line). Arrows point to a DG−/− cell that retained staining for intracellular but not cell-surface laminin. Cells immunostained for laminin in the absence of Tween-20 are shown below the line, with corresponding phase images. (B) Immunofluorescent images of DG+/+ and partial-DG−/− cell populations treated with 10 nM exogenously added laminin-111-FITC for 4 hours. Samples were co-stained using C-terminal β-DG antibody and Rhodamine-labeled secondary antibody. Corresponding phase images are shown in the bottom panel. Arrows point to a DG−/− cell lacking laminin-111-FITC staining. (C) Immunofluorescent images of DG+/+ cells treated with 10 nM exogenous laminin-111-FITC for 24 hours in the absence or presence of α6 and/or β1 integrin function blocking antibodies (upper panel). Corresponding phase images are shown in the bottom panel. Insets show cells incubated only with 10 nM AEBSF-treated laminin-111 for 24 hours, followed by immunostaining for laminin as described for upper images in A. Bars, 10 μm.
To observe the assembly of laminin-111 exclusively, cells were exposed to exogenous laminin-111-FITC for 4 hours and imaged without the use of antibodies. Examination of DG+/+ cell monolayers revealed punctate patterns and extensive patches of surface laminin-111-FITC (Fig. 4B, left panel). This binding was found to be time- and concentration-dependent with initial punctate patterns evolving into progressively larger, connected patches (data not shown). By contrast, cells lacking DG in the partial-DG−/− cell population did not show laminin binding at any time (4–24 hours) or even at a high concentration of 10 nM laminin-111-FITC (Fig. 4B, right panel; data not shown).
These findings demonstrate that DG−/− cells retain the ability to synthesize laminin, but are unable to bind endogenous laminins or exogenous laminin-111 either in monolayers or within a 3D matrix. Hence, DG serves as the crucial link between laminin-111 interaction with MECs and subsequent induction of polarization in a 3D environment.
To test whether DG and integrins cooperate in laminin assembly in MECs, we employed antibodies that block integrin function. DG+/+ cells were exposed to laminin-111-FITC for 24 hours, a time at which extensive polymerization had occurred (Fig. 4C, left panel). Blocking of α6 integrins had no effect on laminin-111 assembly (Fig. 4C, second panel from left). However, inhibition of β1 integrins diminished the extent of laminin-111 polymerization, but still allowed laminin cell-surface binding (Fig. 4C, third panel from left). This pattern was similar to that seen when DG+/+ cells were incubated with non-polymerizing laminin-111 and immunostained for laminin (Fig. 4C, insets in third panel from left). This laminin was generated by treatment with the serine protease inhibitor p-aminoethylbenzene-sulfonyl fluoride (AEBSF) (Colognato et al., 1999). Inclusion of both α6-integrin- and β1-integrin-blocking antibodies produced a result similar to that seen with the β1 antibody alone (Fig. 4C, right panel). These findings indicate that DG and β1 integrins cooperate in laminin-111 assembly on MECs, with DG serving as the initial binding site, enabling β1 integrins to participate in subsequent polymerization and signaling.
Chimeric MEC colonies do not polarize
Cues for epithelial polarization originate from the BM and neighboring cells (Yeaman et al., 1999). To determine whether DG influences polarization of neighboring cells, and the minimal number of DG expressors required for colony polarization, we analyzed the polarity of chimeric colonies containing both DG+/+ and DG−/− cells. Such colonies were produced in polarity assays using the partial-DG−/− MEpG population, which contained a subpopulation of DG+/+ cells (Fig. 1). Growth of cells within the 3D matrix led to a non-random distribution of DG+/+ and DG−/− cells in the final chimeric colonies. Staining of chimeric colonies in collagen-I–laminin-111 for actin and α6 integrin revealed an apolar phenotype, even when half or more of the cells in the colony were DG+/+ (Fig. 5A). Quantification using α6 integrin staining showed minimal levels of overall polarization in chimeric colonies, even when the majority of cells in a colony were DG-expressing (data are given as the mean ± s.d. and were 0.98±1.34%; n=5 counts × 40 colonies per count). Interestingly, laminin staining was visible only on the surface of DG+/+ cells, where it co-localized with DG in an extensive reticular network (Fig. 5B,C). These observations suggest that global DG expression in MEC colonies is essential for laminins to assemble around the entire colony and trigger cooperative participation of all cells in colony polarization.
Fig. 5.
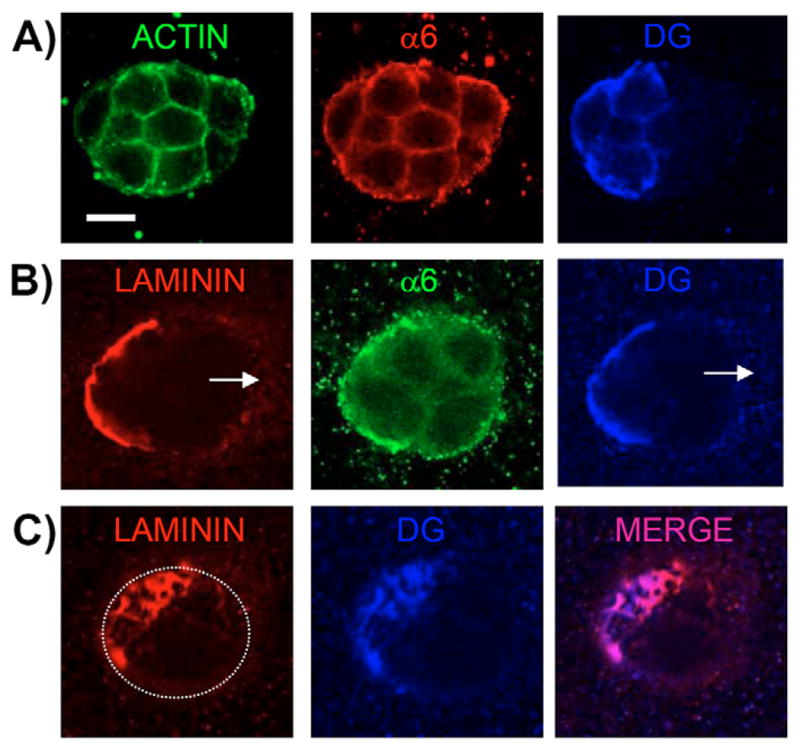
Partial-DG+/+ colonies grown in a 3D matrix of collagen-I–laminin-111 retain laminin and DG colocalization on the surface of DG+/+ cells only, but fail to polarize. (A,B) Confocal immunofluorescent images taken at the center of partial-DG+/+ MEpG colonies grown in collagen-I–laminin-111 and co-immunostained as follows: (A) α6 integrin and C-terminal β-DG antibodies were detected using Rhodamine- (red) and Cy5- (blue) labeled secondary antibodies, respectively. Actin was visualized with Alexa Fluor-488–phalloidin (green). (B) Laminin, α6 integrin and C-terminal β-DG antibodies were detected using Rhodamine- (red), FITC- (green), and Cy5- (blue) labeled secondary antibodies, respectively. Arrows show part of colony surface lacking laminin and β-DG staining. (C) Confocal immunofluorescent images were taken at the cell surface of the colony shown in B to reveal co-staining for laminin and β-DG, and their extent of co-localization. Dotted outline represents outer edge of colony. Bar, 10 μm.
DG loss disrupts β-casein production in MECs
Previous results indicated that BM-induced β-casein expression in MECs required β1 integrins, α6β4 integrin, and a laminin receptor binding the LG4 and LG5 domains (Faraldo et al., 1998; Muschler et al., 1999; Streuli et al., 1991). To determine directly the role of DG in laminin-111-induced β-casein production, DG+/+ and partial-DG−/− MEC populations (derived from the MEpL cell line) were tested in β-casein assays using lactogenic hormones and a laminin-111 overlay (Streuli et al., 1995).
DG+/+ and the partial-DG−/− cell populations produced β-casein protein in response to laminin-111 in the presence, but not absence, of lactogenic hormones, as expected (Fig. 6A). However, the partial-DG−/− cell population showed a drastic reduction in laminin-111-induced β-casein levels. As expected, no β-casein was detected in either cell population upon omission of the laminin-111 overlay (Fig. 6A).
Fig. 6.

Loss of β-casein production in response to laminin-111 in DG−/− cells. (A) Western blot of cell extracts (10 μg protein) prepared from DG+/+ and partial-DG−/− MEpL cell populations incubated with laminin-111 overlay in the absence (–) or presence (+) of prolactin and hydrocortisone. Blots were incubated with antibodies specific for β-casein or E-cadherin (loading control), followed by HRP-conjugated secondary antibodies. Sizes of molecular mass markers are shown in kDa. Dotted lines separate non-adjacent lanes derived from the same blot.
(B) Immunofluorescent images of DG+/+ and partial-DG−/− cell population treated with 10 nM exogenously added laminin-111-FITC for 4 hours. Samples were co-stained using C-terminal β-DG antibody and Rhodamine-labeled secondary antibody. Corresponding phase images are shown in the bottom panel. Arrows point to a DG−/− cell lacking laminin-111-FITC binding.
Analysis of the ability of DG+/+ and partial-DG−/− cells to bind laminin revealed results similar to those seen with the MEpG cell line (Fig. 4). DG, but not DG, cell monolayers bound endogenous laminins (data not shown) and exogenous laminin-111-FITC (Fig. 6B). These results show that the decrease in laminin-111-induced β-casein levels in the partial-DG−/− cell population is due to disruption of laminin-111 binding to DG−/− cells.
The DG extracellular domain alone is crucial to laminin assembly
The β-subunit of DG contains cytoplasmic sites potentially recognized by SH3, SH2 and WW domain proteins (Ibraghimov-Beskrovnaya et al., 1992; Pawson, 2004). To investigate whether DG plays an active signaling role in MEC functions, three deletion mutants of the β-DG cytoplasmic domain (DEL A, B and C) were generated and tested (supplementary material Fig. S3). DEL A lacked the entire cytoplasmic domain, except for six amino acids beyond the transmembrane region. DEL B had an internal deletion resulting in retention of the C-terminal 15 amino acids and proximal removal of several potential WW, SH3 and SH2 domain protein recognition sites. DEL C lacked the C-terminal 15 amino acids, which contain proven interaction sites for SH3-, SH2- and WW-domain proteins (Ilsley et al., 2002; Sotgia et al., 2001; Yang et al., 1995).
We generated a pure DG−/− cell line (entirely lacking DG protein expression) by single-cell cloning from the partial-DG−/− MEpG cell population and then infected the DG−/− cell line with either empty retroviral vector (VEC) or vector encoding full-length DG (wtDG), DEL A, DEL B, or DEL C. Western blots showed that VEC cells were deficient in α- and β-DG protein (Fig. 7A, left panels), whereas the other infected cells expressed α-DG protein the same size as DG+/+ cells (Fig. 7A, upper left panel). An N-terminal β-DG antibody verified expression of the full-length β-subunit in wtDG cells and truncated versions in DG-mutant cells (Fig. 7A, upper right panel). A β-DG antibody recognizing an epitope in the C-terminal 15 amino acids detected the β-subunit in wtDG and DEL B cells, but not in DEL A or C cells, verifying the lack of this epitope in the latter two populations (Fig. 7A, middle left panel).
Fig. 7.
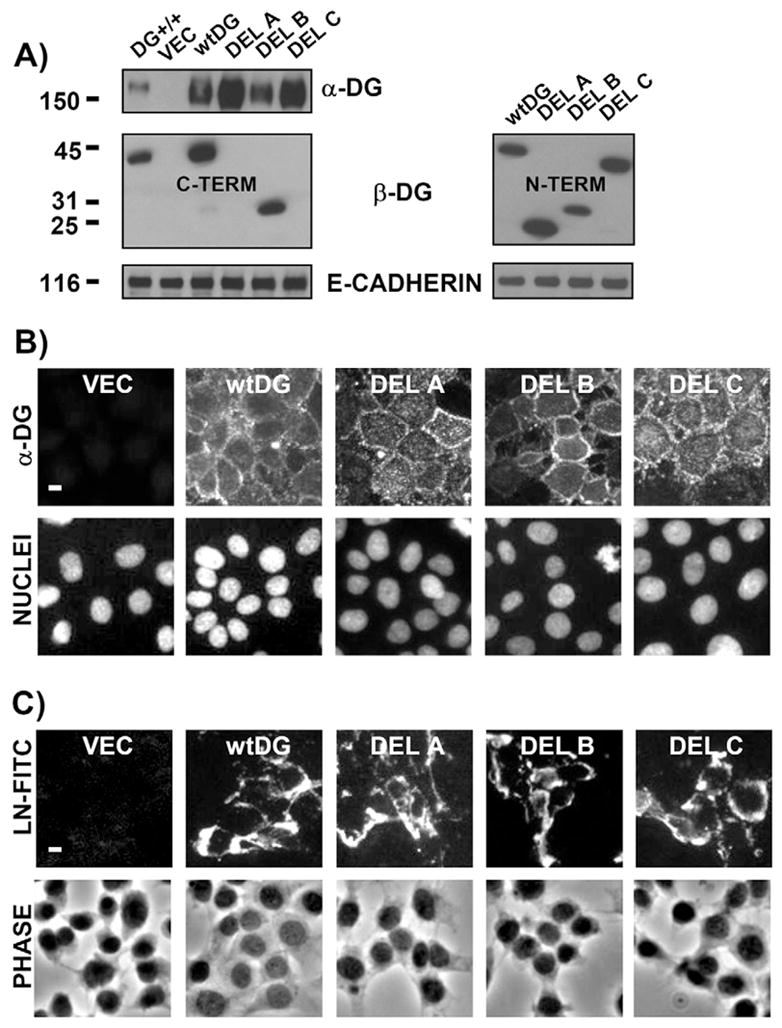
Re-expression of full-length DG or DG mutants in a completely DG−/− cell line restored laminin-111 binding on monolayer cell surfaces. (A) Western blot of cell extracts (10 μg protein) prepared from DG+/+ cells and from a DG−/− cell line (derived from MEpG cells) infected with retroviral vector (VEC) or that encoding full-length DG (wtDG) or various β-DG cytoplasmic deletions (DEL A, B and C). Blots were incubated with antibodies specific for α-DG, N-terminal β-DG (right panel), C-terminal β-DG (left panel), or E-cadherin (loading control), followed by HRP-conjugated secondary antibodies. Sizes of molecular mass markers are shown in kDa. (B) Paired immunofluorescent images of cells in A, co-stained for α-DG and nuclei, using FITC-labeled secondary antibody and propidium iodide, respectively. (C) Immunofluorescent images of cells in A, treated for 4 hours with 10 nM exogenously added laminin-111-FITC. Corresponding phase images are shown in the bottom panel. Bars, 10 μm.
α-DG was correctly localized to the surface of cells infected with wtDG or DG mutant (wtDG and DG-mutant cells, respectively), and was not detected in cells infected with the empty retroviral vector (VEC cells) (Fig. 7B). Laminin-111-FITC is bound and assembled at the surface of wtDG cells but not VEC cells, showing that DG re-expression corrected the laminin assembly defects (Fig. 7C). All DG-mutant cells also assembled cell surface laminin-111-FITC comparable to wtDG cells, revealing that, surprisingly, DG cytoplasmic domains were not required (Fig. 7C). Identical results were obtained upon expression of wtDG or DG mutants in DG−/− cells derived from the MEpL cell line (data not shown).
Analysis of laminin-111-induced polarity in VEC, wtDG and DG-mutant cells demonstrated very few polar colonies in collagen I (Fig. 8A, top panel; 8B). Addition of laminin-111 to the collagen-I gel resulted in significant increases in the number of polar colonies for all but the DG−/− (VEC) cells (Fig. 8A, middle panel; 8B). In addition, immunostaining revealed laminin localization on colony surfaces of all but VEC cells grown in collagen-I–laminin-111 (Fig. 8A, bottom panel). Likewise, laminin-111-induced β-casein levels were restored in wtDG and DG-mutant cells compared with VEC cells (Fig. 9). However, even in the complete absence of DG expression, low levels of β-casein were seen in VEC cells treated with laminin-111. As in Fig. 6A, no β-casein was detected in any of the cell populations in the absence of a BM overlay.
Fig. 8.

Expression of full-length DG and DG mutants in a pure DG−/− MEpG cell line restored polarity and surface laminin in an 3D matrix of collagen-I-laminin-111. (A) Confocal immunofluorescent images taken at the center of colonies grown in collagen I (upper panel) or collagen-I–laminin-111 (middle panel). Samples were co-stained for ZO-1, α6 integrin and nuclei as described in Fig. 2A. Bottom panel shows laminin staining of a second group of colonies grown in collagen-I–laminin-111, visualized with Rhodamine-labeled secondary antibody (red). Cells are described in Fig. 7A. Bars, 10 μm. (B) Quantification of polarity in colonies grown in collagen I (C) or collagen-I–laminin-111 (C/L) using ZO-1 as a polarity marker. Results are shown as the average ± s.e.m. of 3–5 independent experiments, each with triplicate or quadruplicate counts. (^) or (*)=P<0.001 for all paired combinations except with each other.
Fig. 9.

Expression of full-length DG and DG mutants in a pure DG−/− MEpL cell line restored β-casein protein expression in response to laminin-111. Western blot of cell extracts prepared from cells infected with retroviral vector (VEC) or that encoding full-length DG (wtDG) or various β-DG cytoplasmic deletions (DEL A, B and C) and incubated with a laminin-111 overlay in the absence (–) or presence (+) of prolactin and hydrocortisone. Blots were incubated with antibodies specific for β-casein or E-cadherin (loading control), followed by HRP-conjugated secondary antibodies. Sizes of molecular mass markers are shown in kDa.
Additional DG mutants were created to investigate the role of DG transmembrane and extracellular domain sequences in laminin assembly (supplementary material Fig. S3). The cytoplasmic and transmembrane domains of DG were replaced by 44 amino acids encompassing the transmembrane domain of the TNF-α cleaving enzyme (TACE) (Moss et al., 1997) and an unrelated ten amino acid long cytoplasmic tail. When expressed in the DG−/− MEpL cells, this fusion protein (DG-tmf) permitted laminin anchoring and assembly (Fig. 10). A mutant possessing a large deletion in the C-terminal half of the mucin domain (DEL E) also functioned like the wild-type protein. Importantly, only expression of a DG cDNA lacking the entire mucin domain (DEL D) failed to bind and assemble laminin (Fig. 10).
Fig. 10.

The DG extracellular domain alone is crucial to laminin assembly. (A) Western blot of cell extracts prepared from a DG−/− MEpL cell line (entirely DG−/−) infected with retroviral vector (VEC) or that encoding full-length DG (wtDG), a fusion protein comprised of the extracellular DG sequences fused to the transmembrane region of TACE (DG-tmf), or deletions within the α-DG mucin domain (DEL D and E). Blots were incubated with antibodies specific for α-DG, N-terminal β-DG, or E-cadherin (loading control). Sizes of molecular mass markers are shown in kDa. (B) Immunofluorescent images of cells in A, treated for 4 hours with 10 nM exogenously added laminin-111-FITC. Corresponding phase images are shown in the bottom panel. Bar, 10 μm.
Discussion
Laminins are key signaling modulators of cellular architecture, and function during embryonic and post-natal development (Li et al., 2003; Miner and Yurchenco, 2004). In MECs, laminin-111 interaction with cell surface receptors is important for induction and retention of differentiated features, including cellular and tissue polarity and β-casein expression (Gudjonsson et al., 2002; Slade et al., 1999; Streuli et al., 1995). Using a DG genetic deletion in adult MEC lines, we show here that DG plays a crucial role as a laminin-111 co-receptor in MEC functions, appearing to act at an initial and crucial step (see model, Fig. 11). We present evidence that DG acts by mediating laminin-111 anchoring to the MEC surface, such that cell-surface laminin-111 assembly can occur, and induction of signals linked to polarity and β-casein levels can proceed via other co-receptors.
Fig. 11.

Model for the role of DG as a MEC co-receptor in laminin-111 assembly and laminin-111-induced functions. α-DG on the MEC surface serves as the initial anchoring site for laminin-111 (LN) monomers by interacting with their C-terminal LG domains (step 1). The laminin-111–DG complexes recruit β1 integrin (INT) co-receptors, which contribute to laminin-111 polymerization (step 2). Subsequent activation of co-receptors, possibly integrins (INT), influences intracellular signaling pathways leading to polarity and β-casein induction (step 3).
Role of DG in laminin assembly
Although most laminins self-assemble spontaneously, the process is facilitated by interaction with cell surface receptors. This mechanism involves receptor binding of monomeric laminin through its C-terminal G domain and laminin polymerization through resultant interactions between neighboring N-terminal short arms (Colognato et al., 1999). This polymerization is crucial for recruitment of other BM proteins, cytoskeletal reorganization and signaling events (Colognato et al., 1999).
Data presented here demonstrate in an adult epithelial cell type (MECs) that DG is essential for receptor-facilitated laminin anchoring and assembly, with possible cooperation from β1 integrins at the level of assembly. These results help to explain BM defects seen upon DG reduction or loss in brain cells, Reichert’s membrane, and Drosophila epithelia (Deng et al., 2003; Michele et al., 2002; Moore et al., 2002; Williamson et al., 1997). They also support observations made using embryonic stem cells cultured in monolayer (Henry and Campbell, 1998; Henry et al., 2001b; Lohikangas et al., 2001). However, the requirement for DG in laminin-BM assembly may be tissue-specific. In one study, assays for BM assembly in ES-derived embryoid bodies show no BM defects in DG−/− embryoid bodies, but show a dramatic loss of epiblast cell survival (Li et al., 2002), although another study shows loss of laminin and BM assembly in DG−/− embryoid bodies produced by a method that did not generate a differentiated epiblast layer (Henry and Campbell, 1998). In addition, normal BMs are observed upon DG loss in skeletal muscle and some embryonic tissues (Cohn et al., 2002; Michele et al., 2002; Williamson et al., 1997). In Schwann cells and fibroblasts, certain sulfated glycolipids can mediate laminin-BM assembly (Li et al., 2005), raising the possibility that DG and sulfated glycolipids functionally overlap.
Because DG can mediate laminin assembly and signaling in the absence of endogenous transmembrane and cytoplasmic signaling domains, and also function in the presence of a large internal deletion of the extracellular domain, it appears that laminin anchoring to the cell surface is the main role for DG in the initiation of assembly and signaling. This model is consistent with observations in fibroblasts and Schwann cells showing that laminin binding to cell-surface glycolipids is also sufficient to initiate assembly and signaling (Li et al., 2005). Importantly, no exogenous laminin-111 binding was observed at the surface of DG−/− cells, demonstrating that no other molecule compensated for the role of DG in laminin anchoring to MECs. This result also suggests that co-receptors, such as the β1 integrins, require the interaction of DG with laminin-111 prior to recruitment and/or activation. A recent study in intestinal epithelial cells reported direct interaction of DG and β1 integrins by co-immunoprecipitation (Driss et al., 2005), something we have not yet observed in MECs. This same study also reported an enhancement of integrin-laminin-111 interactions that is dependent on DG cytoplasmic sequences, but this observation is inconsistent with our results in MECs, where deletion of DG cytoplasmic sequences did not perturb function.
DG mediates signals for epithelial architecture and function
Our results show that DG also plays an essential role in mediating laminin-111-induced MEC functions, including tissue architecture and tissue-specific gene expression. DG−/− cells failed to polarize and showed markedly reduced β-casein production because of defects in laminin-111 binding. In addition, our finding that laminin-111 and DG signaling pathways linked to polarity and β-casein levels were independent of the β-DG cytoplasmic domain suggests that the functional coupling of DG with co-receptors enacts signaling. Candidate co-receptors include α6β4 or β1 integrins that influence polarity (Faraldo et al., 1998; Slade et al., 1999; Weaver et al., 1997) and β-casein levels (Faraldo et al., 1998; Muschler et al., 1999; Streuli et al., 1991). A partial, albeit weak, receptor compensation for DG loss was seen in laminin-111-induced β-casein assays, suggesting that, in the presence of high laminin-111 levels, some spontaneous laminin self-assembly may take place, or interaction with a less effective laminin receptor may occur. Whatever the case, DG is still needed as a laminin-111 co-receptor to allow efficient β-casein production.
The results reported here provide a molecular mechanism to explain why overexpression of DG is capable of reverting and normalizing breast tumor cells, and why the functional status of DG correlates strongly with a tumor cells ability to polarize (Muschler et al., 2002). In addition, they explain the observed requirement for multiple MEC receptors in β-casein expression, including a receptor for the laminin LG4-5 domain that is likely to be DG (Muschler et al., 1999; Streuli et al., 1995). They explain the loss of β-casein expression upon siRNA knockdown of DG in HC-11 cells (Sgambato et al., 2006). The results can also explain the role of DG in establishing Drosophila epithelial polarity (Deng et al., 2003). However, DG knockout in mouse embryoid bodies does not affect polarization of epiblast cells (Li et al., 2002), demonstrating that DG is not universally required for polarity in mammalian cells. Epiblast differentiation and polarization are affected in mice lacking the laminin α1 LG4 and LG5 modules, hinting at the existence of other receptors for these modules (Scheele et al., 2005).
Our observation that MEC chimeras, composed of DG+/+ and DG−/− cells, did not polarize, stresses the importance of laminin assembly along the entire basal epithelial surface to establish normal tissue architecture; loss of laminin assembly on even a minority of cells is sufficient to disrupt polarity in the entire acinar structure. This result illustrates the required integration of both cell-cell and cell-BM interactions to establish cellular and tissue polarity (Yeaman et al., 1999). DG−/− cells of chimeric colonies lacked the ability to bind surface laminin-111 and did not receive the necessary external BM cue for activation of intracellular polarity pathways, which include the establishment of proper cell-cell junctions. Consequently, with direct contact of DG+/+ and DG−/− cells, the defect of the DG−/− cells was dominant.
Significance of DG in vivo and in disease
Our findings have important implications for understanding the abnormal behavior of carcinomas of the breast and other tissues. In breast, prostate and colon cancers, loss in DG detection correlates with tumor progression (Henry et al., 2001a; Sgambato et al., 2003). In many carcinoma cell lines, including those of the breast, DG lacks laminin binding ability because of glycosylation changes and/or proteolytic processing (Losasso et al., 2000; Muschler et al., 2002; Singh et al., 2004). Our results reveal that localized disruption of the DG–laminin-111 link in MECs leads to losses in laminin-111-induced responses important to normal epithelial architecture and function, with impact on neighboring cells as well. Thus, loss of DG function is a plausible and attractive explanation for some of the aberrant cell responses to the BM that are evident in cancer progression.
Materials and Methods
Production of immortalized floxed DG mouse MEpG and MEpL cell lines
Mammary glands from mid-pregnant (embryonic day 16–18) homozygous floxed DG transgenic mice (Moore et al., 2002) were digested at 37°C with 0.2% trypsin (Invitrogen), 0.2% collagenase A (Roche), DME/F12 (HyClone), 5% FBS (HyClone), 5 μg/ml insulin (Sigma), and 50 μg/ml gentamicin (Invitrogen), followed by centrifugation (400 g, 5 s) until fibroblast-free. Cells were grown in plastic flasks (MEpG cell line) or collagen-I gels (Cellagen; ICN Biomedicals) (Kittrell et al., 1992) for 5 weeks prior to collagenase A digestion of the gel and cell transfer to plastic flasks (MEpL cell line). Cells were grown in complete media [DME/F12, 2% FBS, 10 μg/ml insulin, 5 ng/ml EGF (BD Biosciences), and 50 μg/ml gentamicin] in humidified 5% CO2 at 37°C and passaged using dispase II (Roche) until spontaneously immortalized, after which 0.025% trypsin with 0.27 mM EDTA (Cellgro) were used. Clones were obtained by limiting dilution and screened for expression of epithelial markers by immunofluorescent staining.
Generation of DG+/+ and partial-DG−/− mouse MEC populations
Adenoviral vectors (Microbix) were amplified twice in QBI-293 packaging cells (Quantum Biotechnologies), grown in DMEM (Invitrogen), 2 mM Gln, 10% FBS, and 10 μg/ml gentamicin. Immortalized floxed DG mouse MEC lines (MEpG, MEpL) were infected with either control (Ad.floxlacZ1) or Cre-recombinase-expressing (Ad.creM1) adenoviral supernatants with multiplicity of infection of 40–50.
Expression of full-length DG and mutants in pure DG−/− MEC lines
Human DG coding sequence was subcloned from pLXSN vector (Muschler et al., 2002) into the EcoRI site of the retroviral expression vector, pBMN-IRES-PURO (Kinoshita et al., 1997). From this construct, β-DG cytoplasmic deletion mutants were constructed using the QuikChange XL site-directed mutagenesis kit (Stratagene) and verified by sequencing. DEL A, B, C, D, and E lacked amino acids 780–895, 806–880 and 881–895, 315–485, and 400–485, respectively (Fig. S3). DG-tmf was constructed from the ligation of two PCR products spanning amino acids 1–739 of DG and amino acids 656–699 of the human TNF-α cleaving enzyme (TACE) gene. The reverse primer for the TACE PCR product included the coding sequence for ten additional amino acids at the C-terminus (LDEESILKQE), representing the Myc tag. Retrovirus was generated using Phoenix-ECO packaging cells grown in DME/H21 (UCSF Cell Culture Facility) and 10% FBS, and transfected using calcium phosphate (Sambrook et al., 1989).
DG−/− clones were obtained by limiting dilution of partial-DG−/− MEC populations and screened by immunostaining for lack of DG expression. Clones were seeded in 100-mm dishes, infected with 2 ml of retroviral supernatant, 6 ml of complete media, and 8 μg/ml polybrene, and selected in complete media with 5–10 μg/ml puromycin (Sigma).
3D polarity assays
Trypsinized cells (between 104 and 105 cells) were added to 300 μl of collagen I (Cellagen; ICN Biomedicals) or collagen-I–laminin-111 (35 μg; Sigma) on ice. Matrices were solidified at 37°C and covered with complete media that was changed every 2 days. On days 6 or 7, samples were immunostained. For polarity quantification, colonies with >3 nuclei were considered polar if ZO-1 staining was centrally located within the colony. For statistical analysis, comparisons between groups were subject to one-way analysis of variance and differences between means were determined using Fisher’s least significant difference method.
β-casein and laminin assembly assays
β-casein assays were performed as previously described (Muschler et al., 1999), except that 5 μg/ml prolactin and serum-free complete media were used. To assess laminin assembly, laminin-111-FITC was prepared by dialysing laminin-111 (Sigma) in PBS, 10 μM CaCl2, and incubating with NHS-fluorescein (Pierce) for 2 hours at 4°C in the dark. Dialysis was repeated, and laminin-111-FITC was measured by the Lowry protein assay (Peterson, 1977). Laminin-assembly results observed using lammin-111-FITC were identical to results obtained by immunostaining for laminins after addition of unlabeled laminin-111 (data not shown). Laminin-111 was treated with p-aminoethylbenzene-sulfonyl fluoride (AEBSF) (Calbiochem) as described (Colognato et al., 1999). Cells grown on Lab-Tek II CC2 glass chamber slides (Nalge Nunc) were immunostained following incubation at 37°C in the dark, in serum-free complete media with 10 nM AEBSF-treated laminin-111 for 24 hours or 10 nM laminin-111-FITC for 4 or 24 hours (the latter with or without antibodies that block integrin function).
Immunofluorescent staining
Cells grown on Lab-Tek II CC2 glass chamber slides (Nalge Nunc) or in 3D polarity assays were washed twice in PBS. Some 3D samples were digested with 0.2% collagenase A in complete media at 37°C to remove matrix for easier counting. For actin–α6-integrin, DG and laminin staining, samples were fixed in 2% formaldehyde in PBS for 10 minutes at room temperature, and washed in PBS, 25 mM glycine for 3×10 minutes. For ZO-1–α6-integrin staining, samples were fixed in acetone-methanol (1:1) at −20°C for 5 minutes and air-dried. After blocking in PBS, 10% goat serum (Sigma), 0.1% Tween-20 for 1 hour at room temperature, samples were incubated in blocking solution overnight at 4°C with primary antibodies, followed by 1 hour at room temperature with fluorescent secondary antibodies. For actin staining, Alexa Fluor-488–phalloidin (Molecular Probes) was used for 20 min at room temperature, using a 1:21 dilution in blocking solution. Nuclei were counterstained with 10 μg/ml propidium iodide (Sigma). Washes between antibody incubations were 3×10 minutes in PBS. Samples were mounted in Vectashield mounting media (Vector Laboratories) with glass coverslips.
Microscopy
Immunofluorescent images were obtained with a Nikon Eclipse TE2000-U inverted microscope, Photometrics Cool SNAP HQ camera, MetaMorph 6.1r1 software (Universal Imaging Corporation), and a Nikon Plan Ph1 DL 20× objective (0.40 NA) (Fig. 4C inset obtained with Nikon Plan Apo DIC H 60× oil objective of 1.40 NA). Confocal images were obtained with the same microscope and a Nikon D-Eclipse C1 confocal attachment, Nikon EZ-C1 2.10 software, channel series setup, and the 60× oil objective. Images were cropped and adjusted for contrast using Adobe Photoshop 7.
Western blots
Cell extracts were prepared in 62.6 mM Tris-HCl, pH 6.8, 2% SDS, 5% glycerol, 5 μg/ml pepstatin (Sigma), 500 μM AEBSF, 150 nM aprotinin, 1 μM E-64, 0.5 mM EDTA, 1 μM leupeptin (all from Calbiochem) and measured using the Lowry protein assay (Peterson, 1977). SDS-PAGE was performed under reducing conditions using equal amounts of protein and 4–12% or 4–20% polyacrylamide Tris-glycine gradient gels. Proteins were electrophoretically transferred to Immobilon-P membranes (Millipore). Blots were blocked in 5% non-fat dry milk in TBS-T (50 mM Tris-HCl pH 7.4, 100 mM NaCl, 0.1% Tween-20) for 1 hour at room temperature, followed by incubation in blocking buffer overnight at 4°C with primary antibodies, then 1 hour at room temperature, with HRP-conjugated secondary antibodies.. Blots were washed in TBS-T after antibody incubations, and bands were visualized with the ECL/ECL Plus systems (Amersham Pharmacia).
Antibodies
Mouse monoclonal antibodies (mAbs) specific for C-terminal β-DG (NCL-b-DG; Novocastra), N-terminal β-DG (BD Biosciences), E-cadherin (BD Transduction Labs) and β-casein (Kaetzel and Ray, 1984) were used for immunoblotting at 1:200, 1:500, 1:5000 and 1:2000, respectively. The former antibody was used for immunostaining at 1:50. Rabbit polyclonal Abs (pAbs) specific for ZO-1 (Zymed) or laminin purified from the BM of Engelbreth-Holm-Swarm (EHS) mouse sarcoma (Sigma) were used for immunostaining at 1:100 and 1:40, respectively. Rat mAb GoH3 specific for α6 integrin (Chemicon) was used for immunostaining at 1:30. Mouse IgM mAb IIH6C4 specific for α-DG (Ervasti and Campbell, 1991) (Upstate, Inc.) was used for immunostaining at 1:200 and immunoblotting at 1:300. Function blocking antibodies for α6- and β1-integrins were used at 10 and 50 μg/ml, respectively (PharMingen). The anti-α6 integrin antibody was later tested at 100 μg/ml, and produced the same result.
Cy5-, FITC- or Rhodamine-conjugated, affinity-absorbed antibodies specific for mouse, rat or rabbit IgG and mouse IgM (Amersham Pharmacia; Chemicon; Caltag) were used at a 1:50 dilution. HRP-conjugated antibodies specific for mouse IgG (Amersham Pharmacia) and mouse IgM (Sigma) were used for western blots at 1:2000 and 1:3000, respectively.
Supplementary Material
{kind=link}
Acknowledgments
The authors gratefully acknowledge Ken Yamada, Jimmie Fata, Masahiko Itoh and Peter Yurchenco for helpful discussions and Patrick Hardy, Yoko Itahana, Jae-Young Seo, Sara Cohen, Armin Akhavan and Hui Zhang for expert technical assistance. We thank Garry Nolan and Lee Opresko for providing the pBMN-IRES-PURO vector with modified cloning site and Phoenix-ECO retroviral packaging cells. This work was supported by NIH Grant R01 CA10957-01 and California Breast Cancer Research Program Grant 7KB-0017A (to J.M.). K.P.C. is an investigator of the Howard Hughes Medical Institute. M.J.B. is a recipient of an Innovator award from the Department of Defense Breast Cancer Research Program and a Distinguished Medical Science Fellow of the Office of Biological and Environmental Research of the Department of Energy.
Footnotes
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/119/19/4047/DC1
References
- Aumailley M, Bruckner-Tuderman L, Carter WG, Deutzmann R, Edgar D, Ekblom P, Engel J, Engvall E, Hohenester E, Jones JC, et al. A simplified laminin nomenclature. Matrix Biol. 2005;24:326–332. doi: 10.1016/j.matbio.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Cohn RD, Henry MD, Michele DE, Barresi R, Saito F, Moore SA, Flanagan JD, Skwarchuk MW, Robbins ME, Mendell JR, et al. Disruption of DAG1 in differentiated skeletal muscle reveals a role for dystroglycan in muscle regeneration. Cell. 2002;110:639–648. doi: 10.1016/s0092-8674(02)00907-8. [DOI] [PubMed] [Google Scholar]
- Colognato H, Winkelmann DA, Yurchenco PD. Laminin polymerization induces a receptor-cytoskeleton network. J Cell Biol. 1999;145:619–631. doi: 10.1083/jcb.145.3.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng WM, Schneider M, Frock R, Castillejo-Lopez C, Gaman EA, Baumgartner S, Ruohola-Baker H. Dystroglycan is required for polarizing the epithelial cells and the oocyte in Drosophila. Development. 2003;130:173–184. doi: 10.1242/dev.00199. [DOI] [PubMed] [Google Scholar]
- Driss A, Charrier L, Yan Y, Nduati V, Sitaraman S, Merlin D. Dystroglycan receptor is involved in integrin activation in intestinal epithelia. Am J Physiol Gastrointest Liver Physiol. 2005;290:G1228–G1242. doi: 10.1152/ajpgi.00378.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbeej M, Campbell KP. Muscular dystrophies involving the dystrophin-glycoprotein complex: an overview of current mouse models. Curr Opin Genet Dev. 2002;12:349–361. doi: 10.1016/s0959-437x(02)00309-x. [DOI] [PubMed] [Google Scholar]
- Durbeej M, Larsson E, Ibraghimov BO, Roberds SL, Campbell KP, Ekblom P. Non-muscle alpha-dystroglycan is involved in epithelial development. J Cell Biol. 1995;130:79–91. doi: 10.1083/jcb.130.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbeej M, Henry MD, Ferletta M, Campbell KP, Ekblom P. Distribution of dystroglycan in normal adult mouse tissues. J Histochem Cytochem. 1998;46:449–457. doi: 10.1177/002215549804600404. [DOI] [PubMed] [Google Scholar]
- Durbeej M, Talts JF, Henry MD, Yurchenco PD, Campbell KP, Ekblom P. Dystroglycan binding to laminin alpha1LG4 module influences epithelial morphogenesis of salivary gland and lung in vitro. Differentiation. 2001;69:121–134. doi: 10.1046/j.1432-0436.2001.690206.x. [DOI] [PubMed] [Google Scholar]
- Ervasti JM, Campbell KP. Membrane organization of the dystrophin-glycoprotein complex. Cell. 1991;66:1121–1131. doi: 10.1016/0092-8674(91)90035-w. [DOI] [PubMed] [Google Scholar]
- Ervasti JM, Campbell KP. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol. 1993;122:809–823. doi: 10.1083/jcb.122.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraldo MM, Deugnier MA, Lukashev M, Thiery JP, Glukhova MA. Perturbation of beta1-integrin function alters the development of murine mammary gland. EMBO J. 1998;17:2139–2147. doi: 10.1093/emboj/17.8.2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee SH, Blacher RW, Douville PJ, Provost PR, Yurchenco PD, Carbonetto S. Laminin-binding protein 120 from brain is closely related to the dystrophin-associated glycoprotein, dystroglycan, and binds with high affinity to the major heparin binding domain of laminin. J Biol Chem. 1993;268:14972–14980. [PubMed] [Google Scholar]
- Gudjonsson T, Ronnov-Jessen L, Villadsen R, Rank F, Bissell MJ, Petersen OW. Normal and tumor-derived myoepithelial cells differ in their ability to interact with luminal breast epithelial cells for polarity and basement membrane deposition. J Cell Sci. 2002;115:39–50. doi: 10.1242/jcs.115.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry MD, Campbell KP. A role for dystroglycan in basement membrane assembly. Cell. 1998;95:859–870. doi: 10.1016/s0092-8674(00)81708-0. [DOI] [PubMed] [Google Scholar]
- Henry MD, Cohen MB, Campbell KP. Reduced expression of dystroglycan in breast and prostate cancer. Hum Pathol. 2001a;32:791–795. doi: 10.1053/hupa.2001.26468. [DOI] [PubMed] [Google Scholar]
- Henry MD, Satz JS, Brakebusch C, Costell M, Gustafsson E, Fassler R, Campbell KP. Distinct roles for dystroglycan, β1 integrin and perlecan in cell surface laminin organization. J Cell Sci. 2001b;114:1137–1144. doi: 10.1242/jcs.114.6.1137. [DOI] [PubMed] [Google Scholar]
- Ibraghimov-Beskrovnaya O, Ervasti JM, Leveille CJ, Slaughter CA, Sernett SW, Campbell KP. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature. 1992;355:696–702. doi: 10.1038/355696a0. [DOI] [PubMed] [Google Scholar]
- Ilsley JL, Sudol M, Winder SJ. The WW domain: linking cell signalling to the membrane cytoskeleton. Cell Signal. 2002;14:183–189. doi: 10.1016/s0898-6568(01)00236-4. [DOI] [PubMed] [Google Scholar]
- Kaetzel CS, Ray DB. Immunochemical characterization with monoclonal antibodies of three major caseins and alpha-lactalbumin from rat milk. J Dairy Sci. 1984;67:64–75. doi: 10.3168/jds.S0022-0302(84)81267-9. [DOI] [PubMed] [Google Scholar]
- Kemler R, Brulet P, Schnebelen MT, Gaillard J, Jacob F. Reactivity of monoclonal antibodies against intermediate filament proteins during embryonic development. J Embryol Exp Morphol. 1981;64:45–60. [PubMed] [Google Scholar]
- Kinoshita S, Su L, Amano M, Timmerman LA, Kaneshima H, Nolan GP. The T cell activation factor NF-ATc positively regulates HIV-1 replication and gene expression in T cells. Immunity. 1997;6:235–244. doi: 10.1016/s1074-7613(00)80326-x. [DOI] [PubMed] [Google Scholar]
- Kittrell FS, Oborn CJ, Medina D. Development of mammary preneoplasias in vivo from mouse mammary epithelial cell lines in vitro. Cancer Res. 1992;52:1924–1932. [PubMed] [Google Scholar]
- Li S, Harrison D, Carbonetto S, Fassler R, Smyth N, Edgar D, Yurchenco PD. Matrix assembly, regulation, and survival functions of laminin and its receptors in embryonic stem cell differentiation. J Cell Biol. 2002;157:1279–1290. doi: 10.1083/jcb.200203073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Edgar D, Fassler R, Wadsworth W, Yurchenco PD. The role of laminin in embryonic cell polarization and tissue organization. Dev Cell. 2003;4:613–624. doi: 10.1016/s1534-5807(03)00128-x. [DOI] [PubMed] [Google Scholar]
- Li S, Liquari P, McKee KK, Harrison D, Patel R, Lee S, Yurchenco PD. Laminin-sulfatide binding initiates basement membrane assembly and enables receptor signaling in Schwann cells and fibroblasts. J Cell Biol. 2005;169:179–189. doi: 10.1083/jcb.200501098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohikangas L, Gullberg D, Johansson S. Assembly of laminin polymers is dependent on beta1-integrins. Exp Cell Res. 2001;265:135–144. doi: 10.1006/excr.2001.5170. [DOI] [PubMed] [Google Scholar]
- Losasso C, Di Tommaso F, Sgambato A, Ardito R, Cittadini A, Giardina B, Petrucci TC, Brancaccio A. Anomalous dystroglycan in carcinoma cell lines. FEBS Lett. 2000;484:194–198. doi: 10.1016/s0014-5793(00)02157-8. [DOI] [PubMed] [Google Scholar]
- Michele DE, Campbell KP. Dystrophin-glycoprotein complex: post-translational processing and dystroglycan function. J Biol Chem. 2003;278:15457–15460. doi: 10.1074/jbc.R200031200. [DOI] [PubMed] [Google Scholar]
- Michele DE, Barresi R, Kanagawa M, Saito F, Cohn RD, Satz JS, Dollar J, Nishino I, Kelley RI, Somer H, et al. Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature. 2002;418:417–422. doi: 10.1038/nature00837. [DOI] [PubMed] [Google Scholar]
- Miner JH, Yurchenco PD. Laminin functions in tissue morphogenesis. Annu Rev Cell Dev Biol. 2004;20:255–284. doi: 10.1146/annurev.cellbio.20.010403.094555. [DOI] [PubMed] [Google Scholar]
- Moore SA, Saito F, Chen J, Michele DE, Henry MD, Messing A, Cohn RD, Ross-Barta SE, Westra S, Williamson RA, et al. Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature. 2002;418:422–425. doi: 10.1038/nature00838. [DOI] [PubMed] [Google Scholar]
- Moss ML, Jin SL, Milla ME, Bickett DM, Burkhart W, Carter HL, Chen WJ, Clay WC, Didsbury JR, Hassler D, et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature. 1997;385:733–736. doi: 10.1038/385733a0. [DOI] [PubMed] [Google Scholar]
- Muschler J, Lochter A, Roskelley CD, Yurchenco P, Bissell MJ. Division of labor among the alpha6beta4 integrin, beta1 integrins, and an E3 laminin receptor to signal morphogenesis and beta-casein expression in mammary epithelial cells. Mol Biol Cell. 1999;10:2817–2828. doi: 10.1091/mbc.10.9.2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muschler J, Levy D, Boudreau R, Henry M, Campbell K, Bissell MJ. A role for dystroglycan in epithelial polarization: loss of function in breast tumor cells. Cancer Res. 2002;62:7102–7109. [PubMed] [Google Scholar]
- Naylor MJ, Li N, Cheung J, Lowe ET, Lambert E, Marlow R, Wang P, Schatzmann F, Wintermantel T, Schuetz G, et al. Ablation of beta1 integrin in mammary epithelium reveals a key role for integrin in glandular morphogenesis and differentiation. J Cell Biol. 2005;171:717–728. doi: 10.1083/jcb.200503144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patarroyo M, Tryggvason K, Virtanen I. Laminin isoforms in tumor invasion, angiogenesis and metastasis. Semin Cancer Biol. 2002;12:197–207. doi: 10.1016/S1044-579X(02)00023-8. [DOI] [PubMed] [Google Scholar]
- Pawson T. Specificity in signal transduction: from phosphotyrosine-SH2 domain interactions to complex cellular systems. Cell. 2004;116:191–203. doi: 10.1016/s0092-8674(03)01077-8. [DOI] [PubMed] [Google Scholar]
- Peterson GL. A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal Biochem. 1977;83:346–356. doi: 10.1016/0003-2697(77)90043-4. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor: Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- Scheele S, Falk M, Franzen A, Ellin F, Ferletta M, Lonaio P, Andersson B, Timpl R, Forsberg E, Ekblom P. Laminin alpha1 globular domains 4–5 induce fetal development but are not vital for embryonic basement membrane assembly. Proc Natl Acad Sci USA. 2005;102:1502–1506. doi: 10.1073/pnas.0405095102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgambato A, Migaldi M, Montanari M, Camerini A, Brancaccio A, Rossi G, Cangiano R, Losasso C, Capelli G, Trentini GP, et al. Dystroglycan expression is frequently reduced in human breast and colon cancers and is associated with tumor progression. Am J Pathol. 2003;162:849–860. doi: 10.1016/S0002-9440(10)63881-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgambato A, Di Salvatore MA, De Paola B, Rettino A, Faraglia B, Boninsegna A, Graziani C, Camerini A, Proietti G, Cittadini A. Analysis of dystroglycan regulation and functions in mouse mammary epithelial cells and implications for mammary tumorigenesis. J Cell Physiol. 2006;207:520–529. doi: 10.1002/jcp.20600. [DOI] [PubMed] [Google Scholar]
- Singh J, Itahana Y, Knight-Krajewski S, Kanagawa M, Campbell KP, Bissell MJ, Muschler J. Proteolytic enzymes and altered glycosylation modulate dystroglycan function in carcinoma cells. Cancer Res. 2004;64:6152–6159. doi: 10.1158/0008-5472.CAN-04-1638. [DOI] [PubMed] [Google Scholar]
- Slade MJ, Coope RC, Gomm JJ, Coombes RC. The human mammary gland basement membrane is integral to the polarity of luminal epithelial cells. Exp Cell Res. 1999;247:267–278. doi: 10.1006/excr.1998.4340. [DOI] [PubMed] [Google Scholar]
- Sotgia F, Lee H, Bedford MT, Petrucci T, Sudol M, Lisanti MP. Tyrosine phosphorylation of beta-dystroglycan at its WW domain binding motif, PPxY, recruits SH2 domain containing proteins. Biochemistry. 2001;40:14585–14592. doi: 10.1021/bi011247r. [DOI] [PubMed] [Google Scholar]
- Streuli CH, Bailey N, Bissell MJ. Control of mammary epithelial differentiation: basement membrane induces tissue-specific gene expression in the absence of cell-cell interaction and morphological polarity. J Cell Biol. 1991;115:1383–1395. doi: 10.1083/jcb.115.5.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streuli CH, Schmidhauser C, Bailey N, Yurchenco P, Skubitz AP, Roskelley C, Bissell MJ. Laminin mediates tissue-specific gene expression in mammary epithelia. J Cell Biol. 1995;129:591–603. doi: 10.1083/jcb.129.3.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver VM, Petersen OW, Wang F, Larabell CA, Briand P, Damsky C, Bissell MJ. Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J Cell Biol. 1997;137:231–245. doi: 10.1083/jcb.137.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir ML, Muschler J. Dystroglycan: emerging roles in mammary gland function. J Mammary Gland Biol Neoplasia. 2003;8:409–419. doi: 10.1023/B:JOMG.0000017428.38034.a7. [DOI] [PubMed] [Google Scholar]
- Williamson RA, Henry MD, Daniels KJ, Hrstka RF, Lee JC, Sunada Y, Ibraghimov BO, Campbell KP. Dystroglycan is essential for early embryonic development: disruption of Reichert’s membrane in Dag1-null mice. Hum Mol Genet. 1997;6:831–841. doi: 10.1093/hmg/6.6.831. [DOI] [PubMed] [Google Scholar]
- Yang B, Jung D, Motto D, Meyer J, Koretzky G, Campbell KP. SH3 domain-mediated interaction of dystroglycan and Grb2. J Biol Chem. 1995;270:11711–11714. doi: 10.1074/jbc.270.20.11711. [DOI] [PubMed] [Google Scholar]
- Yeaman C, Grindstaff KK, Nelson WJ. New perspectives on mechanisms involved in generating epithelial cell polarity. Physiol Rev. 1999;79:73–98. doi: 10.1152/physrev.1999.79.1.73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.